
Activation of LXRs Reduces Oxysterol Lipotoxicity in RPE Cells by Promoting Mitochondrial Function

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Animal Experiments
2.3. Cell Culture and Transfection
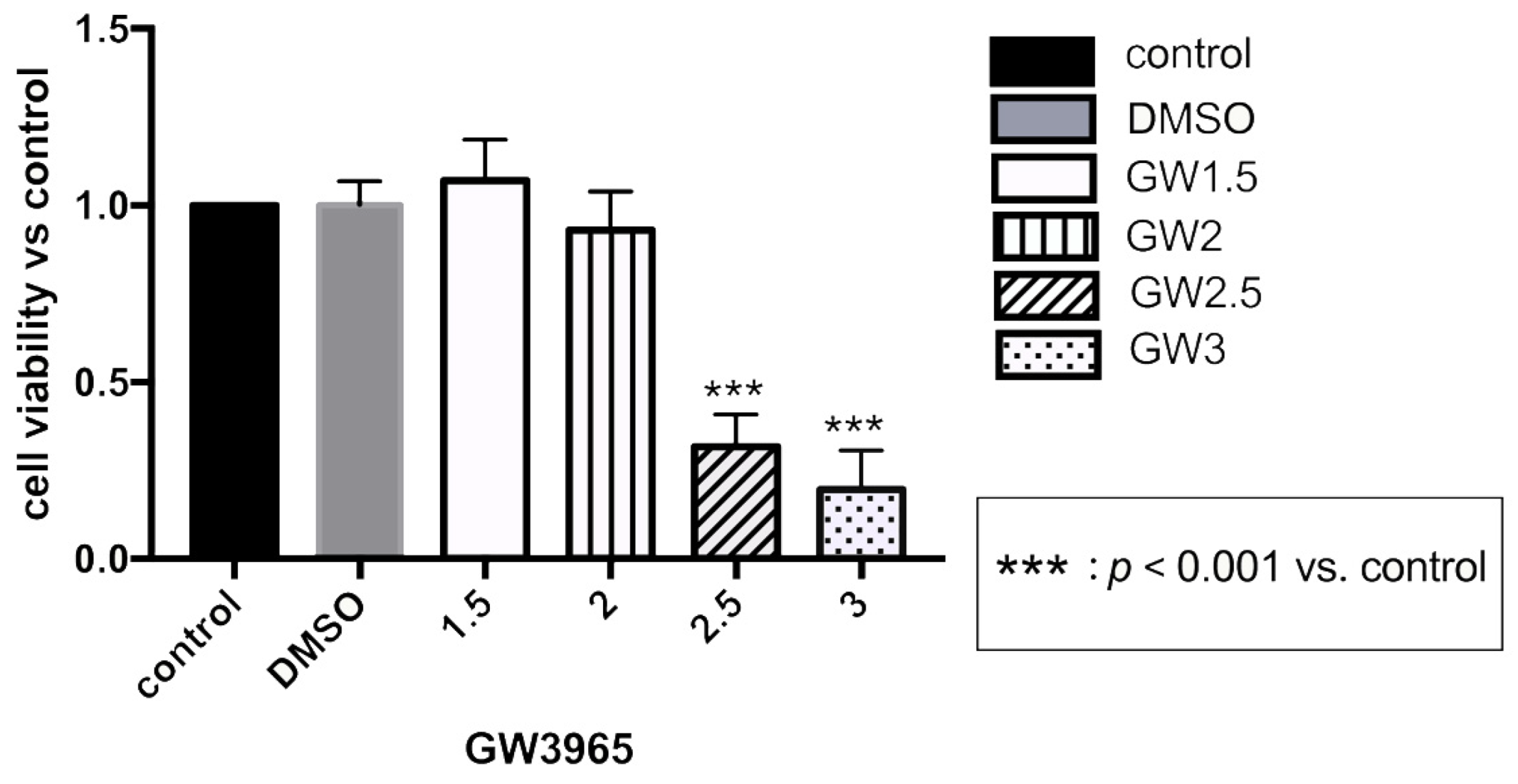
2.4. Cell Counting Kit-8 Assay
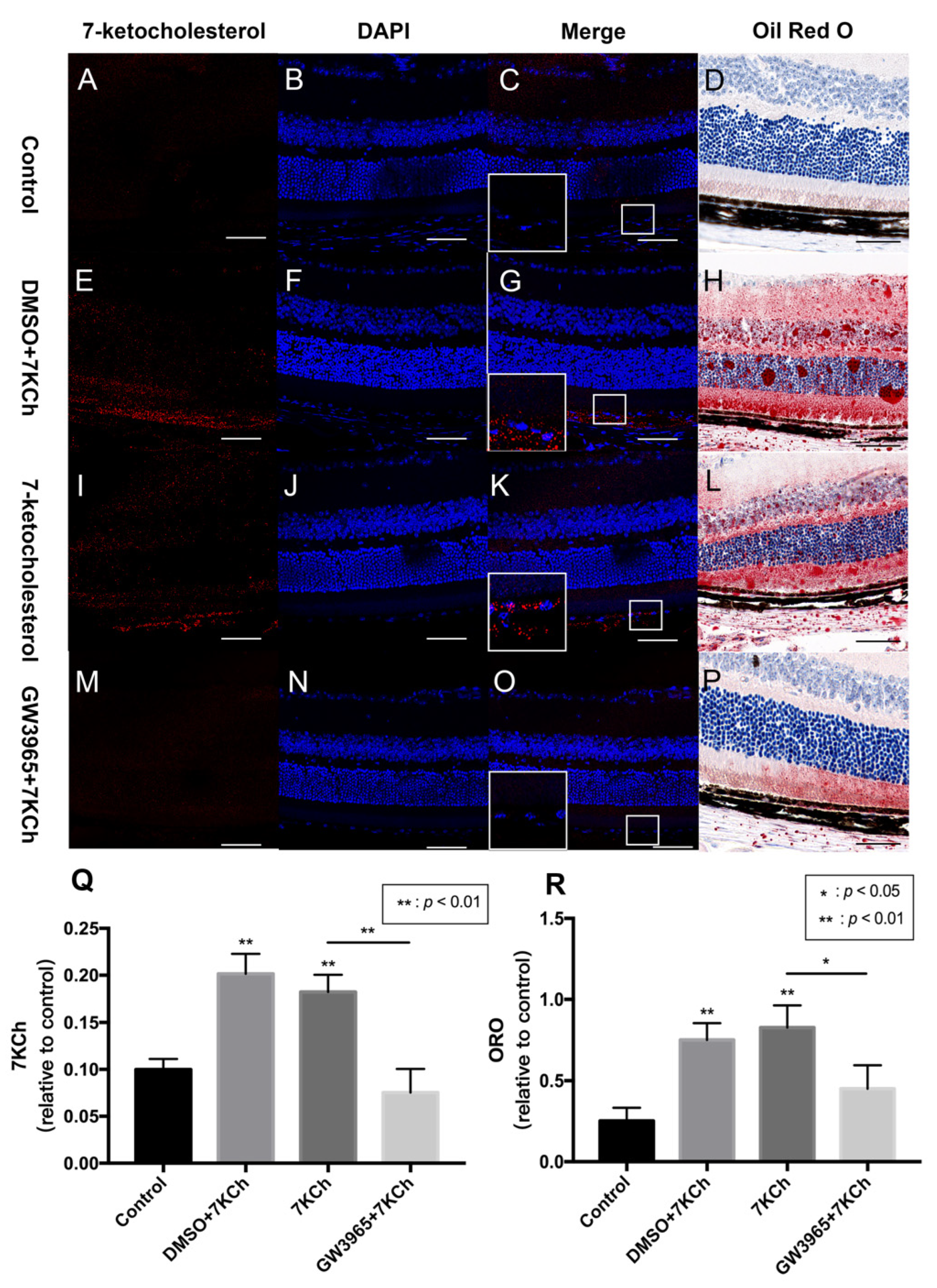
2.5. Immunofluorescence Staining
2.6. Oil Red O Staining of Retinas and ARPE-19 Cells
2.7. Terminal Deoxynucleotidyl Transferase Mediated dUTP Nick end Labeling Assay of Mice Retinas
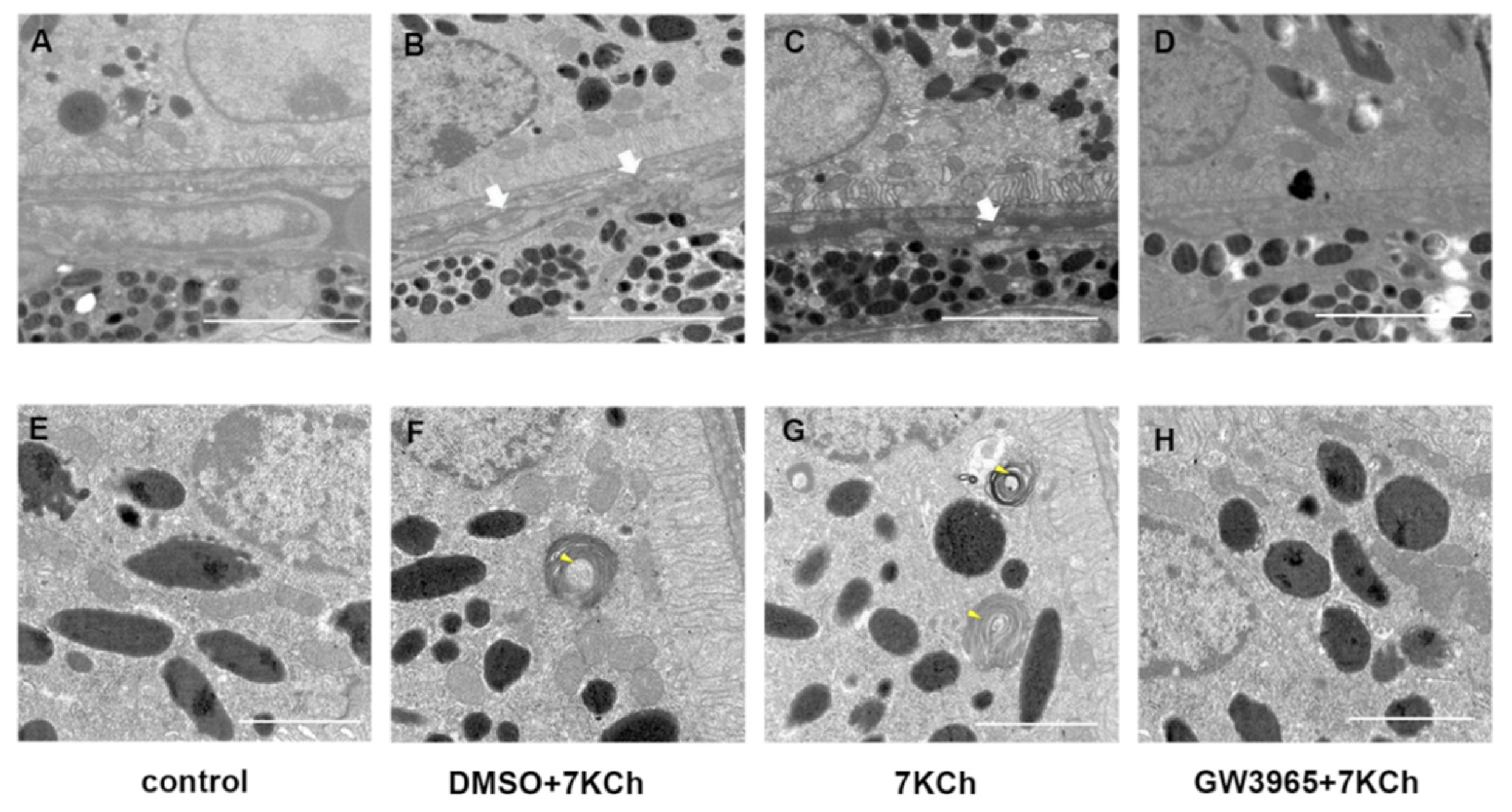
2.8. Transmission Electron Microscope Examination
2.9. Detection of Intracellular Reactive Oxygen Species (ROS)
2.10. JC-1 Assay
2.11. Western Blot
2.12. Statistical Analysis
3. Results
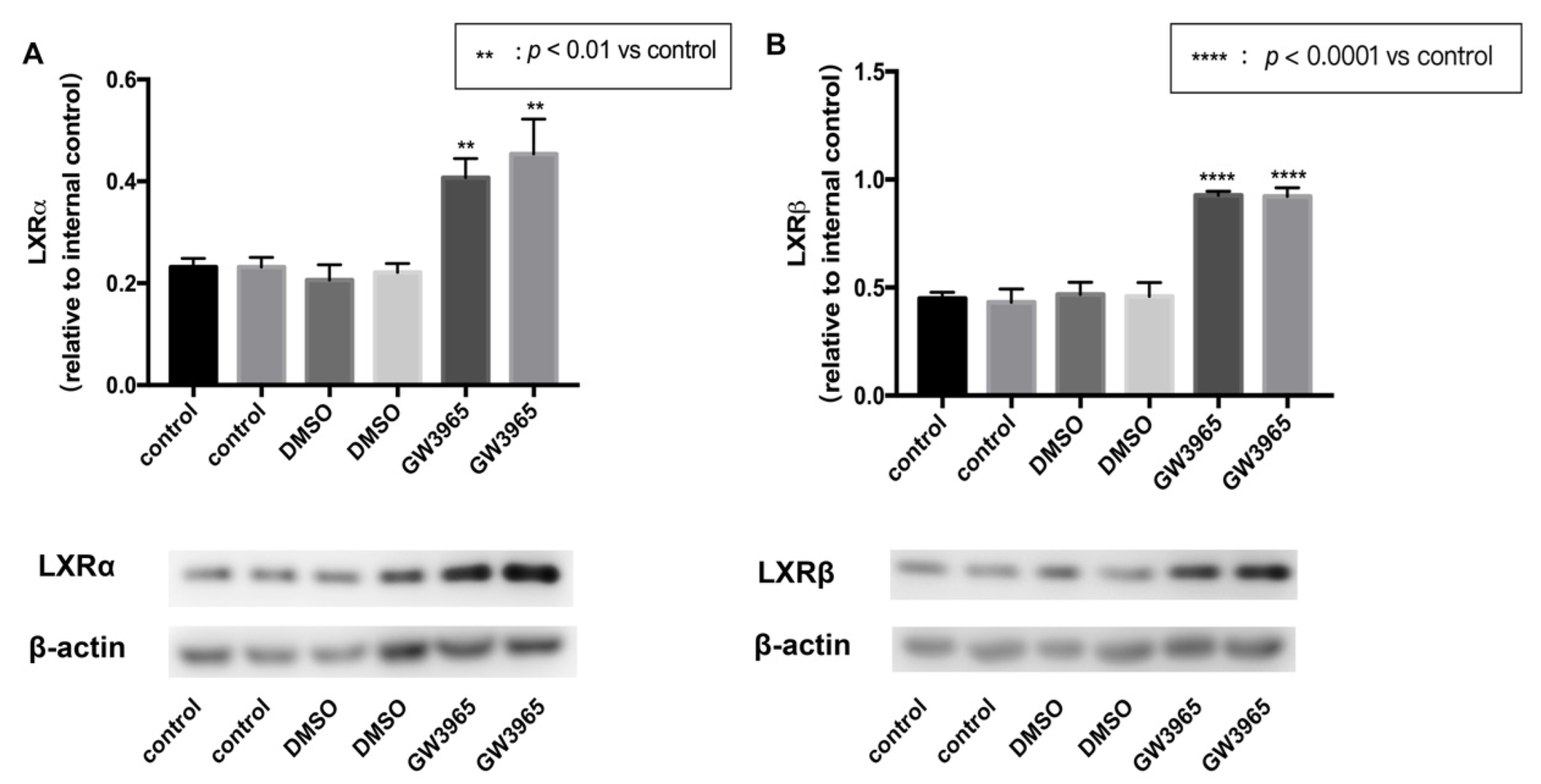
3.1. LXRs Activation in C57BL/6 Mice
3.2. 7KCh Immunofluorescence and Oil Red O Staining of Retinas
3.3. Apoptosis of RPE Cells Detected by TUNEL Assay
3.4. Ultrastructural Observations
3.5. 7KCh Immunocytofluorescence and Oil Red O Staining of ARPE-19 Cells
3.6. Apoptosis of ARPE-19 Cells Measured by ROS Level
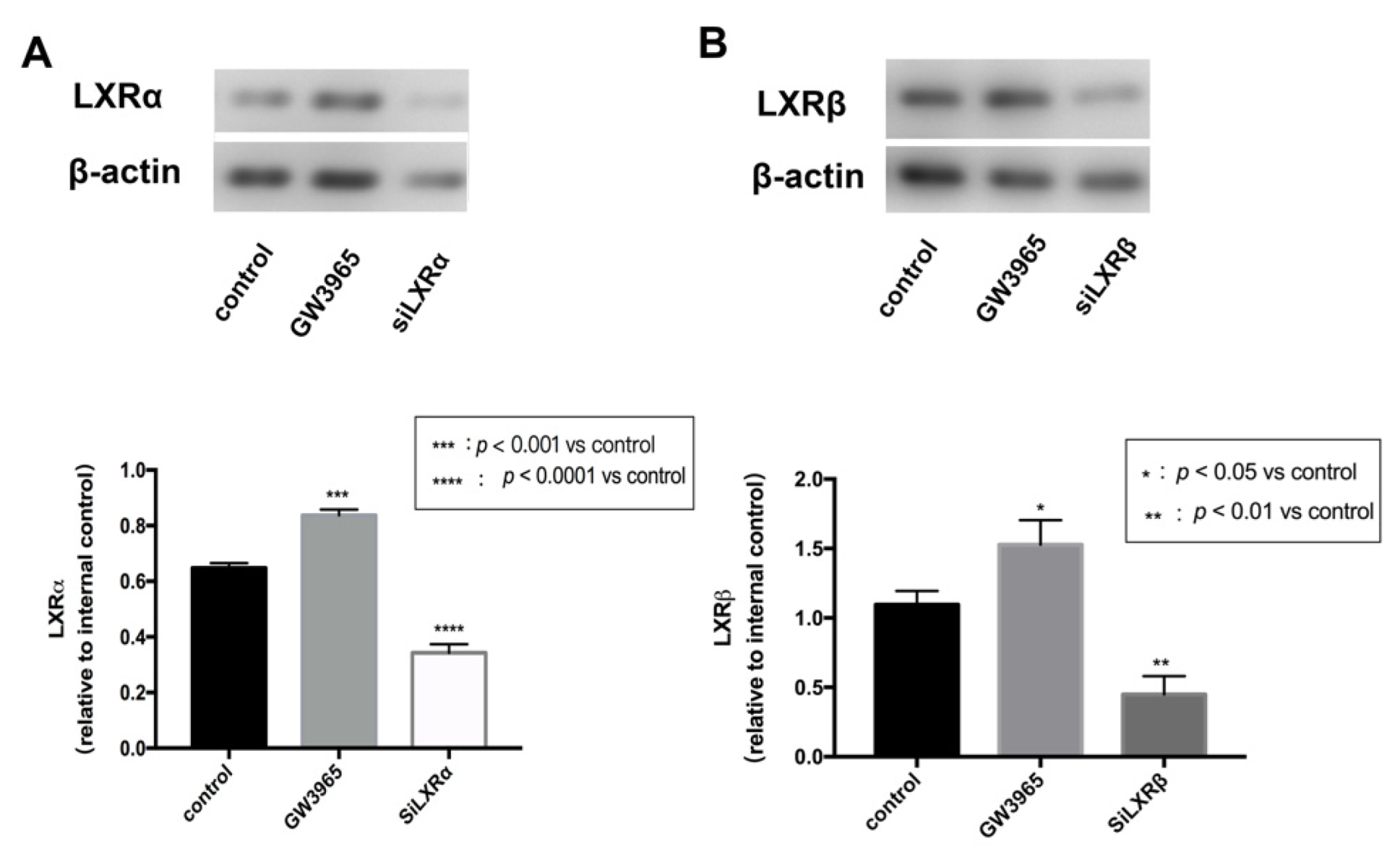
3.7. LXRs Activation in ARPE-19 Cells
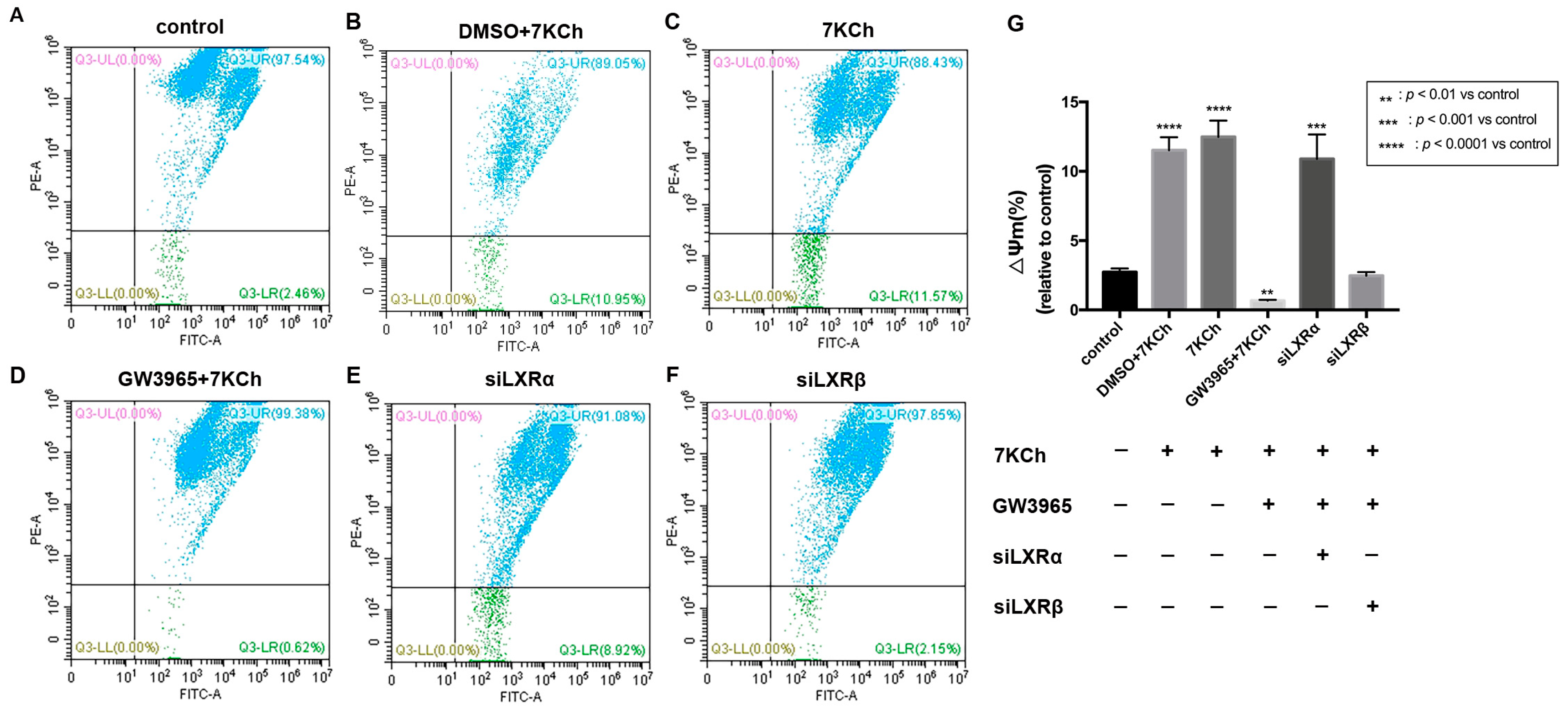
3.8. Flow Cytometric Analysis of Mitochondrial Membrane Potential in ARPE-19 Cells
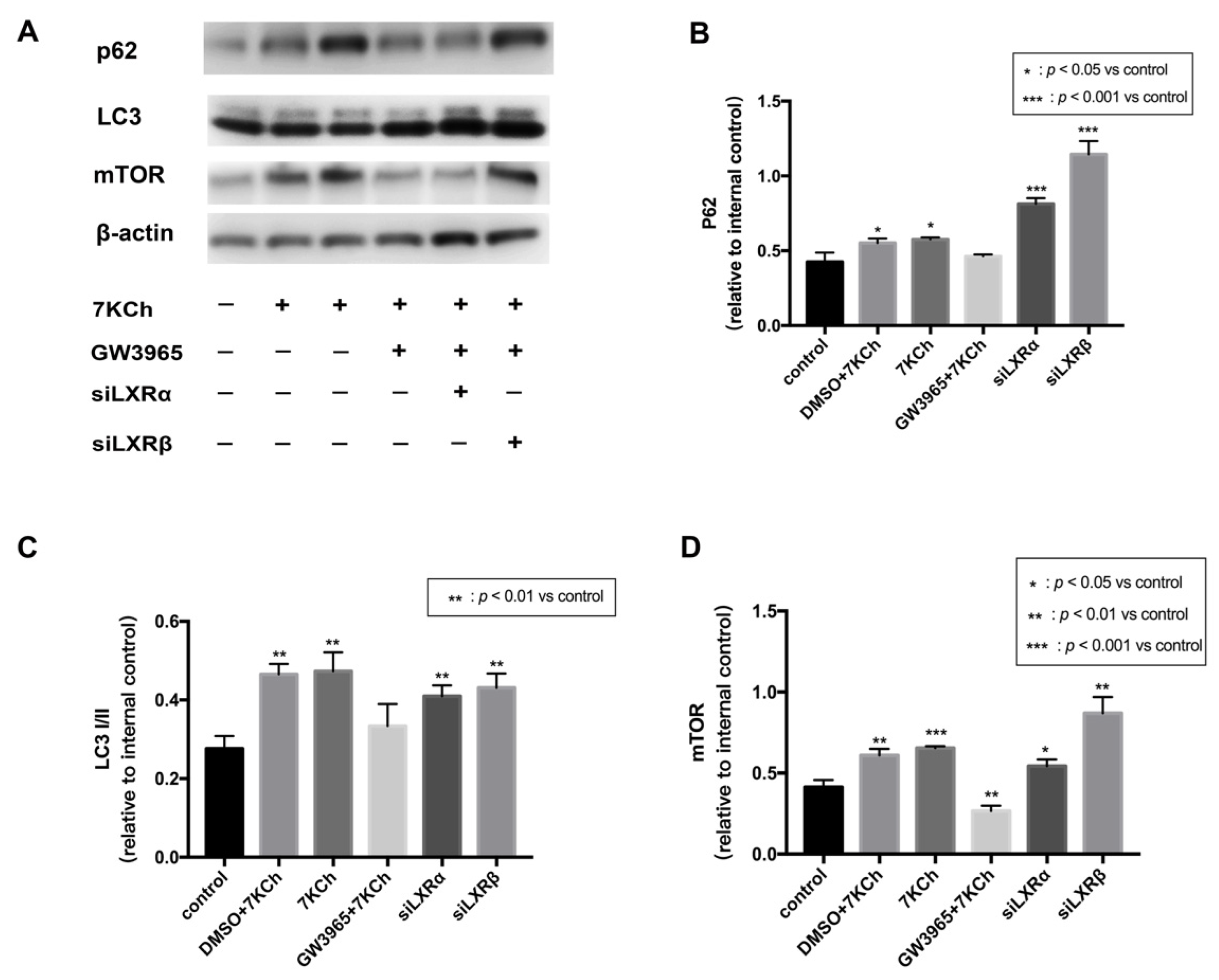
3.9. Western Blot Analysis of Protein in ARPE-19 Cells
