
Vitamin D Derivatives in Acute Myeloid Leukemia: The Matter of Selecting the Right Targets


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
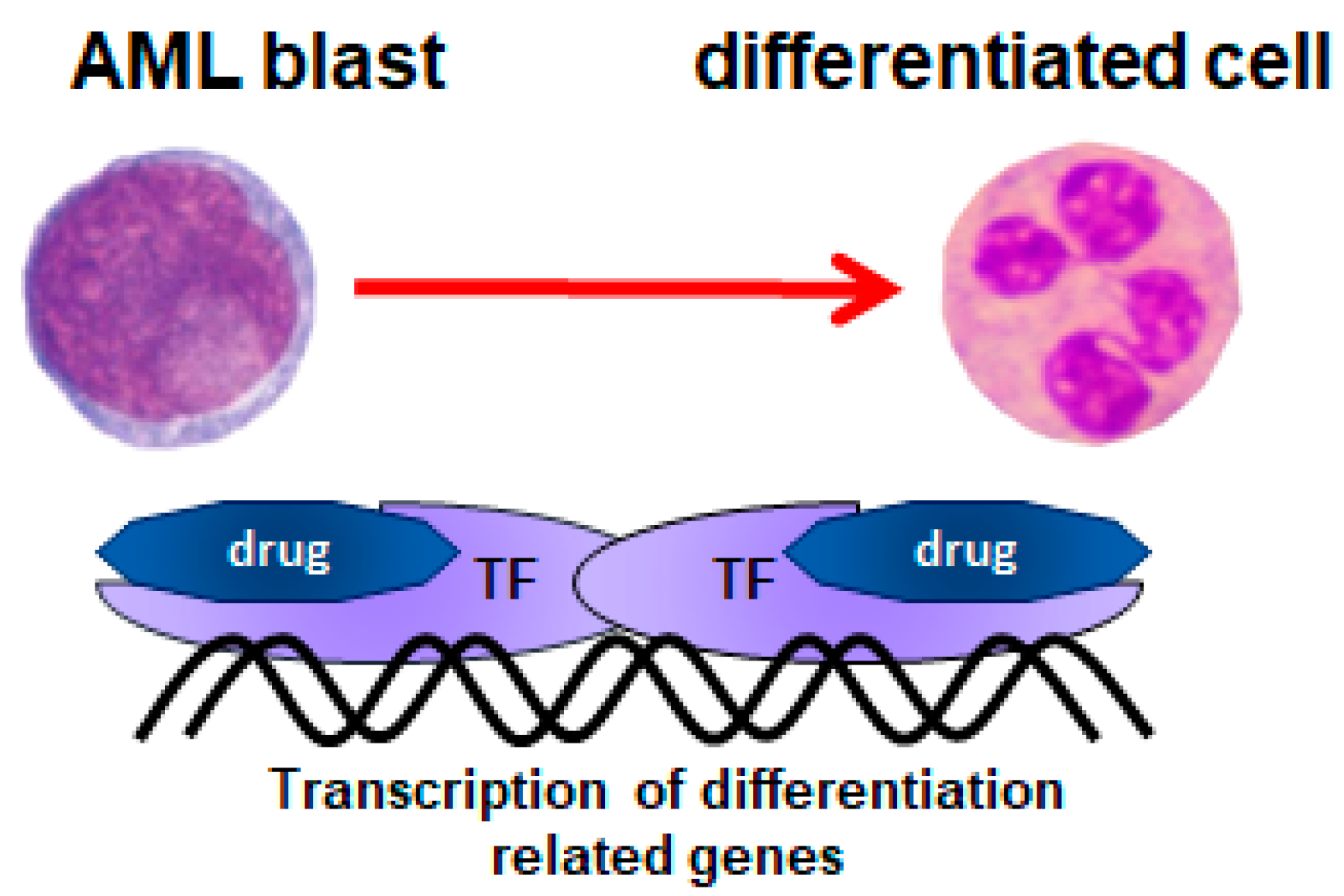
:1. Introduction
2. All-trans-Retinoic Acid (ATRA)
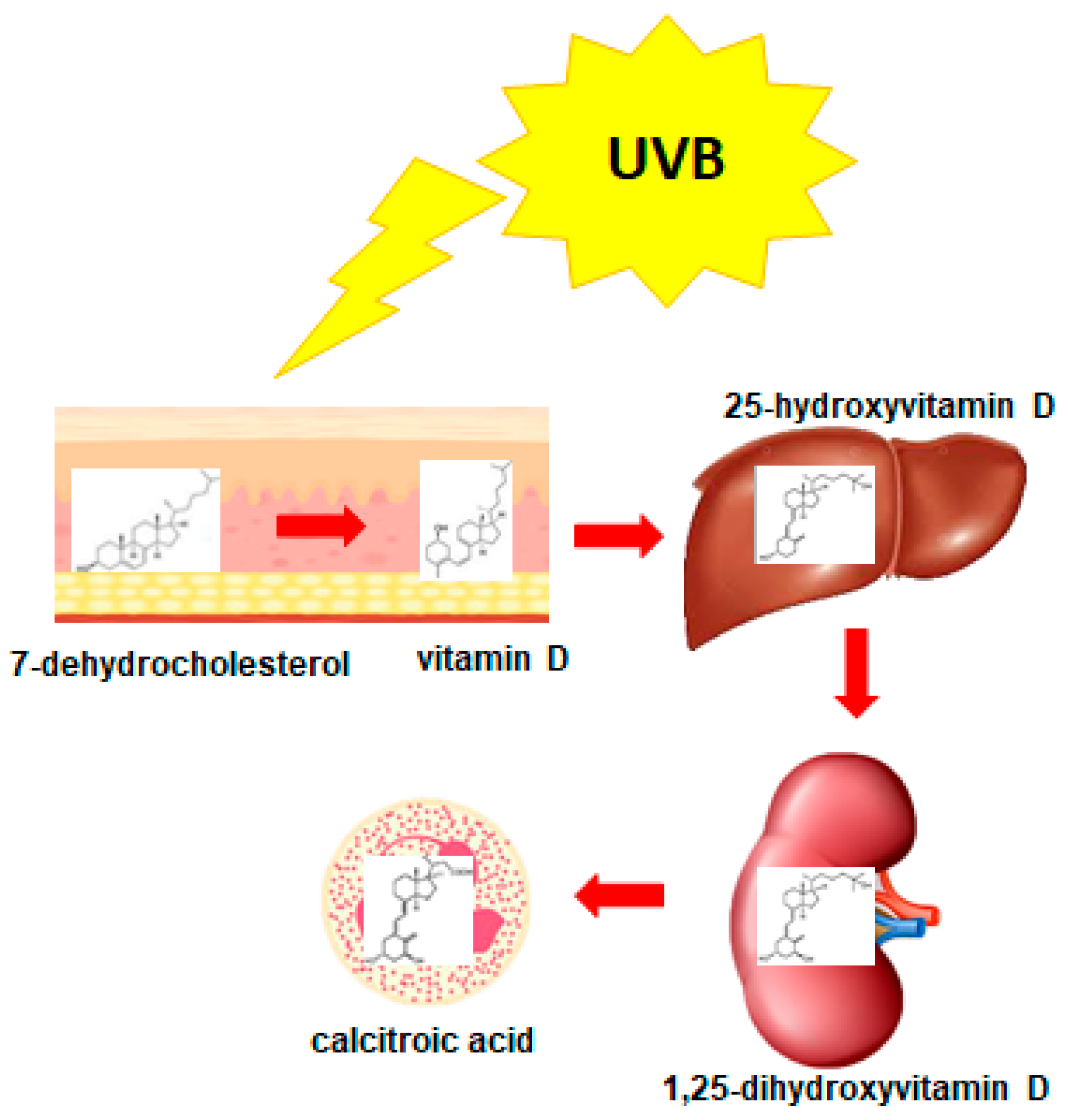
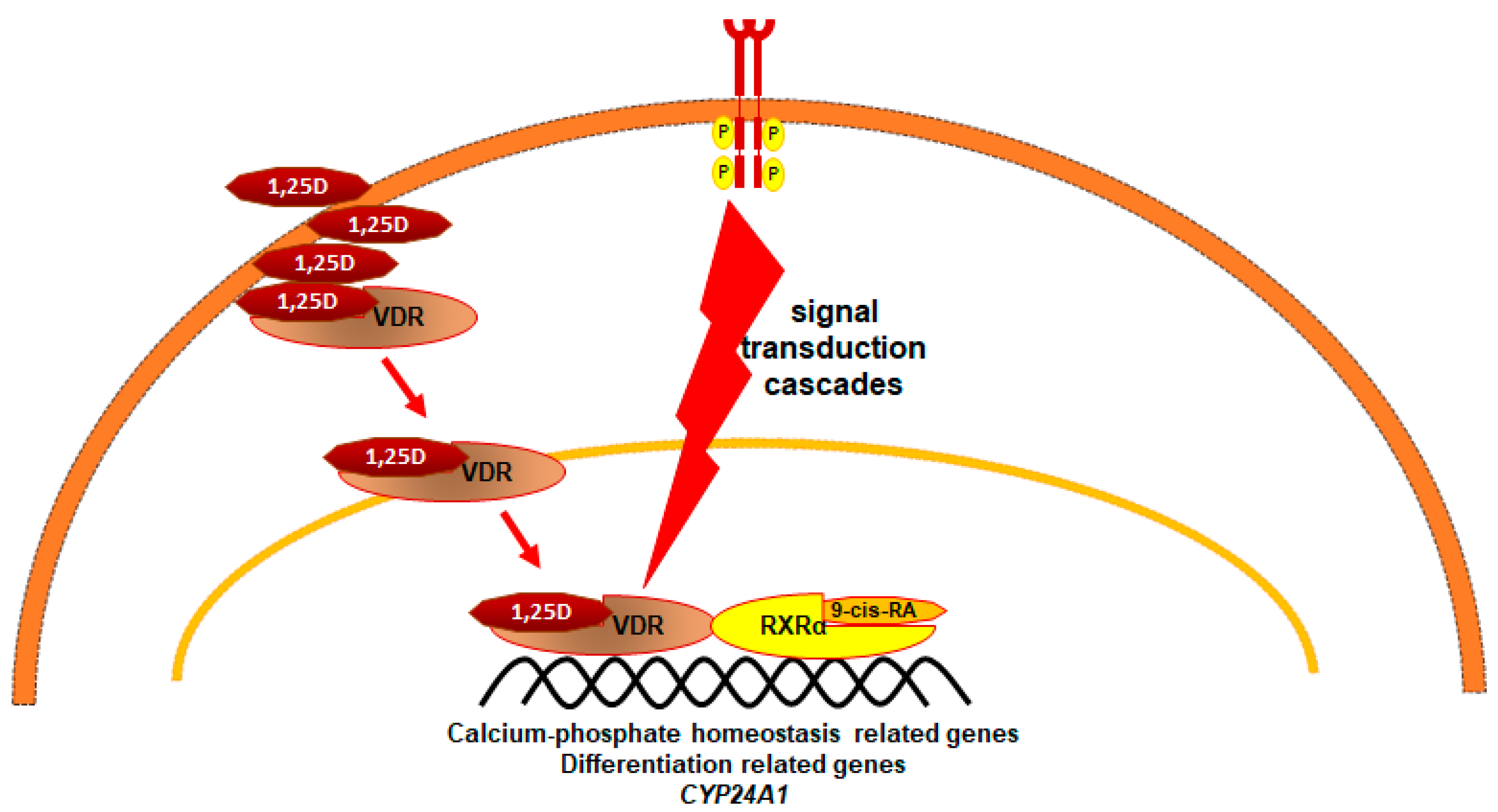
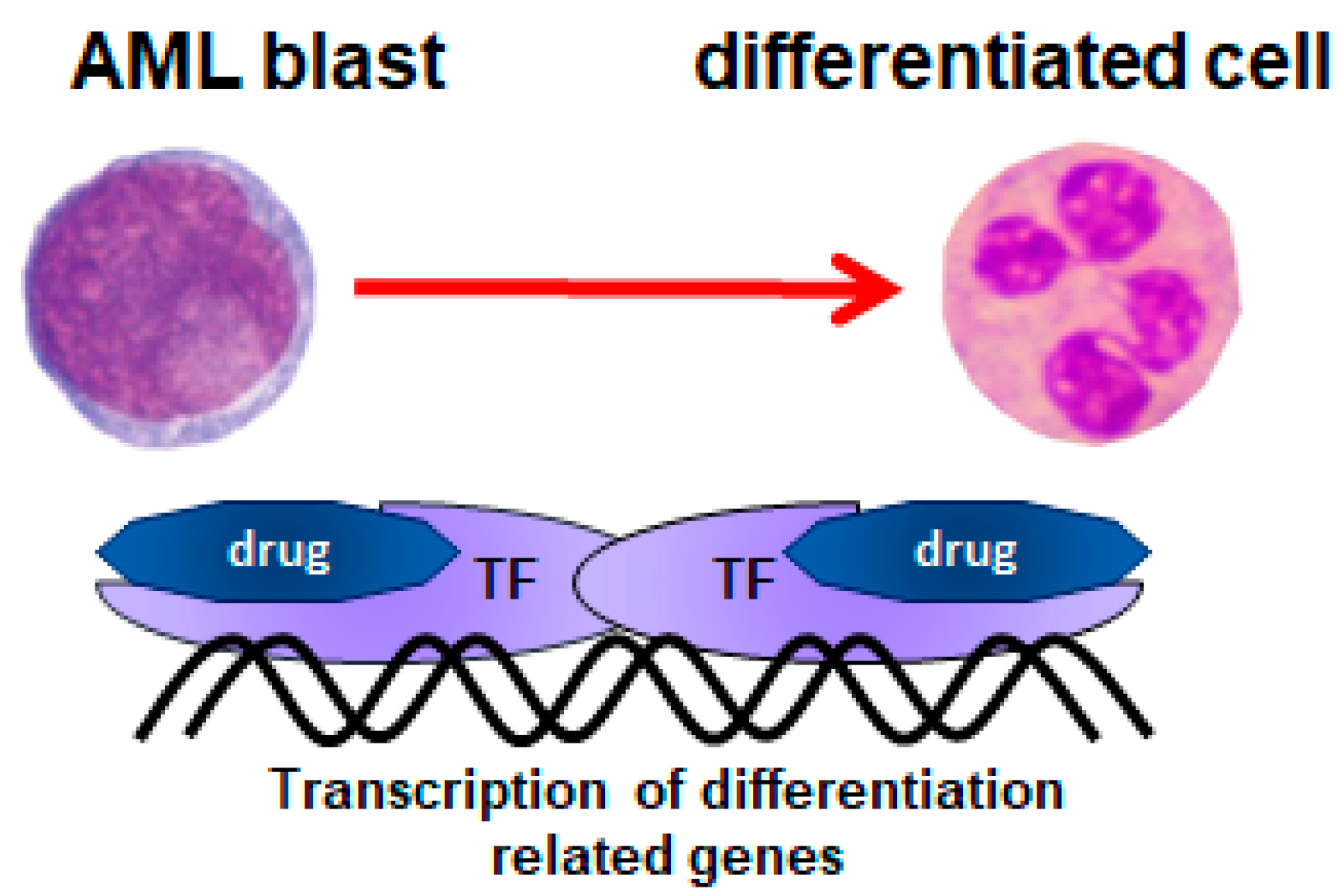
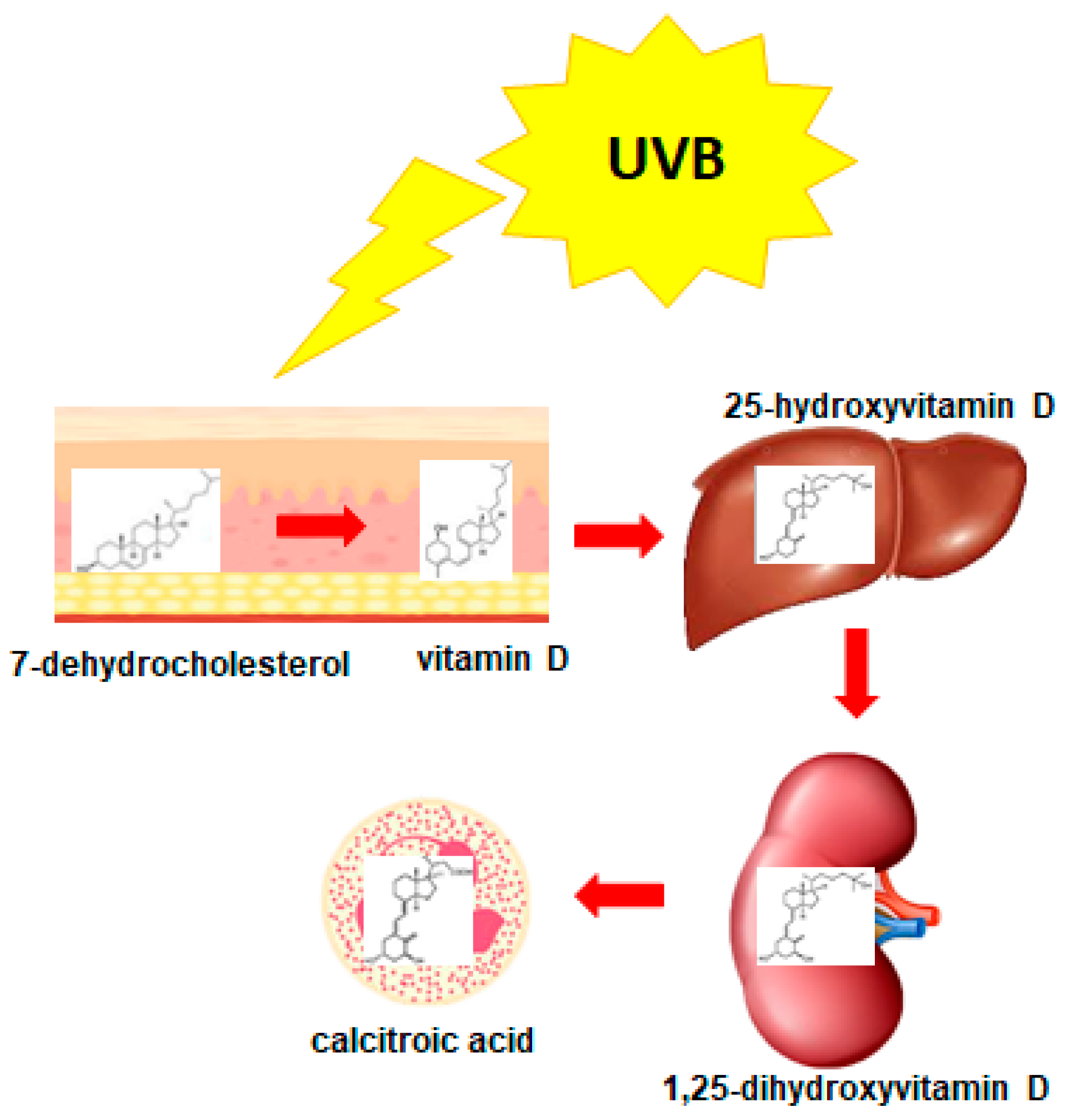
3. 1,25-Dihydroxyvitamin D3 (1,25D)
4. Low-Calcemic Analogs of 1,25D
5. The Heterogeneity of AML
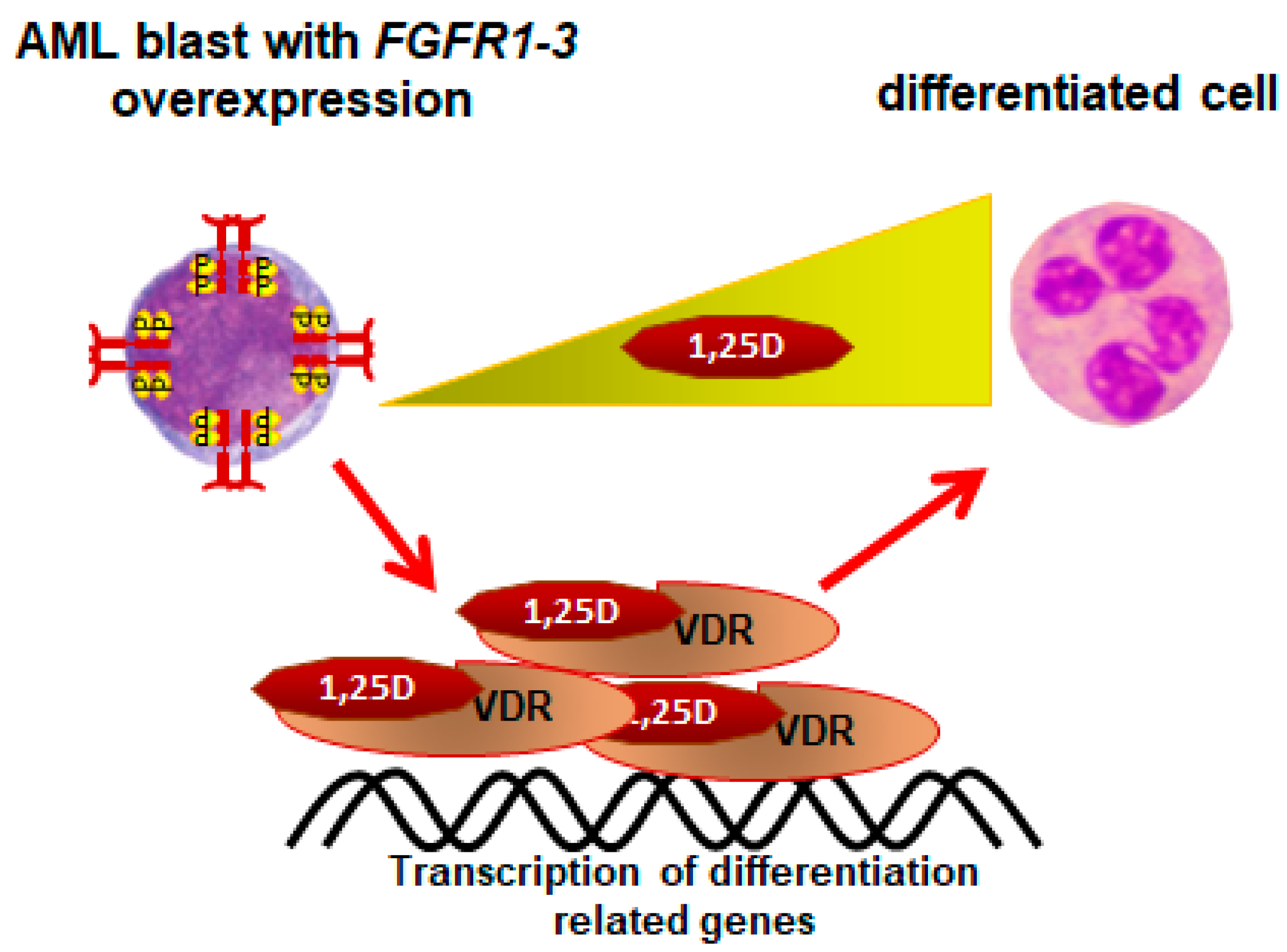
6. AMLs Resistant to 1,25D
7. AMLs Sensitive to 1,25D
8. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Pelcovits, A.; Niroula, R. Acute Myeloid Leukemia: A Review. Rhode Isl. Med. J. 2013, 103, 38–40. [Google Scholar]
- Weycker, D.; Barron, R.; Kartashov, A.; Legg, J.; Lyman, G. Incidence, treatment, and consequences of chemotherapy-induced febrile neutropenia in the inpatient and outpatient settings. J. Oncol. Pharm. Pract. 2014, 20, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Newell, L.F.; Cook, R.J. Advances in acute myeloid leukemia. BMJ 2021, 375, n2026. [Google Scholar] [CrossRef] [PubMed]
- Padmakumar, D.; Chandraprabha, V.R.; Gopinath, P.; Vimala Devi, A.R.T.; Anitha, G.R.J.; Sreelatha, M.M.; Padmakumar, A.; Sreedharan, H. A concise review on the molecular genetics of acute myeloid leukemia. Leuk. Res. 2021, 111, 16. [Google Scholar] [CrossRef]
- Friedman, A. Transcriptional control of granulocyte and monocyte development. Oncogene 2007, 26, 6816–6828. [Google Scholar] [CrossRef] [Green Version]
- Brown, G.; Hughes, P.; Michell, R.; Rolink, A.; Ceredig, R. The sequential determination model of hematopoiesis. Trends Immunol. 2007, 28, 442–448. [Google Scholar] [CrossRef]
- Theilgaard-Mönch, K.; Pundhir, S.; Reckzeh, K.; Su, J.; Tapia, M.; Furtwängler, B.; Jendholm, J.; Jakobsen, J.S.; Hasemann, M.S.; Knudsen, K.J.; et al. Transcription factor-driven coordination of cell cycle exit and lineage-specification in vivo during granulocytic differentiation: In memoriam Professor Niels Borregaard. Nat. Commun. 2022, 13, 022–31332. [Google Scholar] [CrossRef]
- Heidari, N.; Abroun, S.; Bertacchini, J.; Vosoughi, T.; Rahim, F.; Saki, N. Significance of Inactivated Genes in Leukemia: Pathogenesis and Prognosis. Cell J. 2017, 19, 9–26. [Google Scholar]
- Nie, Y.; Su, L.; Li, W.; Gao, S. Novel insights of acute myeloid leukemia with CEBPA deregulation: Heterogeneity dissection and re-stratification. Crit. Rev. Oncol. Hematol. 2021, 163, 1. [Google Scholar] [CrossRef]
- Rowley, J.; Golomb, H.; Dougherty, C. 15/17 translocation, a consistent chromosomal change in acute promyelocytic leukaemia. Lancet 1977, 1, 549–550. [Google Scholar] [CrossRef]
- Hillestad, L. Acute promyelocytic leukemia. Acta Med. Scand. 1957, 159, 189–194. [Google Scholar] [CrossRef]
- Mark, M.; Chambon, P. Functions of RARs and RXRs in vivo: Genetic dissection of the retinoid signaling pathway. Pure Appl. Chem. 2003, 75, 1709–1732. [Google Scholar] [CrossRef]
