
Adherence to Gluten-Free Diet Restores Alpha Diversity in Celiac People but the Microbiome Composition Is Different to Healthy People

 ,
, 
 , ,
, ,  , and
, and
Abstract
:1. Introduction
2. Methods
3. Results
3.1. 16S rRNA V3-V4 Region Sequencing Results
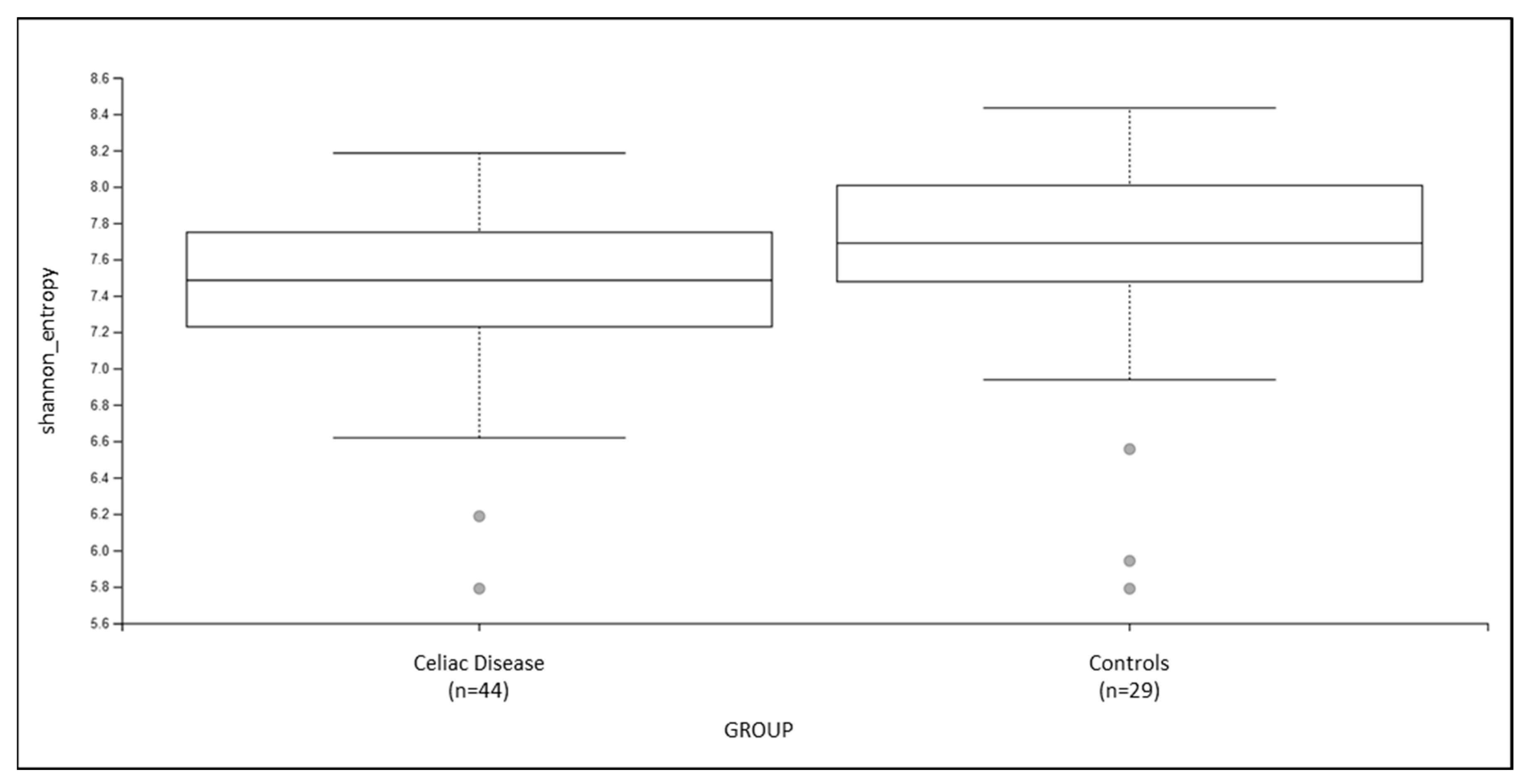
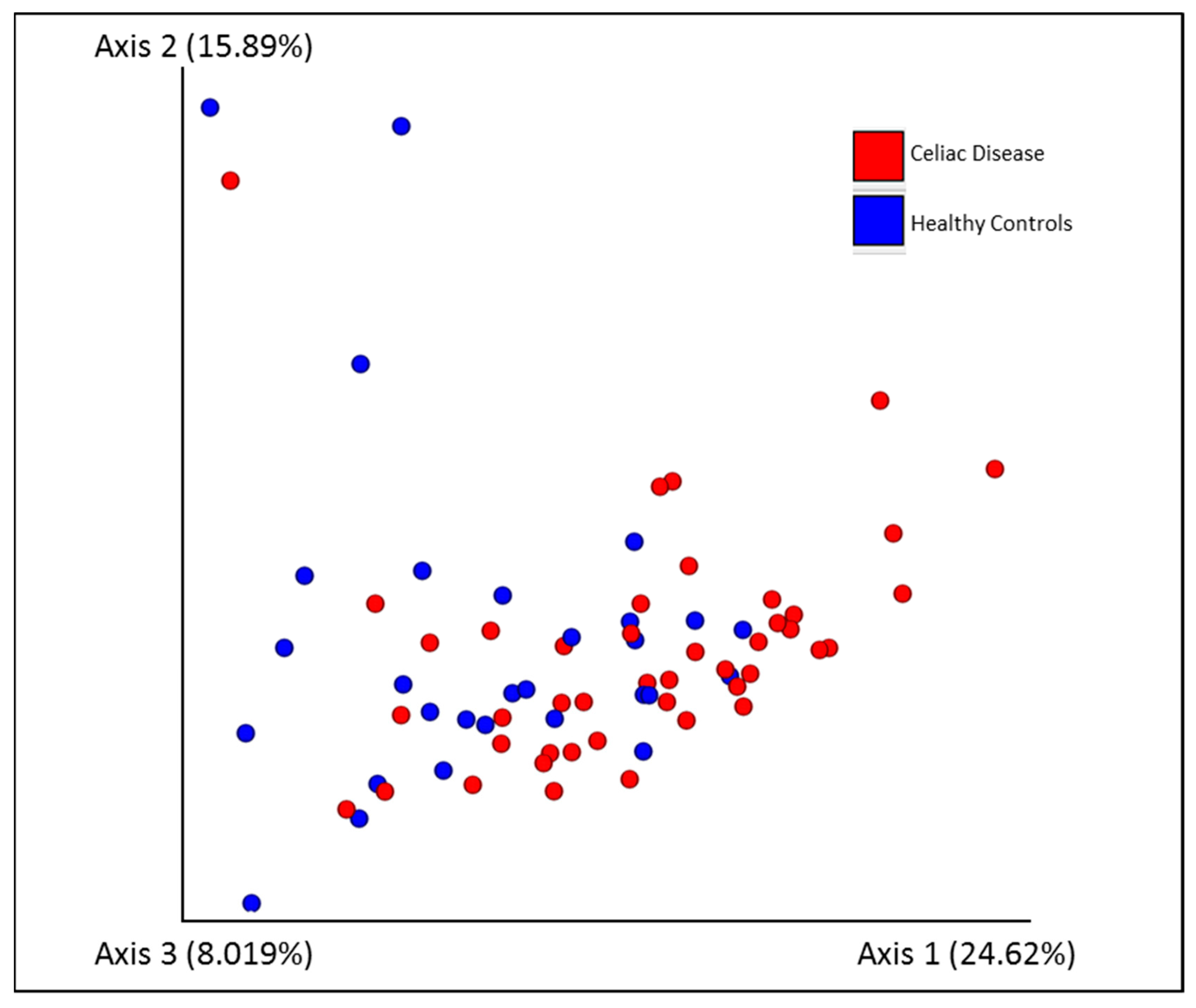
3.2. Fecal Microbiota Diversity Analysis
3.3. Microbial Signature Detection
3.4. Differential Analysis of Predicted Functional Content
3.5. Dysbiosis Analysis of CD Patients vs. Controls
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lebwohl, B.; Rubio-Tapia, A. Epidemiology, Presentation, and Diagnosis of Celiac Disease. Gastroenterology 2021, 160, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Voisine, J.; Abadie, V. Interplay Between Gluten, HLA, Innate and Adaptive Immunity Orchestrates the Development of Coeliac Disease. Front. Immunol. 2021, 12, 674313. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.Y.; Zhang, X.; Zhou, Y.Y.; Jiang, C.M.; Shi, Y.D. Infection, antibiotic exposure, and risk of celiac disease: A systematic review and meta-analysis. J. Gastroenterol. Hepatol. 