
Synergistic Effect of β-Lapachone and Aminooxyacetic Acid on Central Metabolism in Breast Cancer

 ,
,  , ,
, ,
Abstract
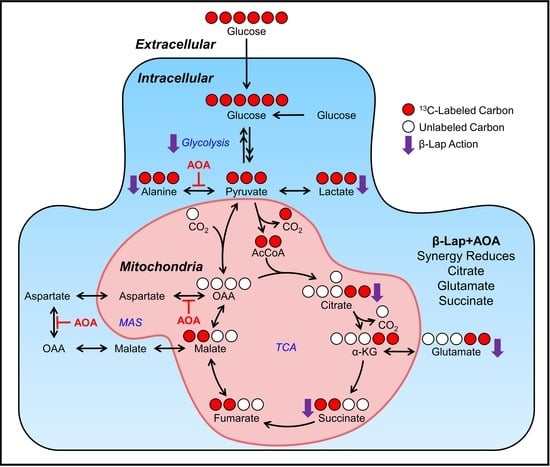
:
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Reagents
2.2. Cell Culture Experiment
2.3. Extraction of Metabolites and Sample Preparation for GC-MS Analysis
2.4. GC-MS Analysis
2.5. Data Processing, Isotopologue Analysis, and Flux Modeling Analysis
2.6. Multivariate Statistical Analysis
2.7. Coefficient of Drug Interaction
2.8. NQO1 Protein Expression Analysis
2.9. Cell Viability Assay
3. Results
3.1. Global Metabolomics
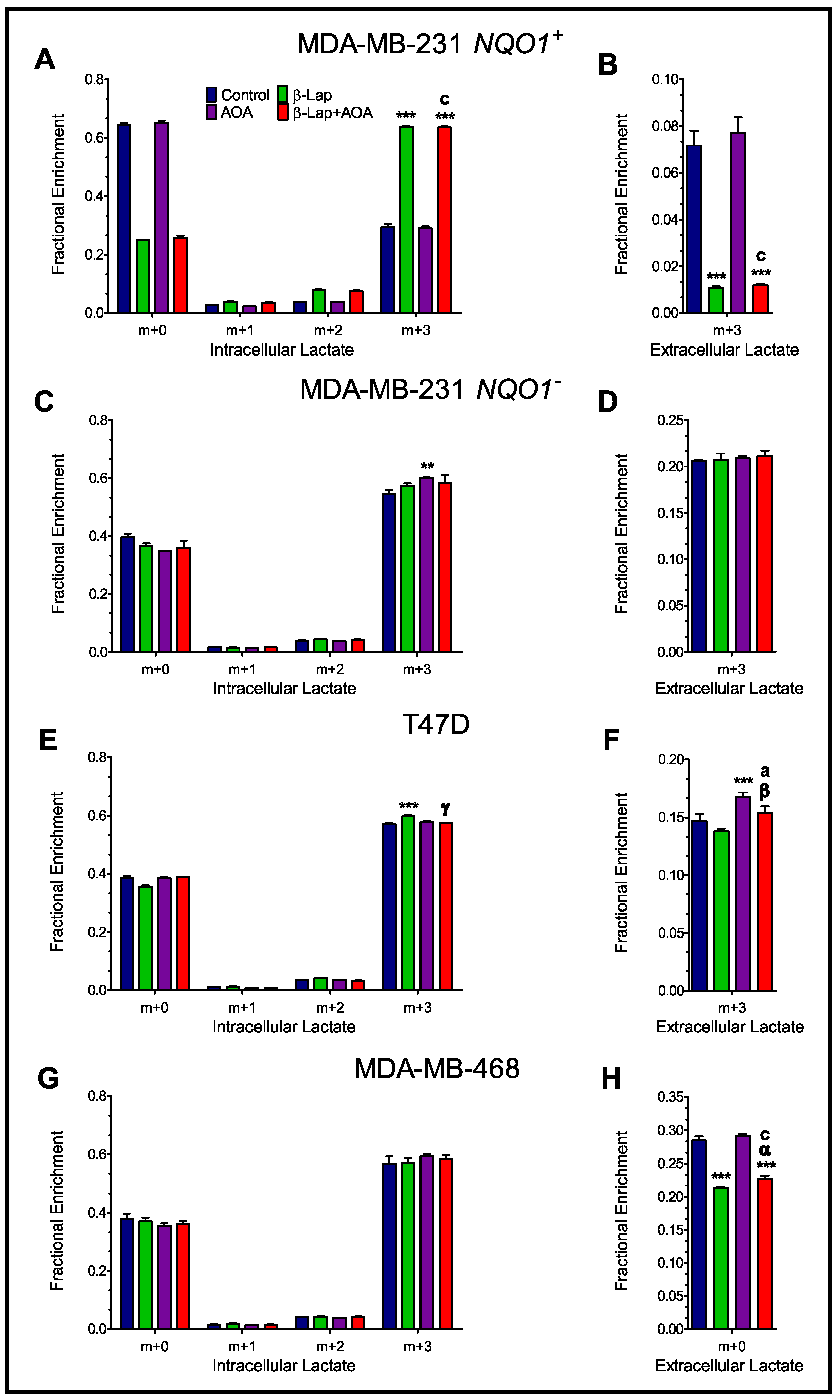
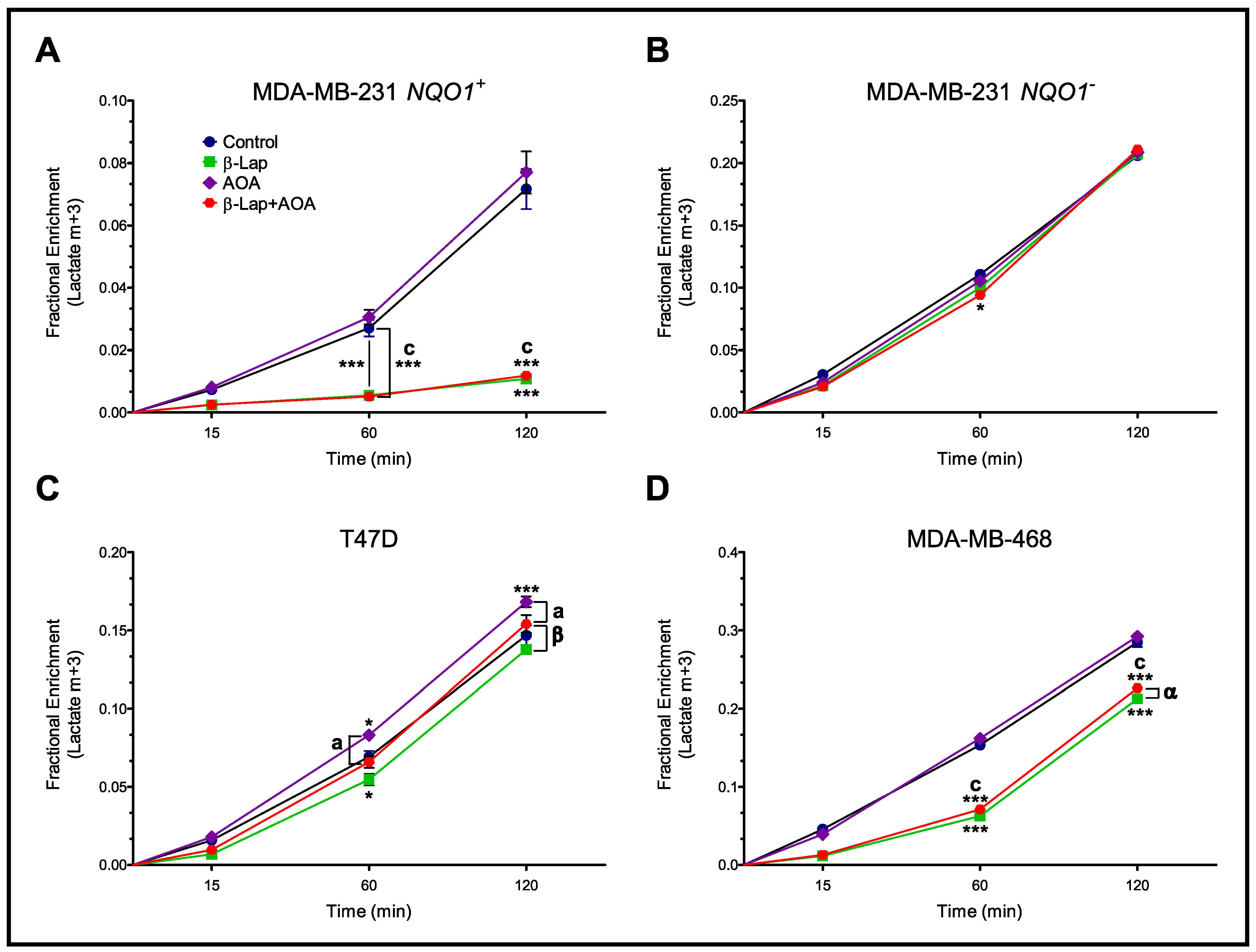
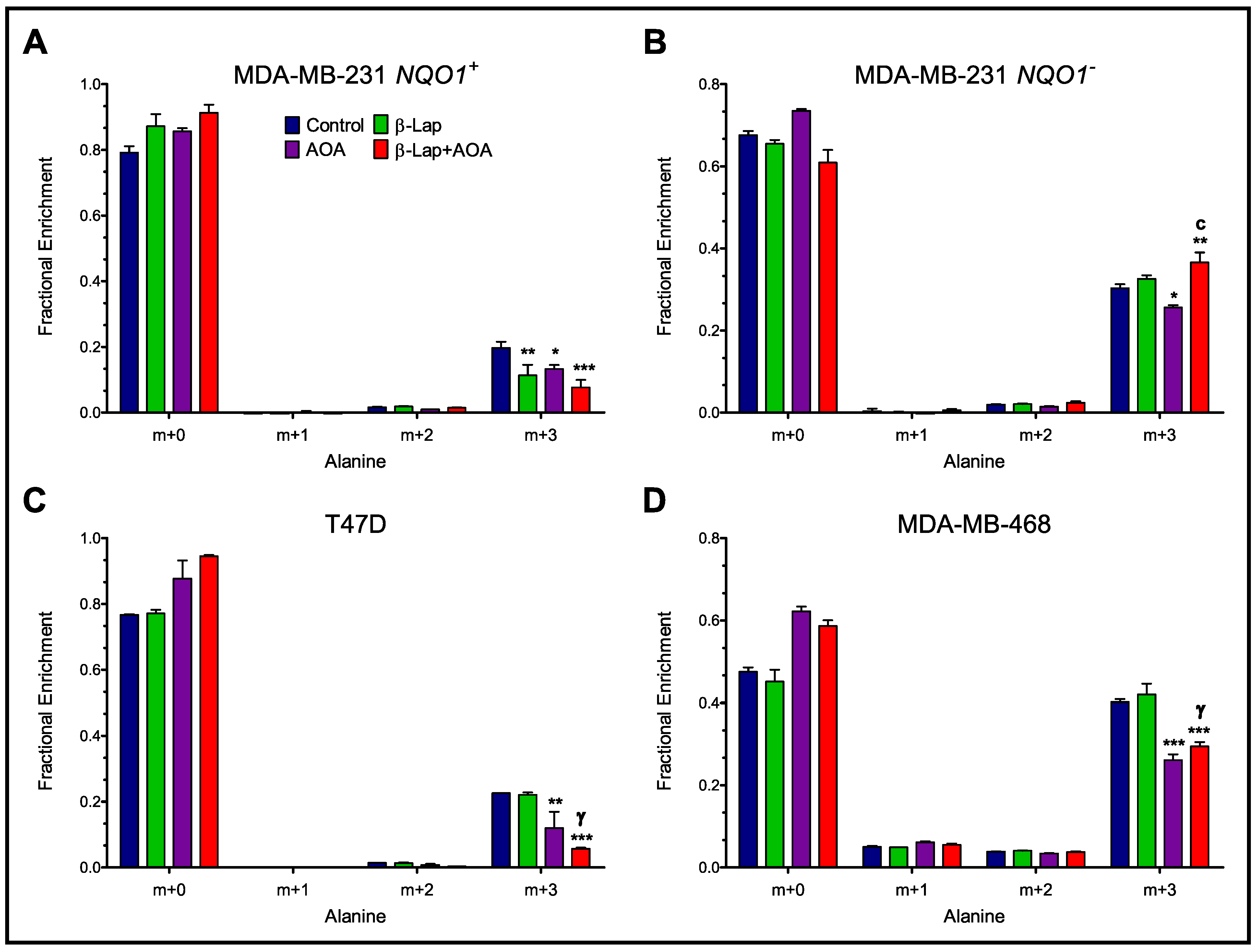
3.2. Fractional Enrichment of 13C-lactate and 13C-alanine Isotopologues
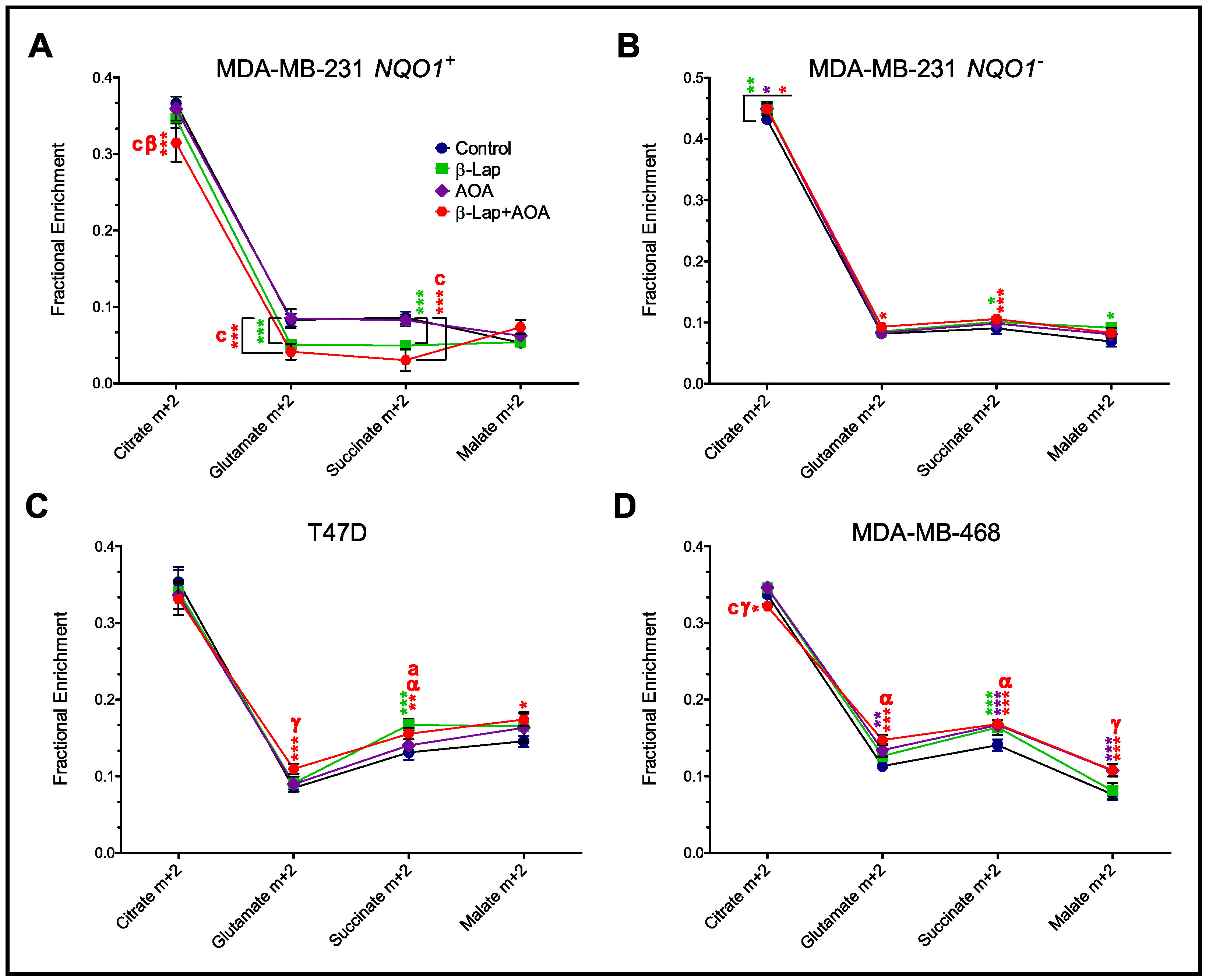
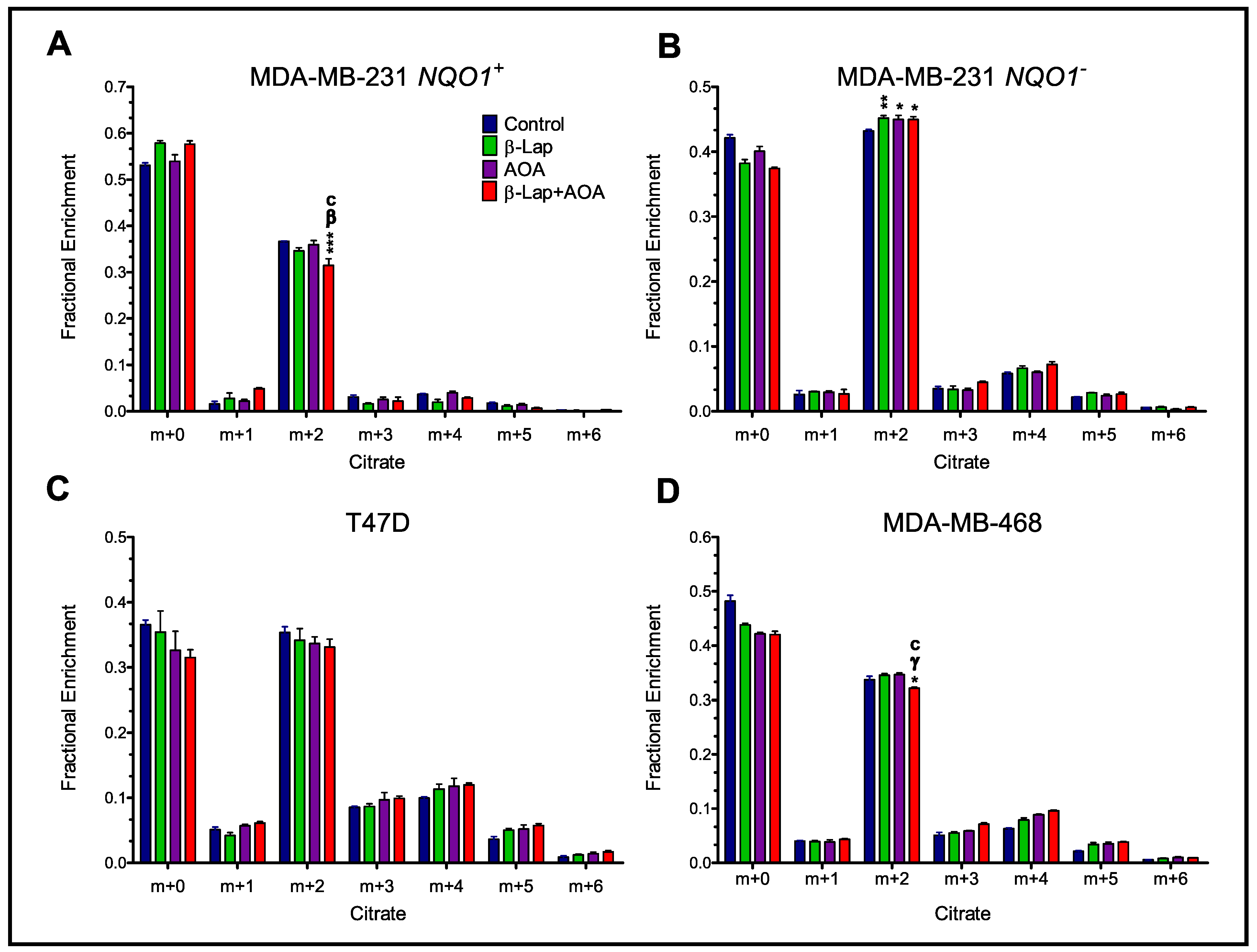
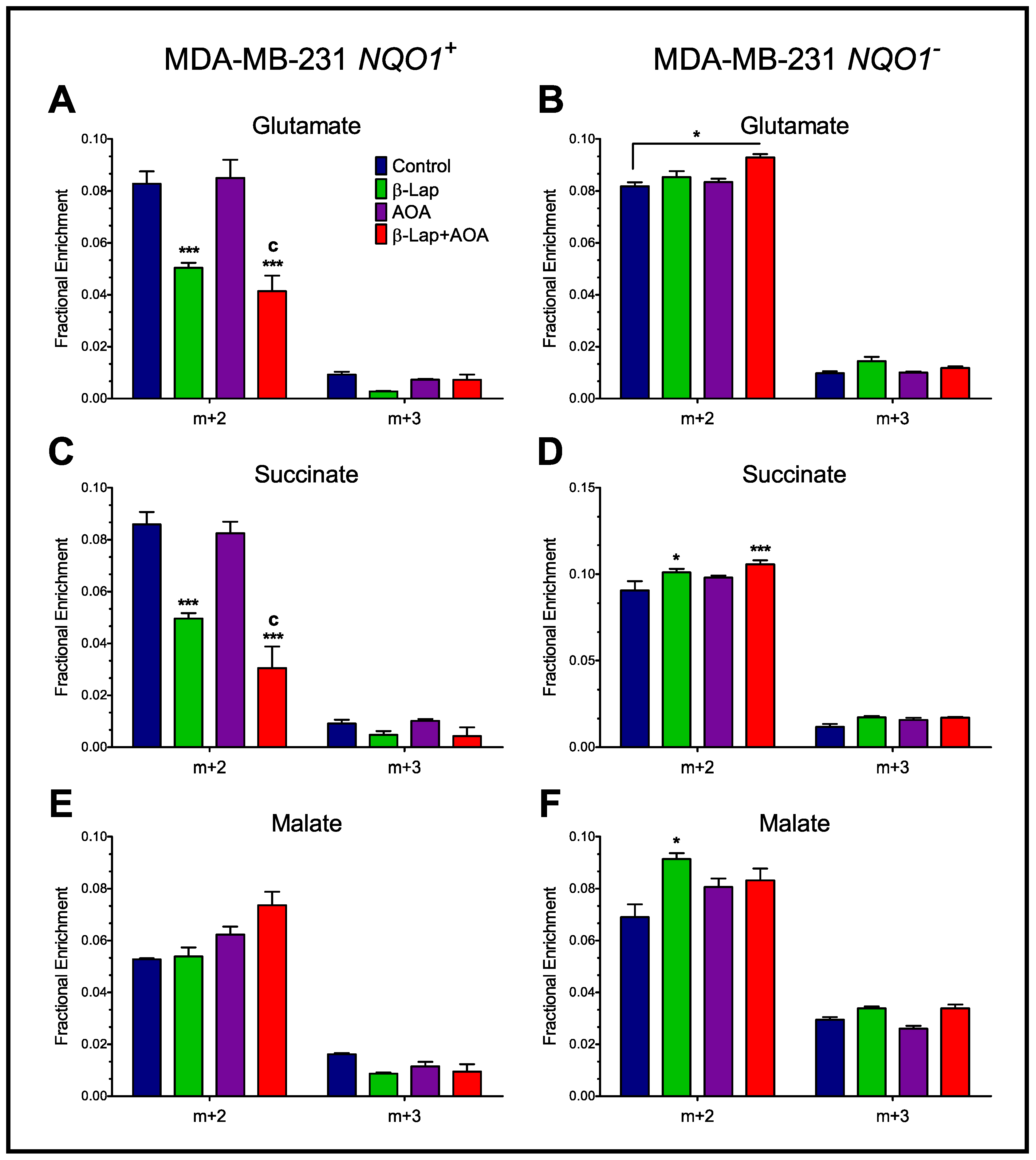
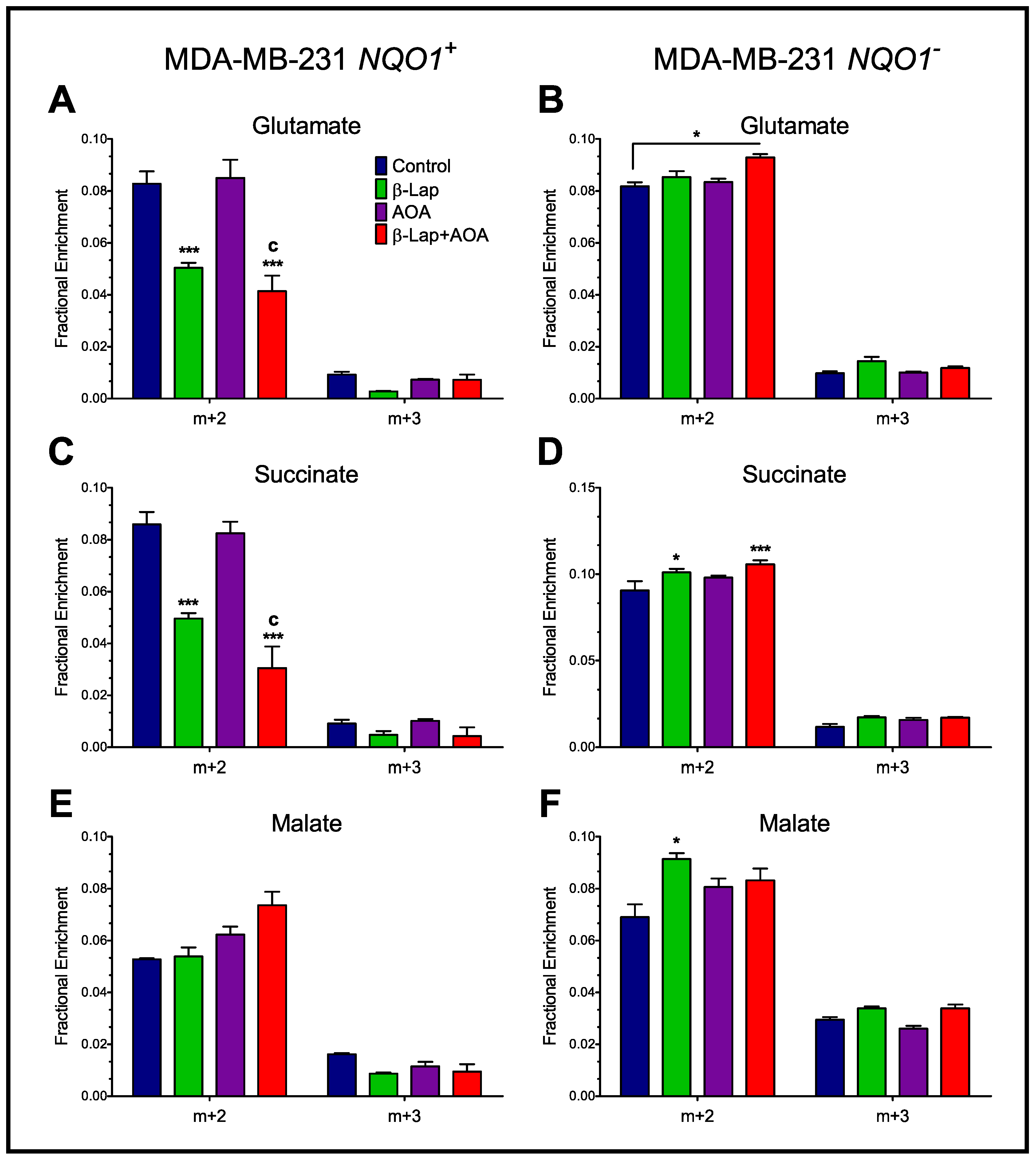
3.3. Fractional Enrichment of TCA Cycle Intermediates
3.4. Metabolic Flux Analysis
3.5. Coefficient of Drug Interaction Analysis
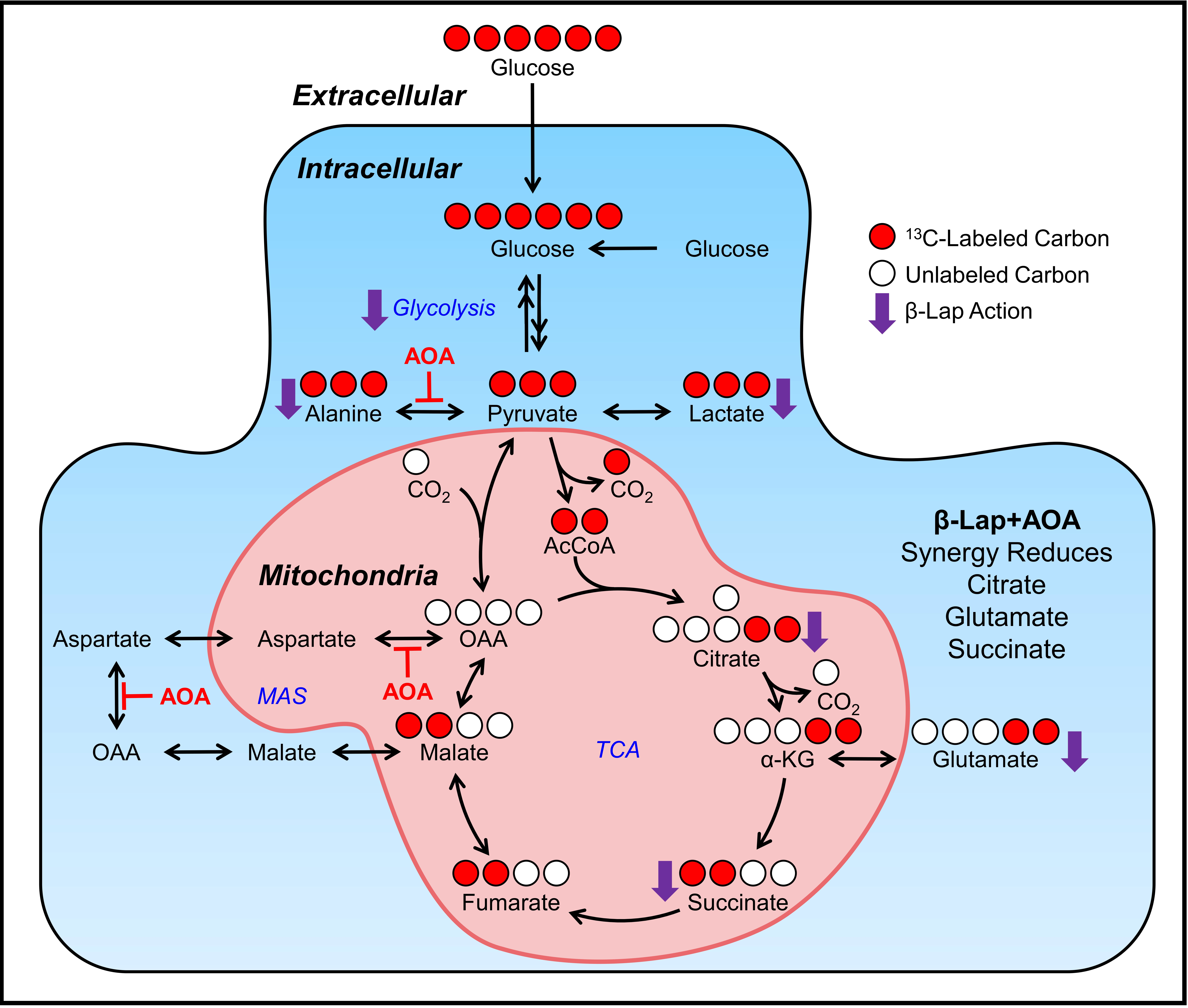
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kim, J.H.; Lee, S.M.; Myung, C.H.; Lee, K.R.; Hyun, S.M.; Lee, J.E.; Park, Y.S.; Jeon, S.R.; Park, J.I.; Chang, S.E.; et al. Melanogenesis Inhibition of β-Lapachone, a Natural Product from Tabebuia Avellanedae, with Effective in Vivo Lightening Potency. Arch. Dermatol. Res. 2015, 307, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Gómez Castellanos, J.R.; Prieto, J.M.; Heinrich, M. Red Lapacho (Tabebuia Impetiginosa)—A Global Ethnopharmacological Commodity? J. Ethnopharmacol. 