
Recommendations for the Diagnosis and Therapeutic Management of Hyperammonaemia in Paediatric and Adult Patients

 ,
,  , and
, and
Abstract
:1. Introduction
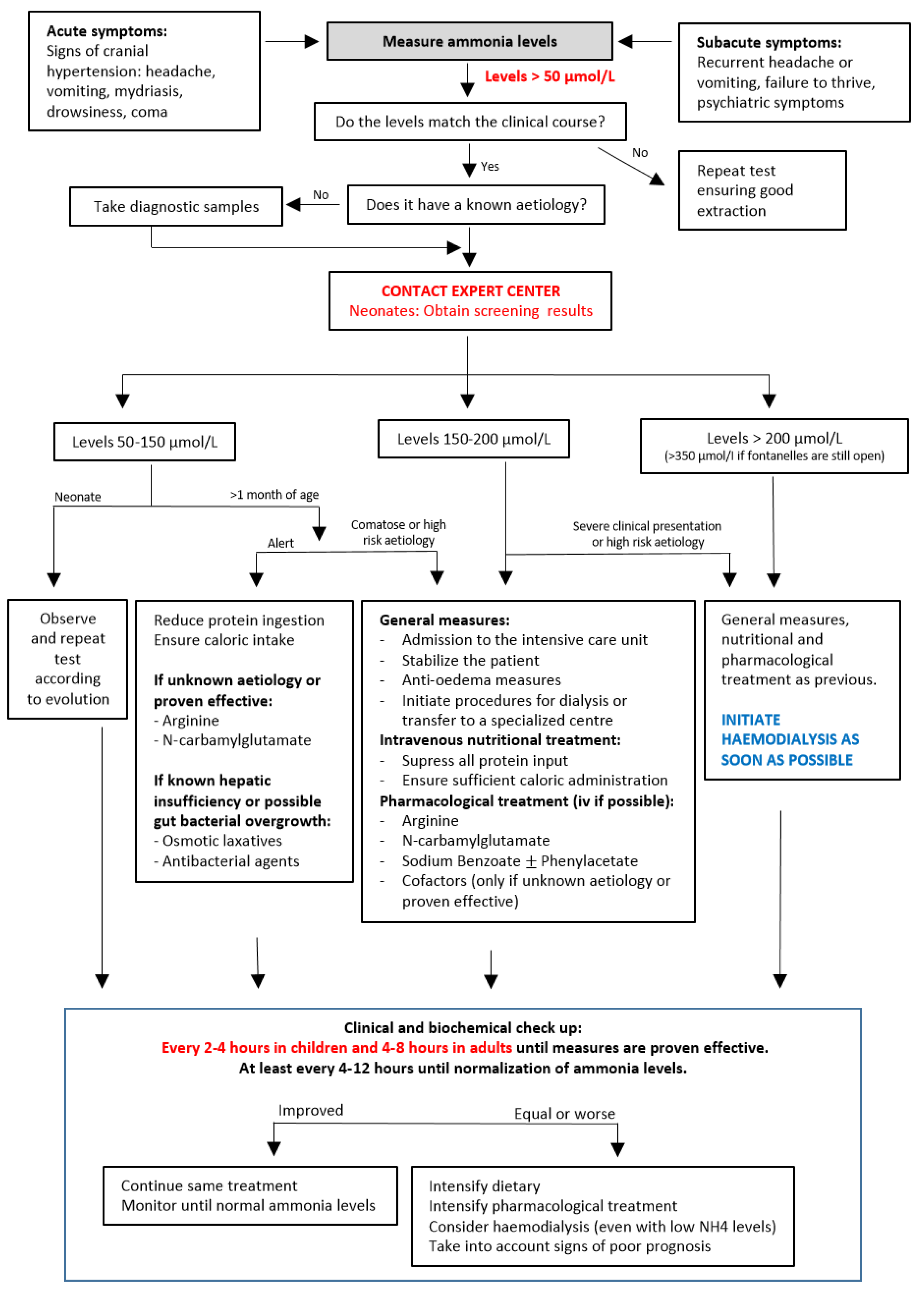
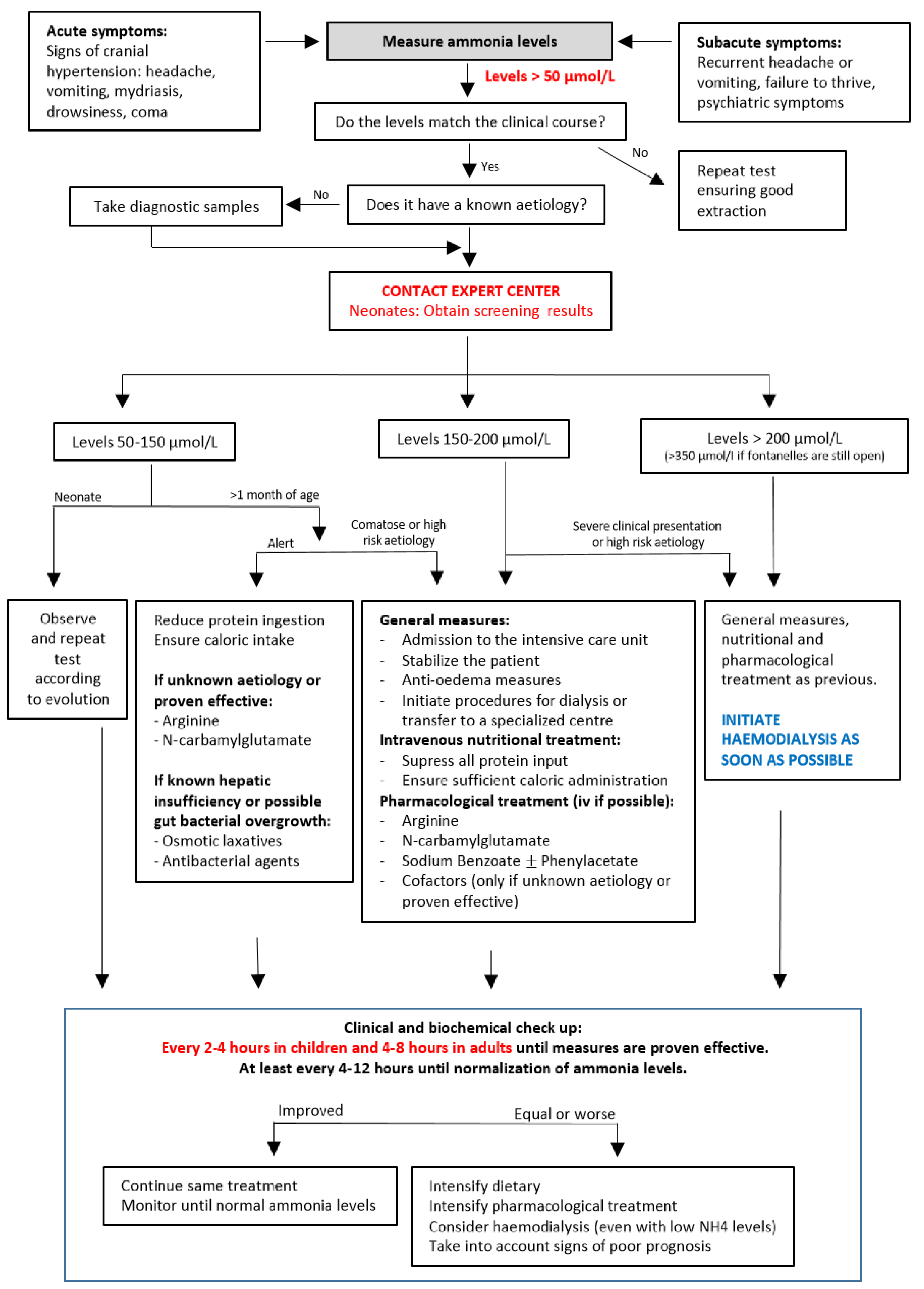
2. Diagnosis of Hyperammonaemia
2.1. Clinical Presentation
2.2. Diagnostic Approach
3. Management of Hyperammonaemia in the Acute Phase
3.1. General and Brain Protection Measures
3.2. Nutritional Treatment
3.3. Pharmacological Treatment
3.4. Extrarenal Clearance Procedures
3.5. Poor Neurological Prognostic Factors
- Ammonia levels >1000 μmol/L at diagnosis, especially when it increases or does not begin to decrease within the next 12 h.