2020, 35, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Kahrs, C.R.; Chuda, K.; Tapia, G.; Stene, L.C.; Mårild, K.; Rasmussen, T.; Rønningen, K.S.; Lundin, K.E.A.; Kramna, L.; Cinek, O.; et al. Enterovirus as trigger of coeliac disease: Nested case-control study within prospective birth cohort. BMJ 2019, 364, l231. [Google Scholar] [CrossRef] [Green Version]
- Valitutti, F.; Cucchiara, S.; Fasano, A. Celiac Disease and the Microbiome. Nutrients 2019, 11, 2403. [Google Scholar] [CrossRef] [Green Version]
- Abdukhakimova, D.; Dossybayeva, K.; Poddighe, D. Fecal and Duodenal Microbiota in Pediatric Celiac Disease. Front. Pediatr. 2021, 9, 652208. [Google Scholar] [CrossRef]
- Caio, G.; Lungaro, L.; Segata, N.; Guarino, M.; Zoli, G.; Volta, U.; De Giorgio, R. Effect of Gluten-Free Diet on Gut Microbiota Composition in Patients with Celiac Disease and Non-Celiac Gluten/Wheat Sensitivity. Nutrients 2020, 12, 1832. [Google Scholar] [CrossRef]
- Schiepatti, A.; Bacchi, S.; Biagi, F.; Panelli, S.; Betti, E.; Corazza, G.R.; Capelli, E.; Ciccocioppo, R. Relationship between duodenal microbiota composition, clinical features at diagnosis, and persistent symptoms in adult Coeliac disease. Dig. Liver Dis. 2021, 53, 972–979. [Google Scholar] [CrossRef]
- Wacklin, P.; Laurikka, P.; Lindfors, K.; Collin, P.; Salmi, T.; Lähdeaho, M.L.; Saavalainen, P.; Mäki, M.; Mättö, J.; Kurppa, K.; et al. Altered duodenal microbiota composition in celiac disease patients suffering from persistent symptoms on a long-term gluten-free diet. Am. J. Gastroenterol. 2014, 109, 1933–1941. [Google Scholar] [CrossRef]
- Andriulli, A.; Bevilacqua, A.; Palmieri, O.; Latiano, A.; Fontana, R.; Gioffreda, D.; Castellana, S.; Mazza, T.; Panza, A.; Menzaghi, C.; et al. Healthy and pro-inflammatory gut ecology plays a crucial role in the digestion and tolerance of a novel Gluten Friendly™ bread in celiac subjects: A randomized, double blind, placebo control in vivo study. Food Funct. 2022, 13, 1299–1315. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020; Available online: https://www.R-project.org/ (accessed on 1 January 2020).