2009, 121, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Guiraud, P.; Steiman, R.; Campos-Takaki, G.-M.; Seigle-Murandi, F.; de Buochberg, M. Comparison of Antibacterial and Antifungal Activities of Lapachol and β-Lapachone. Planta Med. 1994, 60, 373–374. [Google Scholar] [CrossRef] [PubMed]
- Mokarizadeh, N.; Karimi, P.; Kazemzadeh, H.; Fathi Maroufi, N.; Sadigh-Eteghad, S.; Nikanfar, S.; Rashtchizadeh, N. An Evaluation on Potential Anti-Inflammatory Effects of β-Lapachone. Int. Immunopharmacol. 2020, 87, 106810. [Google Scholar] [CrossRef]
- de, M.H.; Choi, H.M.; Hahm, D.-H.; Her, E.; Yang, H.-I.; Yoo, M.C.; Kim, K.S. Analgesic and Anti-Inflammatory Effects in Animal Models of an Ethanolic Extract of Taheebo, the Inner Bark of Tabebuia Avellanedae. Mol. Med. Rep. 2012, 6, 791–796. [Google Scholar] [CrossRef] [Green Version]
- Macedo, L.; Fernandes, T.; Silveira, L.; Mesquita, A.; Franchitti, A.A.; Ximenes, E.A. β-Lapachone Activity in Synergy with Conventional Antimicrobials against Methicillin Resistant Staphylococcus Aureus Strains. Phytomedicine 2013, 21, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.; Ban, J.-J.; Chung, J.-Y.; Im, W.; Kim, M. Amelioration of Huntington’s Disease Phenotypes by Beta-Lapachone Is Associated with Increases in Sirt1 Expression, CREB Phosphorylation and PGC-1α Deacetylation. PLoS ONE 2018, 13, e0195968. [Google Scholar] [CrossRef] [Green Version]