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A
CCK-8 Assay for Evaluation of Cell Viability after GW3965 Treatment
References
- Mitchell, P.; Liew, G.; Gopinath, B.; Wong, T.Y. Age-related macular degeneration. Lancet 2018, 392, 1147–1159. [Google Scholar] [CrossRef]
- Zajac-Pytrus, H.M.; Pilecka, A.; Turno-Krecicka, A.; Adamiec-Mroczek, J.; Misiuk-Hojlo, M. The Dry Form of Age-Related Macular Degeneration (AMD): The Current Concepts of Pathogenesis and Prospects for Treatment. Adv. Clin. Exp. Med. 2015, 24, 1099–1104. [Google Scholar] [CrossRef]
- Fritsche, L.G.; Igl, W.; Bailey, J.N.C.; Grassmann, F.; Sengupta, S.; Bragg-Gresham, J.L.; Burdon, K.P.; Hebbring, S.J.; Wen, C.; Gorski, M.; et al. A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat. Genet. 2016, 48, 134–143. [Google Scholar] [CrossRef] [Green Version]
- Grassmann, F.; Heid, I.M.; Weber, B.H.; International, A.M.D.G.C. Recombinant Haplotypes Narrow the ARMS2/HTRA1 Association Signal for Age-Related Macular Degeneration. Genetics 2017, 205, 919–924. [Google Scholar] [CrossRef]
- Yang, C.; Xie, L.; Gu, Q.; Qiu, Q.; Wu, X.; Yin, L. 7-Ketocholesterol disturbs RPE cells phagocytosis of the outer segment of photoreceptor and induces inflammation through ERK signaling pathway. Exp. Eye Res. 2019, 189, 107849. [Google Scholar] [CrossRef]