- Ammonia levels >2000 μmol/L at any time in the evolution.
- Ammonia levels >700 μmol/L maintained for more than 48–72 h.
- Intracranial pressure >30 mm Hg maintained for more than 24 h.
- Presence of decortication movements.
- Electroencephalographic signs of brain death.
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ah Mew, N.; Simpson, K.L.; Gropman, A.L.; Lanpher, B.C.; Chapman, K.A.; Summar, M.L. Urea cycle disorders overview. In GeneReviews ®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2017. [Google Scholar]
- Matsumoto, S.; Haberle, J.; Kido, J.; Mitsubuchi, H.; Endo, F.; Nakamura, K. Urea cycle disorders-update. J. Hum. Genet. 2019, 64, 833–847. [Google Scholar] [CrossRef] [PubMed]
- Haberle, J.; Burlina, A.; Chakrapani, A.; Dixon, M.; Karall, D.; Lindner, M.; Mandel, H.; Martinelli, D.; Pintos-Morell, G.; Santer, R.; et al. Suggested guidelines for the diagnosis and management of urea cycle disorders: First revision. J. Inherit. Metab. Dis. 2019, 42, 1192–1230. [Google Scholar] [CrossRef]
- Pintos Morel, G.; Castiñeiras Ramos, D.; Puig, R.; Campos, P.; Martin-Hernandez, E. Protocolo de diagnóstico y tratamiento de los trastornos del ciclo de la urea. In Protocolos de Diagnóstico y Tratamiento de los Errores Congénitos del Metabolismo, 2nd ed.; ERGON: Madrid, Spain, 2018; pp. 1–368. [Google Scholar]
- Savy, N.; Brossier, D.; Brunel-Guitton, C.; Ducharme-Crevier, L.; Du Pont-Thibodeau, G.; Jouvet, P. Acute pediatric hyperammonemia: Current diagnosis and management strategies. Hepatic. Med. Evid. Res. 2018, 10, 105–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Back, A.; Tupper, K.Y.; Bai, T.; Chiranand, P.; Goldenberg, F.D.; Frank, J.I.; Brorson, J.R. Ammonia-induced brain swelling and neurotoxicity in an organotypic slice model. Neurol. Res. 2011, 33, 1100–1108. [Google Scholar] [CrossRef]
- Braissant, O.; McLin, V.A.; Cudalbu, C. Ammonia toxicity to the brain. J. Inherit. Metab. Dis. 2013, 36, 595–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiati Kenston, S.S.; Song, X.; Li, Z.; Zhao, J. Mechanistic insight, diagnosis, and treatment of ammonia-induced hepatic encephalopathy. J. Gastroenterol Hepatol. 2019, 34, 31–39. [Google Scholar] [CrossRef] [Green Version]
- Mallet, M.; Weiss, N.; Thabut, D.; Rudler, M. Why and when to measure ammonemia in cirrhosis? Clin. Res. Hepatol Gastroenterol 2018, 42, 505–511. [Google Scholar] [CrossRef]
- Kumar, S.; Asrani, S.K. Non-cirrhotic hyperammonemia—When high ammonia is not always from cirrhosis. Curr Hepatol Rep. 2015, 14, 25–31. [Google Scholar] [CrossRef]
- Palomino Perez, L.M.; Martin-Rivada, A.; Canedo Villaroya, E.; Garcia-Penas, J.J.; Cuervas-Mons Vendrell, M.; Pedron-Giner, C. Use of carglumic acid in valproate-induced hyperammonemia: 25 pediatric cases. JIMD Rep. 2020, 55, 3–11. [Google Scholar] [CrossRef]
- Pedron-Giner, C.; Marin, L.; Fraile, P.; Herguedas, J.; Garcia-Peñas, J.J.; Rodriguez, A. Valproate induced hyperammonaemic encephalopathy syndrome. Treatment with carglumic acid. J. Inherit. Metab. Dis. 2008, 31, 89. [Google Scholar] [CrossRef]
- Arrieta, F.; Belanger-Quintana, A.; Gajate, L.; Grau, J.; Pintor, R. Carglumic acid (Carbaglu®) treatment in hyperammonemia post liver transplantation. Endocrinol. Diabetes Nutr. 2020, 67, 148–149. [Google Scholar] [CrossRef] [PubMed]