- Iwasaki, H.; Somoza, C.; Shigematsu, H.; Duprez, E.; Iwasaki-Arai, J.; Mizuno, S.; Arinobu, Y.; Geary, K.; Zhang, P.; Dayaram, T.; et al. Distinctive and indispensable roles of PU.1 in maintenance of hematopoietic stem cells and their differentiation. Blood 2005, 106, 1590–1600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duprez, E.; Wagner, K.; Koch, H.; Tenen, D. C/EBPbeta: A major PML-RARA-responsive gene in retinoic acid-induced differentiation of APL cells. EMBO J. 2003, 22, 5806–5816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morosetti, R.; Park, D.; Chumakov, A.; Grillier, I.; Shiohara, M.; Gombart, A.; Nakamaki, T.; Weinberg, K.; Koeffler, H. A novel, myeloid transcription factor, C/EBPepsilon, is upregulated during granulocytic, but not monocytic, differentiation. Blood 1997, 90, 2591–2600. [Google Scholar] [CrossRef]
- de Thé, H.; Lavau, C.; Marchio, A.; Chomienne, C.; Degos, L.; Dejean, A. The PML-RAR alpha fusion mRNA generated by the t(15;17) translocation in acute promyelocytic leukemia encodes a functionally altered RAR. Cell 1991, 66, 675–684. [Google Scholar] [CrossRef]
- De Braekeleer, E.; Douet-Guilbert, N.; De Braekeleer, M. RARA fusion genes in acute promyelocytic leukemia: A review. Expert Rev. Hematol. 2014, 7, 347–357. [Google Scholar] [CrossRef]
- Gianni, M.; Ponzanelli, I.; Mologni, L.; Reichert, U.; Rambaldi, A.; Terao, M.; Garattini, E. Retinoid-dependent growth inhibition, differentiation and apoptosis in acute promyelocytic leukemia cells. Expression and activation of caspases. Cell Death Differ. 2000, 7, 447–460. [Google Scholar] [CrossRef] [Green Version]
- Breitman, T.; Selonick, S.; Collins, S. Induction of differentiation of the human promyelocytic leukemia cell line (HL-60) by retinoic acid. Proc. Natl. Acad. Sci. USA 1980, 77, 2936–2940. [Google Scholar] [CrossRef] [Green Version]
- Huang, M.; Ye, Y.; Chen, S.; Chai, J.; Lu, J.; Zhoa, L.; Gu, L.; Wang, Z. Use of all-trans retinoic acid in the treatment of acute promyelocytic leukemia. Blood 1988, 72, 567–572. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.; Shen, Z.; Yan, H.; Chen, J.; Zeng, X.; Li, J.; Li, X.; Wu, W.; Xiong, S.; Zhao, W.; et al. Studies on the clinical efficacy and pharmacokinetics of low-dose arsenic trioxide in the treatment of relapsed acute promyelocytic leukemia: A comparison with conventional dosage. Leukemia 2001, 15, 735–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Z.; Shi, Z.; Fang, J.; Gu, B.; Li, J.; Zhu, Y.; Shi, J.; Zheng, P.; Yan, H.; Liu, Y.; et al. All-trans retinoic acid/As2O3 combination yields a high quality remission and survival in newly diagnosed acute promyelocytic leukemia. Proc. Natl. Acad. Sci. USA 2004, 101, 5328–5335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Thé, H.; Le Bras, M.; Lallemand-Breitenbach, V. The cell biology of disease: Acute promyelocytic leukemia, arsenic, and PML bodies. J. Cell Biol. 2012, 198, 11–21. [Google Scholar] [CrossRef] [Green Version]
- McCulloch, D.; Brown, C.; Iland, H. Retinoic acid and arsenic trioxide in the treatment of acute promyelocytic leukemia: Current perspectives. Onco Targets Ther. 2017, 10, 1585–1601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Q.; Bhole, A.; Qin, H.; Karp, J.; Malek, S.; Cowell, J.; Ren, M. SCLLTargeting FGFR1 to suppress leukemogenesis in syndromic and de novo AML in murine models. Oncotarget 2016, 7, 49733–49742. [Google Scholar] [CrossRef] [Green Version]