- Riazi-Esfahani, M.; Kuppermann, B.D.; Kenney, M.C. The Role of Mitochondria in AMD: Current Knowledge and Future Applications. J. Ophthalmic. Vis. Res. 2017, 12, 424–428. [Google Scholar] [CrossRef]
- Dernie, F. Mitophagy in Parkinson’s disease: From pathogenesis to treatment target. Neurochem. Int. 2020, 138, 104756. [Google Scholar] [CrossRef]
- Mani, S.; Swargiary, G.; Chadha, R. Mitophagy impairment in neurodegenerative diseases: Pathogenesis and therapeutic interventions. Mitochondrion 2021, 57, 270–293. [Google Scholar] [CrossRef]
- Hyttinen, J.M.T.; Viiri, J.; Kaarniranta, K.; Blasiak, J. Mitochondrial quality control in AMD: Does mitophagy play a pivotal role? Cell Mol. Life Sci. 2018, 75, 2991–3008. [Google Scholar] [CrossRef] [Green Version]
- Hong, C.; Tontonoz, P. Liver X receptors in lipid metabolism: Opportunities for drug discovery. Nat. Rev. Drug Discov. 2014, 13, 433–444. [Google Scholar] [CrossRef]
- Choudhary, M.; Ismail, E.N.; Yao, P.L.; Tayyari, F.; Radu, R.A.; Nusinowitz, S.; Boulton, M.E.; Apte, R.S.; Ruberti, J.W.; Handa, J.T.; et al. LXRs regulate features of age-related macular degeneration and may be a potential therapeutic target. JCI Insight 2020, 5, e131928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Bortnick, A.E.; Nickel, M.; Dhanasekaran, P.; Subbaiah, P.V.; Lund-Katz, S.; Rothblat, G.H.; Phillips, M.C. Effects of apolipoprotein A-I on ATP-binding cassette