- Pink, J.J.; Planchon, S.M.; Tagliarino, C.; Varnes, M.E.; Siegel, D.; Boothman, D.A. NAD(P)H:Quinone Oxidoreductase Activity Is the Principal Determinant of β-Lapachone Cytotoxicity. J. Biol. Chem. 2000, 275, 5416–5424. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Park, A.H.; Lee, S.-H.; Lee, S.-H.; Kim, J.-H.; Yang, S.-J.; Yeom, Y.I.; Kwak, T.H.; Lee, D.; Lee, S.-J.; et al. Beta-Lapachone, a Modulator of NAD Metabolism, Prevents Health Declines in Aged Mice. PLoS ONE 2012, 7, e47122. [Google Scholar] [CrossRef]
- Moon, S.-J.; Jeong, J.Y.; Kim, J.-H.; Choi, D.-H.; Choi, H.; Chang, Y.-K.; Na, K.R.; Lee, K.W.; Lee, C.-H.; Choi, D.E.; et al. The Potential Roles of NAD(P)H:Quinone Oxidoreductase 1 in the Development of Diabetic Nephropathy and Actin Polymerization. Sci. Rep. 2020, 10, 17735. [Google Scholar] [CrossRef]
- Lu, C.Y. β-Lapachone Ameliorates Murine Cisplatin Nephrotoxicity: NAD+, NQO1, and SIRT1 at the Crossroads of Metabolism, Injury, and Inflammation. Kidney Int. 2014, 85, 496–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sitônio, M.M.; de Carvalho Júnior, C.H.; Campos, I.D.A.; Silva, J.B.N.F.; de Lima, M.D.C.A.; Góes, A.J.S.; Maia, M.B.S.; Rolim Neto, P.J.; Silva, T.G. Anti-Inflammatory and Anti-Arthritic Activities of 3,4-Dihydro-2,2-Dimethyl-2H-Naphthol[1,2-b]Pyran-5,6-Dione (β-Lapachone). Inflamm. Res. 2013, 62, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Kung, H.