- Schlenk, R.; Dohner, K.; Kneba, M.; Götze, K.; Hartmann, F.; del Valle, F.; Kirchen, H.; Koller, E.; Fischer, J.T.; Bullinger, L.; et al. Gene mutations and response to treatment with all-trans retinoic acid in elderly patients with acute myeloid leukemia. Results from the AMLSG Trial AML HD98B. Haematologica 2009, 94, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Burnett, A.K.; Hills, R.K.; Green, C.; Jenkinson, S.; Koo, K.; Patel, Y.; Guy, C.; Gilkes, A.; Milligan, D.W.; Goldstone, A.H.; et al. The impact on outcome of the addition of all-trans retinoic acid to intensive chemotherapy in younger patients with nonacute promyelocytic acute myeloid leukemia: Overall results and results in genotypic subgroups defined by mutations in NPM1, FLT3, and CEBPA. Blood 2010, 115, 948–956. [Google Scholar]
- Schlenk, R.F.; Lübbert, M.; Benner, A.; Lamparter, A.; Krauter, J.; Herr, W.; Martin, H.; Salih, H.R.; Kündgen, A.; Horst, H.A.; et al. All-trans retinoic acid as adjunct to intensive treatment in younger adult patients with acute myeloid leukemia: Results of the randomized AMLSG 07-04 study. Ann. Hematol. 2016, 95, 1931–1942. [Google Scholar] [CrossRef] [Green Version]
- Watts, J.; Perez, A.; Pereira, L.; Fan, Y.-S.; Brown, G.; Vega, F.; Petrie, K.; Swords, R.; Zelent, A. A case of AML characterized by a novel t(4;15)(q31;q22) translocation that confers a growth-stimulatory response to retinoid-based therapy. Int. J. Mol. Sci. 2017, 18, 1492. [Google Scholar] [CrossRef] [Green Version]
- Abe, E.; Miamura, C.; Sakagami, H.; Takeda, M.; Konno, K.; Yamazaki, T.; Yoshiki, S.; Suda, T. Differentiation of mouse myeloid leukemia cells induced by 1-alpha,25-dihydroxyvitamin D3. Proc. Natl. Acad. Sci. USA 1981, 78, 4990–4994. [Google Scholar] [CrossRef] [Green Version]
- Studzinski, G.; Bhandal, A.; Brelvi, Z. Cell cycle sensitivity of HL-60 cells to the differentiation-inducing effects of 1-alpha,25-dihydroxyvitamin D3. Cancer Res. 1985, 45, 3898–3905. [Google Scholar]
- Studzinski, G.P.; Bhandal, A.K.; Brelvi, Z.S. A system for monocytic differentiation of leukemic cells HL60 by a short exposure to 1,25-dihydroxycholecalciferol. Proc. Soc. Exp. Biol. Med. 1985, 179, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Sharabani, H.; Izumchenko, E.; Wang, Q.; Kreinin, R.; Steiner, M.; Barvish, Z.; Kafka, M.; Sharoni, Y.; Levy, J.; Uskokovic, M.; et al. Cooperative antitumor effects of vitamin D3 derivatives and rosemary preparations in a mouse model of myeloid leukemia. Int. J. Cancer 2006, 118, 3012–3021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nachliely, M.; Sharony, E.; Bolla, N.; Kutner, A.; Danilenko, M. Prodifferentiation Activity of Novel Vitamin D2 Analogs PRI-1916 and PRI-1917 and Their Combinations with a Plant Polyphenol in Acute Myeloid Leukemia Cells. Int. J. Mol. Sci. 2016, 17, 1068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandler, P.; Buring, J.; Manson, J.; Giovannucci, E.; Moorthy, M.; Zhang, S.; Lee, I.; Lin, J. Circulating Vitamin D Levels and Risk of Colorectal Cancer in Women. Cancer Prev. Res. 2015, 8, 675–682. [Google Scholar] [CrossRef] [Green Version]