transporter A1-mediated efflux of macrophage phospholipid and cholesterol: Formation of nascent high density lipoprotein particles. J. Biol. Chem. 2003, 278, 42976–42984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sallam, T.; Jones, M.C.; Gilliland, T.; Zhang, L.; Wu, X.; Eskin, A.; Sandhu, J.; Casero, D.; Vallim, T.Q.; Hong, C.; et al. Feedback modulation of cholesterol metabolism by the lipid-responsive non-coding RNA LeXis. Nature 2016, 534, 124–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Storti, F.; Raphael, G.; Griesser, V.; Klee, K.; Drawnel, F.; Willburger, C.; Scholz, R.; Langmann, T.; von Eckardstein, A.; Fingerle, J.; et al. Regulated efflux of photoreceptor outer segment-derived cholesterol by human RPE cells. Exp. Eye Res. 2017, 165, 65–77. [Google Scholar] [CrossRef]
- A-Gonzalez, N.; Bensinger, S.J.; Hong, C.; Beceiro, S.; Bradley, M.N.; Zelcer, N.; Deniz, J.; Ramirez, C.; Diaz, M.; Gallardo, G.; et al. Apoptotic cells promote their own clearance and immune tolerance through activation of the nuclear receptor LXR. Immunity 2009, 31, 245–258. [Google Scholar] [CrossRef] [Green Version]
- Pascual-Garcia, M.; Valledor, A.F. Biological roles of liver X receptors in immune cells. Arch. Immunol. Ther. Exp. 2012, 60, 235–249. [Google Scholar] [CrossRef]