- Raina, R.; Bedoyan, J.K.; Lichter-Konecki, U.; Jouvet, P.; Picca, S.; Mew, N.A.; Machado, M.C.; Chakraborty, R.; Vemuganti, M.; Grewal, M.K.; et al. Consensus guidelines for management of hyperammonaemia in paediatric patients receiving continuous kidney replacement therapy. Nat. Rev. Nephrol 2020, 16, 471–482. [Google Scholar] [CrossRef]
- Seethapathy, H.; Fenves, A.Z. Pathophysiology and management of hyperammonemia in organ transplant patients. Am. J. Kidney Dis. 2019, 74, 390–398. [Google Scholar] [CrossRef] [PubMed]
- Martin-Hernandez, E.; Aldamiz-Echevarria, L.; Castejon-Ponce, E.; Pedron-Giner, C.; Couce, M.L.; Serrano-Nieto, J.; Pintos-Morell, G.; Belanger-Quintana, A.; Martinez-Pardo, M.; Garcia-Silva, M.T.; et al. Urea cycle disorders in Spain: An observational, cross-sectional and multicentric study of 104 cases. Orphanet J. Rare Dis. 2014, 9, 187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waisbren, S.E.; Gropman, A.L.; Batshaw, M.L. Improving long term outcomes in urea cycle disorders-report from the Urea Cycle Disorders Consortium. J. Inherit. Metab. Dis. 2016, 39, 573–584. [Google Scholar] [CrossRef] [Green Version]
- Grupo de Consenso reunido en Lisboa 2006 y Madrid 2007. Protocolo Hispano-Luso de Diagnostico Y Tratamiento de Las Hiperamoniemias en Pacientes Neonatos Y de Más de 30 Días de Vida, 2nd ed.; ERGON: Madrid, Spain, 2009. [Google Scholar]
- Arrieta Blanco, F.; Bergua Martínez, A.; Cañedo Villarroya, E.; García Silva, M.T.; Lama More, R.; Martín Hernández, E.; Martínez-Pardo Casanova, M.; Moráis López, A.; Pedrón Giner, C.; Quijada Fraile, P.; et al. Recomendaciones Para El Enfoque Diagnóstico Y Terpéutico de la Hiperamoniemia en Pacientes Pediátricos Y Adultos; ERGON: Majadahonda, Spain, 2017. [Google Scholar]
- Belanger-Quintana, A.; Martinez-Pardo, M.; Garcia, M.J.; Wermuth, B.; Torres, J.; Pallares, E.; Ugarte, M. Hyperammonaemia as a cause of psychosis in an adolescent. Eur. J. Pediatrics 2003, 162, 773–775. [Google Scholar] [CrossRef]
- Gropman, A.L.; Summar, M.; Leonard, J.V. Neurological implications of urea cycle disorders. J. Inherit. Metab Dis 2007, 30, 865–879. [Google Scholar] [CrossRef]
- Serrano, M.; Martins, C.; Perez-Duenas, B.; Gomez-Lopez, L.; Murgui, E.; Fons, C.; Garcia-Cazorla, A.; Artuch, R.; Jara, F.; Arranz, J.A.; et al. Neuropsychiatric manifestations in late-onset urea cycle disorder patients. J. Child. Neurol. 2010, 25, 352–358. [Google Scholar] [CrossRef]
- Rose, C.F.; Amodio, P.; Bajaj, J.S.; Dhiman, R.K.; Montagnese, S.; Taylor-Robinson, S.D.; Vilstrup, H.; Jalan, R. Hepatic encephalopathy: Novel insights into classification, pathophysiology and therapy. J. Hepatol. 2020, 73, 1526–1547. [Google Scholar] [CrossRef]
- Baumgartner, M.R.; Horster, F.; Dionisi-Vici, C.; Haliloglu, G.; Karall, D.; Chapman, K.A.; Huemer, M.; Hochuli, M.; Assoun, M.; Ballhausen, D.; et al. Proposed guidelines for the diagnosis and management of methylmalonic and propionic acidemia. Orphanet J. Rare Dis 2014, 9, 130. [Google Scholar] [CrossRef] [Green Version]
- Ozanne, B.; Nelson, J.; Cousineau, J.; Lambert, M.; Phan, V.; Mitchell, G.; Alvarez, F.; Ducruet, T.; Jouvet, P. Threshold for toxicity from hyperammonemia in critically ill children. J. Hepatol. 2012, 56, 123–128. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Genetic Aetiologies | Non-Genetic Aetiologies | |
|---|---|---|