- Mohr, S.; Gorham, E.; Kim, J.; Hofflich, H.; Cuomo, R.; Garland, C. Could vitamin D sufficiency improve the survival of colorectal cancer patients? J. Steroid Biochem. Mol. Biol. 2015, 148, 239–244. [Google Scholar] [CrossRef]
- Mohr, S.; Gorham, E.; Kim, J.; Hofflich, H.; Garland, C. Meta-analysis of vitamin D sufficiency for improving survival of patients with breast cancer. Anticancer Res. 2014, 34, 1163–1166. [Google Scholar]
- Maalmi, H.; Ordóñez-Mena, J.; Schöttker, B.; Brenner, H. Serum 25-hydroxyvitamin D levels and survival in colorectal and breast cancer patients: Systematic review and meta-analysis of prospective cohort studies. Eur. J. Cancer 2014, 50, 1510–1521. [Google Scholar] [CrossRef]
- Muñoz, A.; Grant, W.B. Vitamin D and Cancer: An Historical Overview of the Epidemiology and Mechanisms. Nutrients 2022, 14, 1448. [Google Scholar] [CrossRef]
- Carlberg, C. The physiology of vitamin D-far more than calcium and bone. Front. Physiol. 2014, 5, 335. [Google Scholar] [CrossRef] [Green Version]
- Prosser, D.; Jones, G. Enzymes involved in the activation and inactivation of vitamin D. Trends Biochem. Sci. 2004, 29, 664–673. [Google Scholar] [CrossRef]
- Vaisanen, S.; Dunlop, T.; Sinkkonen, L.; Frank, C.; Carlberg, C. Spatio-temporal activation of chromatin on the human CYP24 gene promoter in the presence of 1alpha,25-dihydroxyvitamin D3. J. Mol. Biol. 2005, 350, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Holick, M. Vitamin D and bone health. J. Nutr. 1996, 126, 1159S–1164S. [Google Scholar] [CrossRef] [PubMed]
- Aranda, A.; Pascual, A. Nuclear hormone receptors and gene expression. Physiol. Rev. 2001, 81, 1269–1304. [Google Scholar] [CrossRef] [PubMed]
- Pike, J.; Meyer, M. Fundamentals of vitamin D hormone-regulated gene expression. J. Steroid Biochem. Mol. Biol. 2014, 144 Pt A, 5–11. [Google Scholar] [CrossRef] [Green Version]
- Carlberg, C.; Seuter, S.; de Mello, V.; Schwab, U.; Voutilainen, S.; Pulkki, K.; Nurmi, T.; Virtanen, J.; Tuomainen, T.; Uusitupa, M. Primary vitamin D target genes allow a categorization of possible benefits of vitamin D3 supplementation. PLoS ONE 2013, 8, e71042. [Google Scholar] [CrossRef] [Green Version]
- Gocek, E.; Kielbinski, M.; Marcinkowska, E. Activation of intracellular signaling pathways is necessary for an increase in VDR expression and its nuclear translocation. FEBS Lett. 2007, 581, 1751–1757. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, J.; Dang, H.; Galligan, M.; Whitfield, G.; Haussler, C.; Jurutka, P.; Haussler, M. Phosphorylation of human vitamin D receptor serine-182 by PKA suppresses 1,25(OH)2D3-dependent transactivation. Biochem. Biophys. Res. Commun. 2004, 324, 801–809. [Google Scholar] [CrossRef]
- Hsieh, J.; Jurutka, P.; Galligan, M.; Terpening, C.; Haussler, C.; Samuels, D.; Shimizu, Y.; Shimizu, N.; Haussler, M. Human vitamin D receptor is selectively phosphorylated by protein kinase C on serine 51, a residue crucial to its trans-activation function. Proc. Natl. Acad. Sci. USA 1991, 88, 9315–9319. [Google Scholar] [CrossRef] [Green Version]