- Jakobsson, T.; Treuter, E.; Gustafsson, J.A.; Steffensen, K.R. Liver X receptor biology and pharmacology: New pathways, challenges and opportunities. Trends Pharmacol. Sci. 2012, 33, 394–404. [Google Scholar] [CrossRef]
- Graham, A. Mitochondrial regulation of macrophage cholesterol homeostasis. Free Radic. Biol. Med. 2015, 89, 982–992. [Google Scholar] [CrossRef]
- Lemasters, J.J. Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res. 2005, 8, 3–5. [Google Scholar] [CrossRef]
- Saliba-Gustafsson, P.; Pedrelli, M.; Gertow, K.; Werngren, O.; Janas, V.; Pourteymour, S.; Baldassarre, D.; Tremoli, E.; Veglia, F.; Rauramaa, R.; et al. Subclinical atherosclerosis and its progression are modulated by PLIN2 through a feed-forward loop between LXR and autophagy. J. Intern. Med. 2019, 286, 660–675. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; He, P.; Huang, Y.; Li, Y.F.; Lu, J.; Li, M.; Kurihara, H.; Luo, Z.; Meng, T.; Onishi, M.; et al. Selective autophagy of intracellular organelles: Recent research advances. Theranostics 2021, 11, 222–256. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, Y.; Sou, Y.S.; Kageyama, S.; Takahashi, T.; Ueno, T.; Tanaka, K.; Komatsu, M.; Ichimura, Y. LC3B is indispensable for selective autophagy of p62 but not basal autophagy. Biochem. Biophys. Res. Commun. 2014, 446, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Zhang, Y.J.; Cai, Y.; Xu, M.H. The role of mitochondria in mTOR-regulated longevity. Biol. Rev. Camb. Philos. Soc. 2015, 90, 167–181. [Google Scholar] [CrossRef] [PubMed]