-N.; Yang, M.-J.; Chang, C.-F.; Chau, Y.-P.; Lu, K.-S. In Vitro and in Vivo Wound Healing-Promoting Activities of Beta-Lapachone. Am. J. Physiol. Cell Physiol. 2008, 295, C931–C943. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, G.; Silvers, M.A.; Ilcheva, M.; Liu, Y.; Moore, Z.R.; Luo, X.; Gao, J.; Anderson, G.; Liu, L.; Sarode, V.; et al. Tumor-Selective Use of DNA Base Excision Repair Inhibition in Pancreatic Cancer Using the NQO1 Bioactivatable Drug, β-Lapachone. Sci. Rep. 2015, 5, 17066. [Google Scholar] [CrossRef] [Green Version]
- Joseph, P.; Xie, T.; Xu, Y.; Jaiswal, A.K. NAD(P)H:Quinone Oxidoreductase1 (DT-Diaphorase): Expression, Regulation, and Role in Cancer. Oncol. Res. 1994, 6, 525–532. [Google Scholar]
- Belinsky, M.; Jaiswal, A.K. NAD(P)H:Quinone Oxidoreductase1 (DT-Diaphorase) Expression in Normal and Tumor Tissues. Cancer Metast. Rev. 1993, 12, 103–117. [Google Scholar] [CrossRef]
- Bey, E.A.; Bentle, M.S.; Reinicke, K.E.; Dong, Y.; Yang, C.-R.; Girard, L.; Minna, J.D.; Bornmann, W.G.; Gao, J.; Boothman, D.A. An NQO1- and PARP-1-Mediated Cell Death Pathway Induced in Non-Small-Cell Lung Cancer Cells by Beta-Lapachone. Proc. Natl. Acad. Sci. USA 2007, 104, 11832–11837. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Zhou, X.; Xu, M.; Piao, J.; Zhang, Y.; Lin, Z.; Chen, L. β-Lapachone Suppresses Tumour Progression by Inhibiting Epithelial-to-Mesenchymal Transition in NQO1-Positive Breast Cancers. Sci. Rep. 2017, 7, 2681. [Google Scholar] [CrossRef]