- Irino, S.; Taoka, T. Treatment of myelodysplastic syndrome and acute myelogenous leukemia with vitamin D3 [1 alpha(OH)D3]. Gan Kagaku Ryoho Cancer Chemother. 1988, 15, 1183–1190. [Google Scholar]
- Nakayama, S.; Ishikawa, T.; Yabe, H.; Nagai, K.; Kasakura, S.; Uchino, H. Successful treatment of a patient with acute myeloid leukemia with 1 alpha(OH)D3. Nihon Ketsueki Gakkai Zasshi 1988, 51, 1026–1030. [Google Scholar]
- Hellström, E.; Robèrt, K.; Gahrton, G.; Mellstedt, H.; Lindemalm, C.; Einhorn, S.; Björkholm, M.; Grimfors, G.; Udén, A.; Samuelsson, J. Therapeutic effects of low-dose cytosine arabinoside, alpha-interferon, 1 alpha-hydroxyvitamin D3 and retinoic acid in acute leukemia and myelodysplastic syndromes. Eur. J. Haematol. 1988, 40, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Hellström, E.; Robèrt, K.; Samuelsson, J.; Lindemalm, C.; Grimfors, G.; Kimby, E.; Oberg, G.; Winqvist, I.; Billström, R.; Carneskog, J. Treatment of myelodysplastic syndromes with retinoic acid and 1 alpha-hydroxy-vitamin D3 in combination with low-dose ara-C is not superior to ara-C alone. Results from a randomized study. The Scandinavian Myelodysplasia Group (SMG). Eur. J. Haematol. 1990, 45, 255–261. [Google Scholar] [CrossRef]
- Ferrero, D.; Campa, E.; Dellacasa, C.; Campana, S.; Foli, C.; Boccadoro, M. Differentiating agents + low-dose chemotherapy in the management of old/poor prognosis patients with acute myeloid leukemia or myelodysplastic syndrome. Haematologica 2004, 89, 619–620. [Google Scholar] [PubMed]
- Donovan, P.J.; Sundac, L.; Pretorius, C.J.; d’Emden, M.C.; McLeod, D.S.A. Calcitriol-Mediated Hypercalcemia: Causes and Course in 101 Patients. J. Clin. Endocrinol. Metab. 2013, 98, 4023–4029. [Google Scholar] [CrossRef] [PubMed]
- Motomura, S.; Kanamori, H.; Maruta, A.; Kodama, F.; Ohkubo, T. The effect of 1-hydroxyvitamin D3 for prolongation of leukemic transformation-free survival in myelodysplastic syndromes. Am. J. Hematol. 1991, 38, 67–68. [Google Scholar] [CrossRef]
- Mellibovsky, L.; Díez, A.; Pérez-Vila, E.; Serrano, S.; Nacher, M.; Aubía, J.; Supervía, A.; Recker, R. Vitamin D treatment in myelodysplastic syndromes. Br. J. Haematol. 1998, 100, 516–520. [Google Scholar] [CrossRef]
- Hathcock, J.N.; Shao, A.; Vieth, R.; Heaney, R. Risk assessment for vitamin D. Am. J. Clin. Nutr. 2007, 85, 6–18. [Google Scholar] [CrossRef] [Green Version]
- Nadkarni, S.; Chodynski, M.; Corcoran, A.; Marcinkowska, E.; Brown, G.; Kutner, A. Double point modified analogs of vitamin D as potent activators of vitamin D receptor. Curr. Pharm. Des. 2015, 21, 1741–1763. [Google Scholar] [CrossRef]
- Norman, A.W.; Zhou, J.Y.; Henry, H.L.; Uskokovic, M.R.; Koeffler, H.P. Structure-function studies on analogues of 1 alpha,25-dihydroxyvitamin D3: Differential effects on leukemic cell growth, differentiation, and intestinal calcium absorption. Cancer Res. 1990, 50, 6857–6864. [Google Scholar]