Defects of the urea cycle
| Drugs
| Liver function related
|
| Samples/Determinations | Considerations |
|---|---|
| Urgently processed samples | |
| Ammonia | Careful extraction: no compression and through a large-calibre route. Keep the tube cold. Process within 1 h. |
| Ketone bodies | Using a blood reflective device and/or urine test strip. |
| Blood gases and anion gap | 0.3 mL arterial or venous blood |
| Lactate | Careful extraction: no compression and through a large-calibre route. |
| Urgent biochemistry profile | Glycaemia, uric acid, urea, creatinine, total proteins, AST, ALT, gamma-glutamyltransferase, creatine kinase, sodium, potassium, chloride, calcium. |
| Other | Hemogram, coagulation profile, C-reactive protein and procalcitonin |
| Samples obtained in the acute phase and sent to a specialized laboratory as soon as possible | |
| Blood aminogram and acylcarnitines | Serum or plasma samples (separate from whole blood). Liquid samples might need refrigeration or freezing for their conservation. Dried blood samples when it is not possible to obtain or adequately process liquid samples. Must differentiate into isolated peaks: glutamate, glutamine, alanine, citrulline, arginosuccinate, cystine, lysine and arginine. |
| Urine aminogram, orotic acid and organic acids | 2–10 mL of the first urine obtained. Liquid samples might need refrigeration or freezing for their conservation. Dried blood samples when it is not possible to obtain liquid samples. |
| Hormone determination (insulin, C peptide and growth hormone) | If concomitant hypoglycaemia. |
| Bacterial cultures (blood, urine) | To rule out possible triggering infection. |
| Samples that can be obtained later but might be considered in an acute phase in cases with bad prognosis | |
| Genetic testing samples | Preferably whole blood samples. Dried blood samples when it is not possible to obtain or adequately process liquid samples. |
| Mild Hyperammonaemia (<150 μmol/L) Alert Patient Good Oral Enteral Tolerance | Severe Hyperammonaemia (>150 μmol/L) Decreased Conscience Level Decreased Tolerance |
|---|---|
|
1–3 years: 7–8 mg/kg/min (4–5 mL/kg/h) 4–6 years: 6–7 mg/kg/min (3.5–4 mL/kg/h) 7–12 years: 5–6 mg/kg/min (3–3.5 mL/kg/h) Adolescents: 3–5 mg/kg/min (2.5–3 mL/kg/h) Adults: 3–5 mg/kg/min (2–3 mL/kg/h)
|
| Drug | First Dose | Maintenance | Considerations |
|---|---|---|---|
| Urea cycle enhancers: useful in cases of hyperammonaemia due to any cause but primary urea cycle deficiencies. Should be always included in cases of unknown aetiology. | |||
| N-carbamylglutamate | 100 mg/kg | 100–250 mg/kg/day in 2–4 doses | Oral (or crushed through feeding tube): tablets. |
| Useful in most genetic and non-genetic disorders. Not useful in most known primary urea cycle disorders (only NAGS deficiency). | |||
| Maximum dose not stablished. In adults use weight for lean body mass. | |||
| L-Arginine | <20 kg: 250–400 mg/kg | <20 kg: 250 mg/kg/day | Oral: powder, sachets. IV: diluted in 10% glucose solution. Can be administered together with benzoate. |
| >20 kg: 250 mg/kg (max 12 g) | >20 kg: 200 mg/kg/day (max 12 g/day) | ||
| Urea cycle scavengers: useful in all cases of hyperammonaemia. Should be included in hyperammonaemia due to primary urea cycle disorders or severe cases of unknown aetiology. | |||