- Atia, A. Azman Abdullah NQO1 Enzyme and Its Role in Cellular Protection; an Insight. Iberoam. J. Med. 2020, 4, 306–313. [Google Scholar] [CrossRef]
- Ross, D.; Siegel, D. Functions of NQO1 in Cellular Protection and CoQ10 Metabolism and Its Potential Role as a Redox Sensitive Molecular Switch. Front. Physiol. 2017, 8, 595. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.M.; Hwang, S.W.; Wang, T.; Park, C.W.; Ryu, Y.; Jung, J.; Shin, J.H.; Kim, S.; Lee, J.L.; Kim, C.W.; et al. Increased Nicotinamide Adenine Dinucleotide Pool Promotes Colon Cancer Progression by Suppressing Reactive Oxygen Species Level. Cancer Sci. 2019, 110, 629–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silvers, M.A.; Deja, S.; Singh, N.; Egnatchik, R.A.; Sudderth, J.; Luo, X.; Beg, M.S.; Burgess, S.C.; DeBerardinis, R.J.; Boothman, D.A.; et al. The NQO1 Bioactivatable Drug, β-Lapachone, Alters the Redox State of NQO1+ Pancreatic Cancer Cells, Causing Perturbation in Central Carbon Metabolism. J. Biol. Chem. 2017, 292, 18203–18216. [Google Scholar] [CrossRef] [Green Version]
- Mahar, R.; Chang, M.C.; Merritt, M.E. Measuring NQO1 Bioactivation Using [2H7]Glucose. Cancers 2021, 13, 4165. [Google Scholar] [CrossRef]