- Zmijewski, M.A.; Carlberg, C. Vitamin D receptor(s): In the nucleus but also at membranes? Exp. Dermatol. 2020, 29, 876–884. [Google Scholar] [CrossRef] [PubMed]
- Maestro, M.A.; Molnár, F.; Carlberg, C. Vitamin D and Its Synthetic Analogs. J. Med. Chem. 2019, 62, 6854–6875. [Google Scholar] [CrossRef] [PubMed]
- Gaikwad, S.; González, C.M.; Vilariño, D.; Lasanta, G.; Villaverde, C.; Mouriño, A.; Verlinden, L.; Verstuyf, A.; Peluso-Iltis, C.; Rochel, N.; et al. Lithocholic acid-based design of noncalcemic vitamin D receptor agonists. Bioorg. Chem. 2021, 111, 30. [Google Scholar] [CrossRef] [PubMed]
- González, C.M.; Gaikwad, S.; Lasanta, G.; Loureiro, J.; Nilsson, N.; Peluso-Iltis, C.; Rochel, N.; Mouriño, A. Design, synthesis and evaluation of side-chain hydroxylated derivatives of lithocholic acid as potent agonists of the vitamin D receptor (VDR). Bioorg. Chem. 2021, 115, 22. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, H.; Masuno, H.; Kawasaki, H.; Yoshihara, A.; Numoto, N.; Ito, N.; Ishida, H.; Yamamoto, K.; Hirata, N.; Kanda, Y.; et al. Lithocholic Acid Derivatives as Potent Vitamin D Receptor Agonists. J. Med. Chem. 2020, 64, 516–526. [Google Scholar] [CrossRef] [PubMed]
- Harrison, J.; Bershadskiy, A. Clinical experience using vitamin D and analogs in the treatment of myelodysplasia and acute myeloid leukemia: A review of the literature. Leuk. Res. Treat. 2012, 125814, 8. [Google Scholar] [CrossRef] [PubMed]
- Bennett, J.; Catovsky, D.; Daniel, M.; Flandrin, G.; Galton, D.; Gralnick, H.; Sultan, C. Proposals for the classification of the acute leukaemias. French-American-British (FAB) co-operative group. Br. J. Haematol. 1976, 33, 451–458. [Google Scholar] [CrossRef]
- Vardiman, J.W.; Thiele, J.; Arber, D.A.; Brunning, R.D.; Borowitz, M.J.; Porwit, A.; Harris, N.L.; Le Beau, M.M.; Hellström-Lindberg, E.; Tefferi, A.; et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: Rationale and important changes. Blood 2009, 114, 937–951. [Google Scholar] [CrossRef] [Green Version]
- Gralnick, H.R.; Galton, D.A.G.; Catovsky, D.; Sultan, C.; Bennett, J.M. Classification of Acute Leukemia. Ann. Intern. Med. 1977, 87, 740–753. [Google Scholar] [CrossRef]
- Vardiman, J.; Harris, N.; Brunning, R. The World Health Organization (WHO) classification of the myeloid neoplasms. Blood 2002, 100, 2292–2302. [Google Scholar] [CrossRef]
- Li, S.; Mason, C.E.; Melnick, A. Genetic and epigenetic heterogeneity in acute myeloid leukemia. Curr. Opin. Genet. Dev. 2016, 36, 100–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
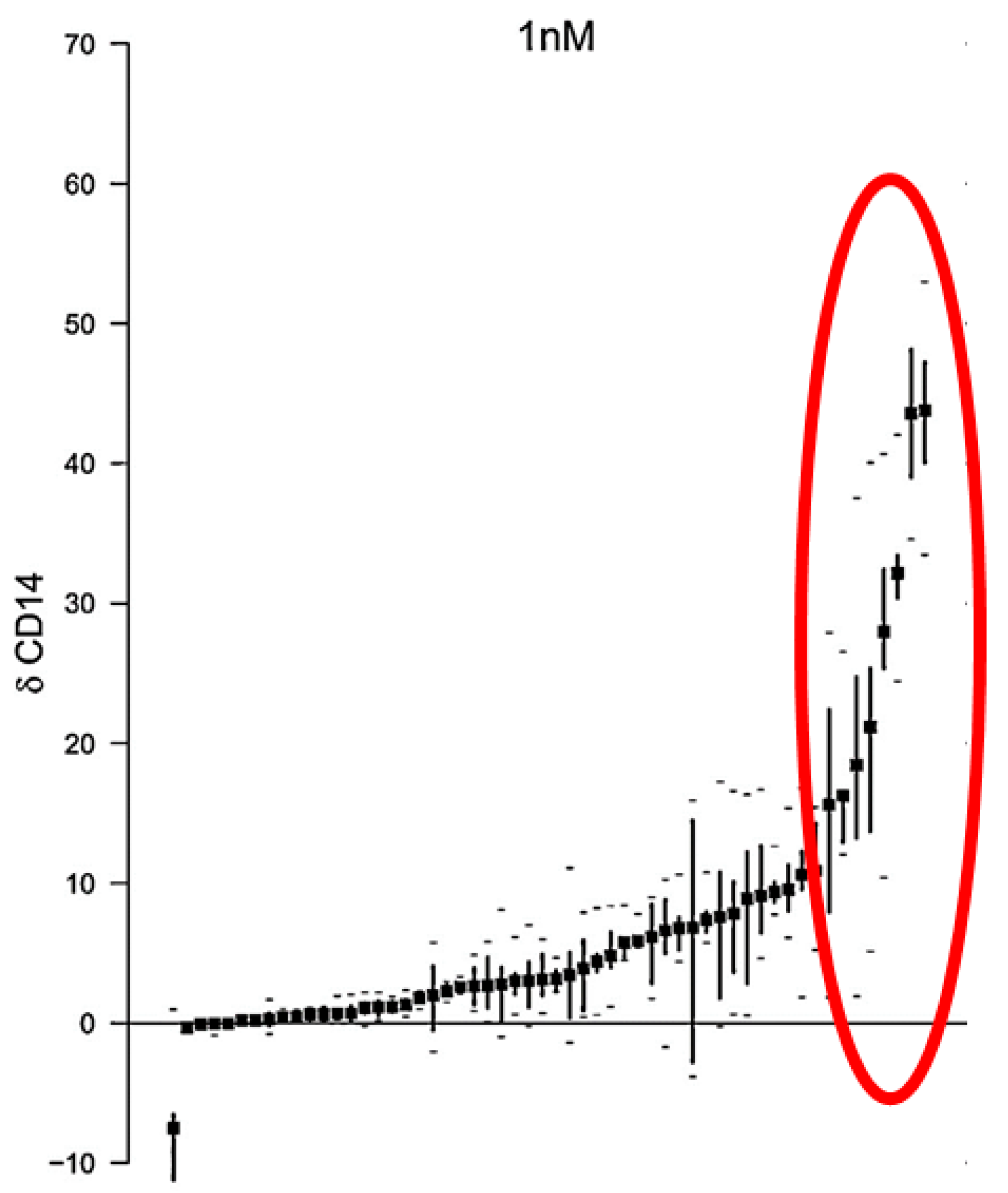
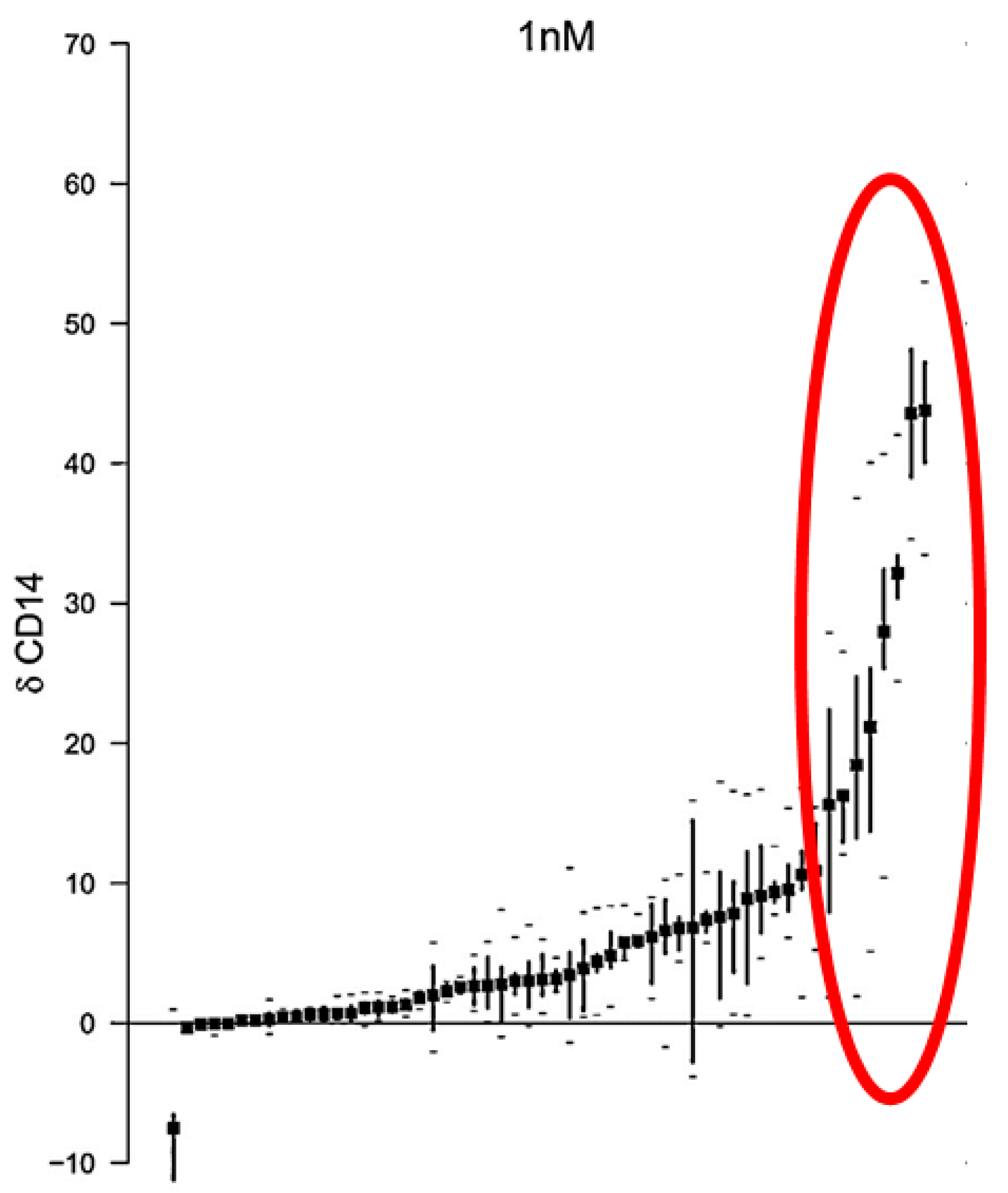
- Baurska, H.; Kiełbiński, M.; Biecek, P.; Haus, O.; Jaźwiec, B.; Kutner, A.; Marcinkowska, E. Monocytic differentiation induced by side-chain modified analogs of vitamin D in ex vivo cells from patients with acute myeloid leukemia. Leuk. Res. 2014, 38, 638–647. [Google Scholar] [CrossRef] [PubMed]
- Gocek, E.; Kielbinski, M.; Baurska, H.; Haus, O.; Kutner, A.; Marcinkowska, E. Different susceptibilities to 1,25-dihydroxyvitamin D3-induced differentiation of AML cells carrying various mutations. Leuk. Res. 2010, 34, 649–657. [Google Scholar] [CrossRef] [PubMed]