- Estaki, M.; Jiang, L.; Bokulich, N.A.; McDonald, D.; González, A.; Kosciolek, T.; Martino, C.; Zhu, Q.; Birmingham, A.; Vázquez-Baeza, Y.; et al. QIIME 2 Enables Comprehensive End-to-End Analysis of Diverse Microbiome Data and Comparative Studies with Publicly Available Data. Curr. Protoc. Bioinform. 2020, 70, e100. [Google Scholar] [CrossRef] [PubMed]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree: Computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 2009, 26, 1641–1650. [Google Scholar] [CrossRef] [PubMed]
- McDonald, D.; Price, M.N.; Goodrich, J.; Nawrocki, E.P.; DeSantis, T.Z.; Probst, A.; Andersen, G.L.; Knight, R.; Hugenholtz, P. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 2012, 6, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Shannon, C.E.; Weaver, W.; Wiener, N. The Mathematical Theory of Communication. Phys. Today 1950, 3, 31–32. [Google Scholar] [CrossRef] [Green Version]
- Faith, D.P. Conservation evaluation and phylogenetic diversity. Biol. Conserv. 1992, 61, 1–10. [Google Scholar] [CrossRef]
- Pielou, E.C. The measurement of diversity in different types of biological collections. J. Theor. Biol. 1966, 13, 131–144. [Google Scholar] [CrossRef]
- Jaccard, P. Nouvelles recherches sur la distribution florale. Bull. Soc. Vaud. Sci. Nat. 1908, 44, 223–270. [Google Scholar] [CrossRef]
- Sørensen, T. A Method of Establishing Groups of Equal Amplitude in Plant Sociology Based on Similarity of Species Content and Its Application to Analyses of the Vegetation on Danish Commons. Biol. Skr. 1948, 5, 1–34. [Google Scholar]
- Lozupone, C.; Knight, R. UniFrac: A new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 2005, 71, 8228–8235. [Google Scholar] [CrossRef] [Green Version]
- Vázquez-Baeza, Y.; Pirrung, M.; Gonzalez, A.; Knight, R. EMPeror: A tool for visualizing high-throughput microbial community data. Gigascience 2013, 2, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandal, S.; Van Treuren, W.; White, R.A.; Eggesbø, M.; Knight, R.; Peddada, S.D. Analysis of composition of microbiomes: A novel method for studying microbial composition. Microb. Ecol. Health Dis. 2015, 26, 27663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, J.; Shi, P.; Li, H. Generalized linear models with linear constraints for microbiome compositional data. Biometrics 2019, 75, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Zou, H.; Hastie, T. Regularization and variable selection via the elastic net. J. R. Stat. Soc. Ser. B Stat. Methodol. 2005, 67, 301–320. [Google Scholar] [CrossRef] [Green Version]
- Rivera-Pinto, J.; Egozcue, J.J.; Pawlowsky-Glahn, V.; Paredes, R.; Noguera-Julian, M.; Calle, M.L. Balances: A New Perspective for Microbiome Analysis. mSystems 2018, 3, e00053-18. [Google Scholar] [CrossRef] [Green Version]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef]
- Caspi, R.; Billington, R.; Keseler, I.M.; Kothari, A.; Krummenacker, M.; Midford, P.E.; Ong, W.K.; Paley, S.; Subhraveti, P.; Karp, P.D. The MetaCyc database of metabolic pathways and enzymes—A 2019 update. Nucleic Acids Res. 2020, 48, D445–D453. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, A.D.; Reid, J.N.; Macklaim, J.M.; McMurrough, T.A.; Edgell, D.R.; Gloor, G.B. Unifying the analysis of high-throughput sequencing datasets: Characterizing RNA-seq, 16S rRNA gene sequencing and selective growth experiments by compositional data analysis. Microbiome 2014, 2, 15. [Google Scholar] [CrossRef] [Green Version]
- Lloyd-Price, J.; Arze, C.; Ananthakrishnan, A.N.; Schirmer, M.; Avila-Pacheco, J.; Poon, T.W.; Andrews, E.; Ajami, N.J.; Bonham, K.S.; Brislawn, C.J.; et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature 2019, 569, 655–662. [Google Scholar] [CrossRef]
- Bashan, A.; Gibson, T.E.; Friedman, J.; Carey, V.J.; Weiss, S.T.; Hohmann, E.L.; Liu, Y.Y. Universality of human microbial dynamics. Nature 2016, 534, 259–262. [Google Scholar] [CrossRef] [Green Version]
- Montassier, E.; Al-Ghalith, G.A.; Hillmann, B.; Viskocil, K.; Kabage, A.J.; McKinlay, C.E.; Sadowsky, M.J.; Khoruts, A.; Knights, D. CLOUD: A non-parametric detection test for microbiome outliers. Microbiome 2018, 6, 137. [Google Scholar] [CrossRef] [PubMed]
- Lahti, L.; Shetty, S. Tools for Microbiome Analysis in R: Version 1.6.0. 2017. Available online: http://microbiome.github.com/microbiome (accessed on 2 May 2019).