- Pikuleva, I.A.; Curcio, C.A. Cholesterol in the retina: The best is yet to come. Prog. Retin. Eye Res. 2014, 41, 64–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spaide, R.F.; Ho-Spaide, W.C.; Browne, R.W.; Armstrong, D. Characterization of peroxidized lipids in Bruch’s membrane. Retina 1999, 19, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Jun, S.; Datta, S.; Wang, L.; Pegany, R.; Cano, M.; Handa, J.T. The impact of lipids, lipid oxidation, and inflammation on AMD, and the potential role of miRNAs on l ipid metabolism in the RPE. Exp. Eye Res. 2019, 181, 346–355. [Google Scholar] [CrossRef]
- Handa, J.T.; Cano, M.; Wang, L.; Datta, S.; Liu, T. Lipids, oxidized lipids, oxidation-specific epitopes, and Age-related Macular Degeneration. Biochim. Et Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 430–440. [Google Scholar] [CrossRef] [Green Version]
- Tserentsoodol, N.; Sztein, J.; Campos, M.; Gordiyenko, N.V.; Fariss, R.N.; Lee, J.W.; Fliesler, S.J.; Rodriguez, I.R. Uptake of cholesterol by the retina occurs primarily via a low density lipoprotein receptor-mediated process. Mol. Vis. 2006, 12, 1306–1318. [Google Scholar]
- Ramachandra Rao, S.; Fliesler, S.J. Cholesterol homeostasis in the vertebrate retina: Biology and pathobiology. J. Lipid Res. 2021, 62, 100057. [Google Scholar] [CrossRef]