- Gocek, E.; Marchwicka, A.; Baurska, H.; Chrobak, A.; Marcinkowska, E. Opposite regulation of vitamin D receptor by ATRA in AML cells susceptible and resistant to vitamin D-induced differentiation. J. Steroid Biochem. Mol. Biol. 2012, 132, 220–226. [Google Scholar] [CrossRef]
- Paubelle, E.; Zylbersztejn, F.; Maciel, T.; Carvalho, C.; Mupo, A.; Cheok, M.; Lieben, L.; Sujobert, P.; Decroocq, J.; Yokoyama, A.; et al. Vitamin D Receptor Controls Cell Stemness in Acute Myeloid Leukemia and in Normal Bone Marrow. Cell Rep. 2020, 30, 739–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munker, R.; Norman, A.; Koeffler, H. Vitamin D compounds. Effect on clonal proliferation and differentiation of human myeloid cells. J. Clin. Investig. 1986, 78, 424–430. [Google Scholar] [CrossRef]
- Sohal, J.; Chase, A.; Mould, S.; Corcoran, M.; Oscier, D.; Iqbal, S.; Parker, S.; Welborn, J.; Harris, R.; Martinelli, G.; et al. Identification of four new translocations involving FGFR1 in myeloid disorders. Genes Chromosomes Cancer 2001, 32, 155–163. [Google Scholar] [CrossRef]
- Gu, T.; Goss, V.; Reeves, C.; Popova, L.; Nardone, J.; Macneill, J.; Walters, D.; Wang, Y.; Rush, J.; Comb, M.; et al. Phosphotyrosine profiling identifies the KG-1 cell line as a model for the study of FGFR1 fusions in acute myeloid leukemia. Blood 2006, 108, 4202–4204. [Google Scholar] [CrossRef]
- Jin, Y.; Zhen, Y.; Haugsten, E.; Wiedlocha, A. The driver of malignancy in KG-1a leukemic cells, FGFR1OP2–FGFR1, encodes an HSP90 addicted oncoprotein. Cell. Signal. 2011, 23, 1758–1766. [Google Scholar] [CrossRef]
- Marchwicka, A.; Corcoran, A.; Berkowska, K.; Marcinkowska, E. Restored expression of vitamin D receptor and sensitivity to 1,25-dihydroxyvitamin D3 in response to disrupted fusion FOP2-FGFR1 gene in acute myeloid leukemia cells. Cell Biosci. 2016, 6, 7. [Google Scholar] [CrossRef] [Green Version]
- Jackson, C.; Medeiros, L.; Miranda, R. 8p11 myeloproliferative syndrome: A review. Hum. Pathol. 2010, 41, 461–476. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.J.; Meng, L. 8p11 Myeloproliferative syndrome with t(8;22)(p11;q11): A case report. Exp. Ther. Med. 2018, 16, 1449–1453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boutzen, H.; Saland, E.; Larrue, C.; de Toni, F.; Gales, L.; Castelli, F.A.; Cathebas, M.; Zaghdoudi, S.; Stuani, L.; Kaoma, T.; et al. Isocitrate dehydrogenase 1 mutations prime the all-trans retinoic acid myeloid differentiation pathway in acute myeloid leukemia. J. Exp. Med. 2016, 213, 483–497. [Google Scholar] [CrossRef]
- Sabatier, M.; Boet, E.; Zaghdoudi, S.; Guiraud, N.; Hucteau, A.; Polley, N.; Cognet, G.; Saland, E.; Lauture, L.; Farge, T.; et al. Activation of Vitamin D Receptor Pathway Enhances Differentiating Capacity in Acute Myeloid Leukemia with Isocitrate Dehydrogenase Mutations. Cancers 2021, 13, 5243. [Google Scholar] [CrossRef]
- Manfredini, R.; Trevisan, F.; Grande, A.; Tagliafico, E.; Montanari, M.; Lemoli, R.; Visani, G.; Tura, S.; Ferrari, S.; Ferrari, S. Induction of a functional vitamin D receptor in all-trans-retinoic acid-induced monocytic differentiation of M2-type leukemic blast cells. Cancer Res. 1999, 59, 3803–3811. [Google Scholar]