- McMurdie, P.J.; Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zafeiropoulou, K.; Nichols, B.; Mackinder, M.; Biskou, O.; Rizou, E.; Karanikolou, A.; Clark, C.; Buchanan, E.; Cardigan, T.; Duncan, H.; et al. Alterations in Intestinal Microbiota of Children With Celiac Disease at the Time of Diagnosis and on a Gluten-free Diet. Gastroenterology 2020, 159, 2039–2051.e20. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Mazcorro, J.F.; Noratto, G.; Remes-Troche, J.M. The Effect of Gluten-Free Diet on Health and the Gut Microbiota Cannot Be Extrapolated from One Population to Others. Nutrients 2018, 10, 1421. [Google Scholar] [CrossRef] [Green Version]
- Collado, M.C.; Donat, E.; Ribes-Koninckx, C.; Calabuig, M.; Sanz, Y. Imbalances in faecal and duodenal Bifidobacterium species composition in active and non-active coeliac disease. BMC Microbiol. 2008, 8, 232. [Google Scholar] [CrossRef] [Green Version]
- McCarville, J.L.; Dong, J.; Caminero, A.; Bermudez-Brito, M.; Jury, J.; Murray, J.A.; Duboux, S.; Steinmann, M.; Delley, M.; Tangyu, M.; et al. A Commensal Bifidobacterium longum Strain Prevents Gluten-Related Immunopathology in Mice through Expression of a Serine Protease Inhibitor. Appl. Environ. Microbiol. 2017, 83, e01323-17. [Google Scholar] [CrossRef] [Green Version]
- Vacca, M.; Celano, G.; Calabrese, F.M.; Portincasa, P.; Gobbetti, M.; De Angelis, M. The Controversial Role of Human Gut Lachnospiraceae. Microorganisms 2020, 8, 573. [Google Scholar] [CrossRef]
- Ugural, A.; Akyol, A. Can pseudocereals modulate microbiota by functioning as probiotics or prebiotics? Crit. Rev. Food Sci. Nutr. 2022, 62, 1725–1739. [Google Scholar] [CrossRef]
- Hansen, L.B.S.; Roager, H.M.; Søndertoft, N.B.; Gøbel, R.J.; Kristensen, M.; Vallès-Colomer, M.; Vieira-Silva, S.; Ibrügger, S.; Lind, M.V.; Mærkedahl, R.B.; et al. A low-gluten diet induces changes in the intestinal microbiome of healthy Danish adults. Nat. Commun. 2018, 9, 4630. [Google Scholar] [CrossRef] [Green Version]
- De Palma, G.; Capilla, A.; Nadal, I.; Nova, E.; Pozo, T.; Varea, V.; Polanco, I.; Castillejo, G.; López, A.; Garrote, J.A.; et al. Interplay between human leukocyte antigen genes and the microbial colonization process of the newborn intestine. Curr. Issues Mol. Biol. 2010, 12, 1–10. [Google Scholar]
- Leonard, M.M.; Karathia, H.; Pujolassos, M.; Troisi, J.; Valitutti, F.; Subramanian, P.; Camhi, S.; Kenyon, V.; Colucci, A.; Serena, G.; et al. Multi-omics analysis reveals the influence of genetic and environmental risk factors on developing gut microbiota in infants at risk of celiac disease. Microbiome 2020, 8, 130. [Google Scholar] [CrossRef] [PubMed]
- Collado, M.C.; Donat, E.; Ribes-Koninckx, C.; Calabuig, M.; Sanz, Y. Specific duodenal and faecal bacterial groups associated with paediatric coeliac disease. J. Clin. Pathol. 2009, 62, 264–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schippa, S.; Iebba, V.; Barbato, M.; Di Nardo, G.; Totino, V.; Checchi, M.P.; Longhi, C.; Maiella, G.; Cucchiara, S.; Conte, M.P. A distinctive ‘microbial signature’ in celiac pediatric patients. BMC Microbiol. 2010, 10, 175. [Google Scholar] [CrossRef] [PubMed]