- Parikh, S.; Le, A.; Davenport, J.; Gorin, M.B.; Nusinowitz, S.; Matynia, A. An Alternative and Validated Injection Method for Accessing the Subretinal Space via a Transcleral Posterior Approach. J. Vis. Exp. 2016, 118, e54808. [Google Scholar] [CrossRef]
- Li, W.; Laskar, A.; Sultana, N.; Osman, E.; Ghosh, M.; Li, Q.; Yuan, X.M. Cell death induced by 7-oxysterols via lysosomal and mitochondrial pathways is p53-dependent. Free Radic. Biol. Med. 2012, 53, 2054–2061. [Google Scholar] [CrossRef] [PubMed]
- Alba, G.; Reyes-Quiroz, M.E.; Saenz, J.; Geniz, I.; Jimenez, J.; Martin-Nieto, J.; Pintado, E.; Sobrino, F.; Santa-Maria, C. 7-Keto-cholesterol and 25-hydroxy-1 cholesterol rapidly enhance ROS production in human neutrophils. Eur. J. Nutr. 2016, 55, 2485–2492. [Google Scholar] [CrossRef] [PubMed]
- Nury, T.; Zarrouk, A.; Vejux, A.; Doria, M.; Riedinger, J.M.; Delage-Mourroux, R.; Lizard, G. Induction of oxiapoptophagy, a mixed mode of cell death associated with oxidative stress, apoptosis and autophagy, on 7-ketocholesterol-treated 158N murine oligodendrocytes: Impairment by alpha-tocopherol. Biochem. Biophys. Res. Commun. 2014, 446, 714–719. [Google Scholar] [CrossRef] [PubMed]