- Marchwicka, A.; Cebrat, M.; Łaszkiewicz, A.; Śnieżewski, Ł.; Brown, G.; Marcinkowska, E. Regulation of vitamin D receptor expression by retinoic acid receptor alpha in acute myeloid leukemia cells. J. Steroid Biochem. Mol. Biol. 2016, 159, 121–130. [Google Scholar] [CrossRef] [Green Version]
- Katoh, M.; Nakagama, H. FGF receptors: Cancer biology and therapeutics. Med. Res. Rev. 2014, 34, 280–300. [Google Scholar] [CrossRef] [PubMed]
- Katoh, M. FGFR inhibitors: Effects on cancer cells, tumor microenvironment and whole-body homeostasis (Review). Int. J. Mol. Med. 2016, 38, 3–15. [Google Scholar] [CrossRef] [Green Version]
- Marchwicka, A.; Jakuszak, A.; Grembowska, A.; Kumari, P.; Marcinkowska, E. The Influence of Overexpressed Fibroblast Growth Factor Receptors Towards Vitamin D Receptor Expression and Activity. Preprints 2021, 2021100304. [Google Scholar] [CrossRef]
- Xie, Y.; Su, N.; Yang, J.; Tan, Q.; Huang, S.; Jin, M.; Ni, Z.; Zhang, B.; Zhang, D.; Luo, F.; et al. FGF/FGFR signaling in health and disease. Signal. Transduct. Target. Ther. 2020, 5, 181. [Google Scholar]
- Eswarakumar, V.P.; Lax, I.; Schlessinger, J. Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev. 2005, 16, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Rao, J.; Studzinski, G.P. Inhibition of p38 MAP kinase activity up-regulates multiple MAP kinase pathways and potentiates 1,25-dihydroxyvitamin D3-induced differentiation of human leukemia HL60 cells. Exp. Cell Res. 2000, 258, 425–437. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Studzinski, G. Inhibition of p38 MAP kinase potentiates the JNK/SAPK pathway and AP-1 activity in monocytic but not in macrophage or granulocytic differentiation of HL60 cells. J. Cell Biochem. 2001, 82, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Hart, K.; Robertson, S.; Kanemitsu, M.; Meyer, A.; Tynan, J.; Donoghue, D. Transformation and Stat activation by derivatives of FGFR1, FGFR3, and FGFR4. Oncogene 2000, 19, 3309–3320. [Google Scholar] [CrossRef] [Green Version]
- Sasieni, P.D.; Shelton, J.; Ormiston-Smith, N.; Thomson, C.S.; Silcocks, P.B. What is the lifetime risk of developing cancer? The effect of adjusting for multiple primaries. Br. J. Cancer 2011, 105, 460–465. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marcinkowska, E. Vitamin D Derivatives in Acute Myeloid Leukemia: The Matter of Selecting the Right Targets. Nutrients 2022, 14, 2851. https://doi.org/10.3390/nu14142851
Marcinkowska E. Vitamin D Derivatives in Acute Myeloid Leukemia: The Matter of Selecting the Right Targets. Nutrients. 2022; 14(14):2851. https://doi.org/10.3390/nu14142851
Chicago/Turabian StyleMarcinkowska, Ewa. 2022. "Vitamin D Derivatives in Acute Myeloid Leukemia: The Matter of Selecting the Right Targets" Nutrients 14, no. 14: 2851. https://doi.org/10.3390/nu14142851
APA StyleMarcinkowska, E. (2022). Vitamin D Derivatives in Acute Myeloid Leukemia: The Matter of Selecting the Right Targets. Nutrients, 14(14), 2851. https://doi.org/10.3390/nu14142851