- Moore, Z.; Chakrabarti, G.; Luo, X.; Ali, A.; Hu, Z.; Fattah, F.J.; Vemireddy, R.; DeBerardinis, R.J.; Brekken, R.A.; Boothman, D.A. NAMPT Inhibition Sensitizes Pancreatic Adenocarcinoma Cells to Tumor-Selective, PAR-Independent Metabolic Catastrophe and Cell Death Induced by β-Lapachone. Cell Death Dis. 2015, 6, e1599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.; Motea, E.A.; Moore, Z.R.; Yao, J.; Dong, Y.; Chakrabarti, G.; Kilgore, J.A.; Silvers, M.A.; Patidar, P.L.; Cholka, A.; et al. Leveraging an NQO1 Bioactivatable Drug for Tumor-Selective Use of Poly(ADP-Ribose) Polymerase Inhibitors. Cancer Cell 2016, 30, 940–952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerber, D.E.; Bisen, A.K.; Beg, M.S.; Frankel, A.E.; Fatunde, O.; Fattah, F.; Arriaga, Y.E.; Dowell, J.; Meek, C.; Bolluyt, J.D.; et al. Phase 1 Study of ARQ 761, a β-Lapachone Analog That Promotes NQO1-Mediated Programmed Cancer Cell Necrosis. JCO 2017, 35, 2517. [Google Scholar] [CrossRef]
- Gerber, D.E.; Beg, M.S.; Fattah, F.; Frankel, A.E.; Fatunde, O.; Arriaga, Y.; Dowell, J.E.; Bisen, A.; Leff, R.D.; Meek, C.C.; et al. Phase 1 Study of ARQ 761, a β-Lapachone Analogue That Promotes NQO1-Mediated Programmed Cancer Cell Necrosis. Br. J. Cancer 2018, 119, 928–936. [Google Scholar] [CrossRef] [Green Version]
- Su, X.; Wang, J.; Jiang, L.; Chen, Y.; Lu, T.; Mendonca, M.S.; Huang, X. PCNA Inhibition Enhances the Cytotoxicity of β-Lapachone in NQO1-Positive Cancer Cells by Augmentation of Oxidative Stress-Induced DNA Damage. Cancer Lett. 2021, 519, 304–314. [Google Scholar] [CrossRef]