| Sodium benzoate ± Sodium phenylacetate | <20 kg: 250 mg/kg | <20 kg: 250–500 mg/kg/day | IV: requires central venous access. |
| >20 kg: 5.5 g/m2 (max 12g) | >20 kg: 5.5 g/m2 (max 12g/day) | Diluted in 10% glucose solution and administered over 2 h. | |
| Attention to the sodium content. | |||
| Precaution in organic acidaemias. | |||
| Phenylbutyrate | <20 kg: 250–500 mg/kg/day in 4 doses | Oral: tablet, powder, or solution presentations. | |
| >20 kg: 9.9–13 g/m2/day in 4 doses | Slow action: not first option in acute hyperammonaemia. | ||
| Cofactor therapy: useful if unknown aetiology or an underlying genetic disease is suspected. If known diagnosis, only start those that have been proven effective. | |||
| L-Carnitine | 50 mg/kg | 100 mg/kg/day in 4 doses | Oral: 10 or 30% solutions. |
| Maximum dose: 4 g | Maximum dose: 6 g/day | IV: 20% solution. | |
| Caution in long-chain fatty acid oxidation deficiencies. | |||
| Biotin | 10 mg | 20–40 mg/day | Oral or iv presentations. |
| Hydroxocobalamin | 1 mg | Repeat only depending on diagnosis | IM or IV. |
| Only one dose required initially. | |||
| Therapies aimed to reduce ammonia gut production: proven effective in cases of hepatic encephalopathy and propionic acidaemia. Slow action: not useful as monotherapy and if high ammonia levels. | |||
| Osmotic laxatives | Lactulose 15–20 mL every 12 h | Titrate until 2–3 stools/day. | |
| Polyethylene glycol 1 dose | |||
| Antimicrobial agents | Rifaximin 2–12 years of age (off-label) 20–30 mg/kg/day 2–4 doses >12 years of age 200–400 mg/day 2–4 doses | Use preferably antibiotics with low absorption rates. Other options: metronidazole, ciprofloxacin, doxycycline, etc. | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bélanger-Quintana, A.; Arrieta Blanco, F.; Barrio-Carreras, D.; Bergua Martínez, A.; Cañedo Villarroya, E.; García-Silva, M.T.; Lama More, R.; Martín-Hernández, E.; López, A.M.; Morales-Conejo, M.; et al. Recommendations for the Diagnosis and Therapeutic Management of Hyperammonaemia in Paediatric and Adult Patients. Nutrients 2022, 14, 2755. https://doi.org/10.3390/nu14132755
Bélanger-Quintana A, Arrieta Blanco F, Barrio-Carreras D, Bergua Martínez A, Cañedo Villarroya E, García-Silva MT, Lama More R, Martín-Hernández E, López AM, Morales-Conejo M, et al. Recommendations for the Diagnosis and Therapeutic Management of Hyperammonaemia in Paediatric and Adult Patients. Nutrients. 2022; 14(13):2755. https://doi.org/10.3390/nu14132755
Chicago/Turabian StyleBélanger-Quintana, Amaya, Francisco Arrieta Blanco, Delia Barrio-Carreras, Ana Bergua Martínez, Elvira Cañedo Villarroya, María Teresa García-Silva, Rosa Lama More, Elena Martín-Hernández, Ana Moráis López, Montserrat Morales-Conejo, and et al. 2022. "Recommendations for the Diagnosis and Therapeutic Management of Hyperammonaemia in Paediatric and Adult Patients" Nutrients 14, no. 13: 2755. https://doi.org/10.3390/nu14132755
APA StyleBélanger-Quintana, A., Arrieta Blanco, F., Barrio-Carreras, D., Bergua Martínez, A., Cañedo Villarroya, E., García-Silva, M. T., Lama More, R., Martín-Hernández, E., López, A. M., Morales-Conejo, M., Pedrón-Giner, C., Quijada-Fraile, P., Stanescu, S., & Casanova, M. M.-P. (2022). Recommendations for the Diagnosis and Therapeutic Management of Hyperammonaemia in Paediatric and Adult Patients. Nutrients, 14(13), 2755. https://doi.org/10.3390/nu14132755