- Kaarniranta, K.; Sinha, D.; Blasiak, J.; Kauppinen, A.; Vereb, Z.; Salminen, A.; Boulton, M.E.; Petrovski, G. Autophagy and heterophagy dysregulation leads to retinal pigment epithelium dysfunction and development of age-related macular degeneration. Autophagy 2013, 9, 973–984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, E.; Cuervo, A.M. Autophagy gone awry in neurodegenerative diseases. Nat. Neurosci. 2010, 13, 805–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rami, A. Review: Autophagy in neurodegeneration: Firefighter and/or incendiarist? Neuropathol. Appl. Neurobiol. 2009, 35, 449–461. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, L.; Gu, Q.; Wu, X.; Yin, L. Activation of LXRs Reduces Oxysterol Lipotoxicity in RPE Cells by Promoting Mitochondrial Function. Nutrients 2022, 14, 2473. https://doi.org/10.3390/nu14122473
Xie L, Gu Q, Wu X, Yin L. Activation of LXRs Reduces Oxysterol Lipotoxicity in RPE Cells by Promoting Mitochondrial Function. Nutrients. 2022; 14(12):2473. https://doi.org/10.3390/nu14122473
Chicago/Turabian StyleXie, Lirong, Qing Gu, Xingwei Wu, and Lili Yin. 2022. "Activation of LXRs Reduces Oxysterol Lipotoxicity in RPE Cells by Promoting Mitochondrial Function" Nutrients 14, no. 12: 2473. https://doi.org/10.3390/nu14122473
APA StyleXie, L., Gu, Q., Wu, X., & Yin, L. (2022). Activation of LXRs Reduces Oxysterol Lipotoxicity in RPE Cells by Promoting Mitochondrial Function. Nutrients, 14(12), 2473. https://doi.org/10.3390/nu14122473