- Chakrabarti, G.; Moore, Z.R.; Luo, X.; Ilcheva, M.; Ali, A.; Padanad, M.; Zhou, Y.; Xie, Y.; Burma, S.; Scaglioni, P.P.; et al. Targeting Glutamine Metabolism Sensitizes Pancreatic Cancer to PARP-Driven Metabolic Catastrophe Induced by ß-Lapachone. Cancer Metab. 2015, 3, 12. [Google Scholar] [CrossRef] [Green Version]
- Easlon, E.; Tsang, F.; Skinner, C.; Wang, C.; Lin, S.-J. The Malate-Aspartate NADH Shuttle Components Are Novel Metabolic Longevity Regulators Required for Calorie Restriction-Mediated Life Span Extension in Yeast. Genes Dev. 2008, 22, 931–944. [Google Scholar] [CrossRef] [Green Version]
- Thornburg, J.M.; Nelson, K.K.; Clem, B.F.; Lane, A.N.; Arumugam, S.; Simmons, A.; Eaton, J.W.; Telang, S.; Chesney, J. Targeting Aspartate Aminotransferase in Breast Cancer. Breast Cancer Res. 2008, 10, R84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, L.; Li, L.S.; Spruell, C.; Xiao, L.; Chakrabarti, G.; Bey, E.A.; Reinicke, K.E.; Srougi, M.C.; Moore, Z.; Dong, Y.; et al. Tumor-Selective, Futile Redox Cycle-Induced Bystander Effects Elicited by NQO1 Bioactivatable Radiosensitizing Drugs in Triple-Negative Breast Cancers. Antioxid. Redox Signal. 2014, 21, 237–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siderius, D. NIST Standard Reference Simulation Website—SRD 173 2017. Available online: https://data.nist.gov/od/id/FF429BC178798B3EE0431A570681E858232 (accessed on 14 September 2021).
- Young, J.D. INCA: A Computational Platform for Isotopically Non-Stationary Metabolic Flux Analysis. Bioinformatics 2014, 30, 1333–1335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahim, M.; Ragavan, M.; Deja, S.; Merritt, M.E.; Burgess, S.C.; Young, J.D. INCA 2.0: A Tool for Integrated, Dynamic Modeling of NMR- and MS-Based Isotopomer Measurements and Rigorous Metabolic Flux Analysis. Metab. Eng. 2022, 69, 275–285. [Google Scholar] [CrossRef]
- Pang, Z.; Chong, J.; Zhou, G.; de Lima Morais, D.A.; Chang, L.; Barrette, M.; Gauthier, C.; Jacques, P.-É.; Li, S.; Xia, J. MetaboAnalyst 5.0: Narrowing the Gap between Raw Spectra and Functional Insights. Nucleic Acids Res. 2021, 49, W388–W396. [Google Scholar] [CrossRef]
- Cantó, C.; Menzies, K.J.; Auwerx, J. NAD+ Metabolism and the Control of Energy Homeostasis: A Balancing Act between Mitochondria and the Nucleus. Cell Metab. 2015, 22, 31–53. [Google Scholar] [CrossRef] [Green Version]
- Xie, N.; Zhang, L.; Gao, W.; Huang, C.; Huber, P.E.; Zhou, X.; Li, C.; Shen, G.; Zou, B. NAD+ Metabolism: Pathophysiologic Mechanisms and Therapeutic Potential. Signal Transduct. Target. Ther. 2020, 5, 227. [Google Scholar] [CrossRef]
- Borst, P. The Malate–Aspartate Shuttle (Borst Cycle): How It Started and Developed into a Major Metabolic Pathway. IUBMB Life 2020, 72, 2241–2259. [Google Scholar] [CrossRef]
- Indiveri, C.; Krämer, R.; Palmieri, F. Reconstitution of the Malate/Aspartate Shuttle from Mitochondria. J. Biol. Chem. 1987, 262, 15979–15983. [Google Scholar] [CrossRef]
- Bremer, J.; Davis, E.J. Studies on the Active Transfer of Reducing Equivalents into Mitochondria via the Malate-Aspartate Shuttle. Biochim. Biophys. Acta (BBA) Bioenerg. 1975, 376, 387–397. [Google Scholar] [CrossRef]
- Amoedo, N.D.; Punzi, G.; Obre, E.; Lacombe, D.; De Grassi, A.; Pierri, C.L.; Rossignol, R. AGC1/2, the Mitochondrial Aspartate-Glutamate Carriers. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2016, 1863, 2394–2412. [Google Scholar] [CrossRef] [PubMed]
- Antoniewicz, M.R. A Guide to 13C Metabolic Flux Analysis for the Cancer Biologist. Exp. Mol. Med. 2018, 50, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandberg, T.E.; Long, C.P.; Gonzalez, J.E.; Feist, A.M.; Antoniewicz, M.R.; Palsson, B.O. Evolution of E. Coli on [U-13C]Glucose Reveals a Negligible Isotopic Influence on Metabolism and Physiology. PLoS ONE 2016, 11, e0151130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gheni, G.; Ogura, M.; Iwasaki, M.; Yokoi, N.; Minami, K.; Nakayama, Y.; Harada, K.; Hastoy, B.; Wu, X.; Takahashi, H.; et al. Glutamate Acts as a Key Signal Linking Glucose Metabolism to Incretin/CAMP Action to Amplify Insulin Secretion. Cell Rep. 2014, 9, 661–673. [Google Scholar] [CrossRef] [Green Version]
- Linder, S.J.; Mostoslavsky, R. Put Your Mark Where Your Damage Is: Acetyl-CoA Production by ACLY Promotes DNA Repair. Mol. Cell 2017, 67, 165–167. [Google Scholar] [CrossRef] [Green Version]
- Deja, S.; Crawford, P.A.; Burgess, S.C. Krebs Takes a Turn at Cell Differentiation. Cell Metab. 2022, 34, 658–660. [Google Scholar] [CrossRef]
- Mahar, R.; Zeng, H.; Giacalone, A.; Ragavan, M.; Mareci, T.H.; Merritt, M.E. Deuterated Water Imaging of the Rat Brain Following Metabolism of [ 2 H 7 ]Glucose. Magn. Reson. Med. 2021, 85, 3049–3059. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Metabolite | Isotopologue Fractional Enrichment (T/C) | CDI | Effect | ||
|---|---|---|---|---|---|---|
| β-Lap | AOA | β-Lap+AOA | ||||
| NQO1+ | Citrate | 0.94 ± 0.01 | 0.98 ± 0.01 | 0.86 ± 0.01 | 0.93 ± 0.04 | Decrease |
| Glutamate | 0.60 ± 0.00 | 1.03 ± 0.01 | 0.50 ± 0.01 | 0.80 ± 0.12 | Decrease | |
| Succinate | 0.58 ± 0.00 | 0.96 ± 0.00 | 0.35 ± 0.01 | 0.64 ± 0.18 | Decrease | |
| NQO1− | Citrate | 1.05 ± 0.00 | 1.04 ± 0.01 | 1.04 ± 0.00 | 0.96 ± 0.01 | Increase |
| Glutamate | 1.04 ± 0.00 | 1.02 ± 0.00 | 1.14 ± 0.00 | 1.07 ± 0.01 | Increase | |
| Succinate | 1.11 ± 0.00 | 1.08 ± 0.00 | 1.17 ± 0.00 | 0.97 ± 0.02 | Increase | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, M.C.; Mahar, R.; McLeod, M.A.; Giacalone, A.G.; Huang, X.; Boothman, D.A.; Merritt, M.E. Synergistic Effect of β-Lapachone and Aminooxyacetic Acid on Central Metabolism in Breast Cancer. Nutrients 2022, 14, 3020. https://doi.org/10.3390/nu14153020
Chang MC, Mahar R, McLeod MA, Giacalone AG, Huang X, Boothman DA, Merritt ME. Synergistic Effect of β-Lapachone and Aminooxyacetic Acid on Central Metabolism in Breast Cancer. Nutrients. 2022; 14(15):3020. https://doi.org/10.3390/nu14153020
Chicago/Turabian StyleChang, Mario C., Rohit Mahar, Marc A. McLeod, Anthony G. Giacalone, Xiumei Huang, David A. Boothman, and Matthew E. Merritt. 2022. "Synergistic Effect of β-Lapachone and Aminooxyacetic Acid on Central Metabolism in Breast Cancer" Nutrients 14, no. 15: 3020. https://doi.org/10.3390/nu14153020
APA StyleChang, M. C., Mahar, R., McLeod, M. A., Giacalone, A. G., Huang, X., Boothman, D. A., & Merritt, M. E. (2022). Synergistic Effect of β-Lapachone and Aminooxyacetic Acid on Central Metabolism in Breast Cancer. Nutrients, 14(15), 3020. https://doi.org/10.3390/nu14153020