- Rubio-Tapia, A.; Hill, I.D.; Kelly, C.P.; Calderwood, A.H.; Murray, J.A. American College of Gastroenterology ACG clinical guidelines: Diagnosis and management of celiac disease. Am. J. Gastroenterol. 2013, 108, 656–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Downey, L.; Houten, R.; Murch, S.; Longson, D.; Guideline Development Group. Recognition, assessment, and management of coeliac disease: Summary of updated NICE guidance. BMJ 2015, 351, h4513. [Google Scholar] [CrossRef] [PubMed]
- Rondanelli, M.; Faliva, M.A.; Gasparri, C.; Peroni, G.; Naso, M.; Picciotto, G.; Riva, A.; Nichetti, M.; Infantino, V.; Alalwan, T.A.; et al. Micronutrients Dietary Supplementation Advices for Celiac Patients on Long-Term Gluten-Free Diet with Good Compliance: A Review. Medicina 2019, 55, 337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, L.H.; Miller, J.W.; de Groot, L.; Rosenberg, I.H.; Smith, A.D.; Refsum, H.; Raiten, D.J. Biomarkers of Nutrition for Development (BOND): Vitamin B-12 Review. J. Nutr. 2018, 148, 1995S–2027S. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
| N of patients | 46 |
| Female | 28 |
| Age in years, mean ± SD | 38.6 ± 18.2 |
| Measurement | |
| Height, means ± SD in cm | 168.6 ± 9.5 |
| BMI mean ± SD | 22.7 ± 3.5 |
| Duration of GFD in years mean ± SD | 11.1 ± 7.4 |
| 2–3.9 | 4 |
| 4–5.9 | 10 |
| 6–7.9 | 7 |
| >8 | 25 |
| HLA | |
| DQ2 | 40 |
| DQ8 | 2 |
| DQ2–DQ8 | 4 |
| Healthy Control vs. Celiac Disease | |
|---|---|
| Shannon’s Entropy | 0.07 |
| Pielou’s evenness | 0.75 |
| Number of Observed Features | 0.09 |
| Faith’s Phylogenetic Distance | 0.34 |
| Healthy Control vs. Celiac Disease | |
|---|---|
| Jaccard similarity | 0.004 |
| Bray-Curtis dissimilarity | 0.005 |
| Unweighted UniFrac dissimilarity | 0.06 |
| Weighted UniFrac dissimilarity | 0.001 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palmieri, O.; Castellana, S.; Bevilacqua, A.; Latiano, A.; Latiano, T.; Panza, A.; Fontana, R.; Ippolito, A.M.; Biscaglia, G.; Gentile, A.; et al. Adherence to Gluten-Free Diet Restores Alpha Diversity in Celiac People but the Microbiome Composition Is Different to Healthy People. Nutrients 2022, 14, 2452. https://doi.org/10.3390/nu14122452
Palmieri O, Castellana S, Bevilacqua A, Latiano A, Latiano T, Panza A, Fontana R, Ippolito AM, Biscaglia G, Gentile A, et al. Adherence to Gluten-Free Diet Restores Alpha Diversity in Celiac People but the Microbiome Composition Is Different to Healthy People. Nutrients. 2022; 14(12):2452. https://doi.org/10.3390/nu14122452
Chicago/Turabian StylePalmieri, Orazio, Stefano Castellana, Antonio Bevilacqua, Anna Latiano, Tiziana Latiano, Anna Panza, Rosanna Fontana, Antonio Massimo Ippolito, Giuseppe Biscaglia, Annamaria Gentile, and et al. 2022. "Adherence to Gluten-Free Diet Restores Alpha Diversity in Celiac People but the Microbiome Composition Is Different to Healthy People" Nutrients 14, no. 12: 2452. https://doi.org/10.3390/nu14122452
APA StylePalmieri, O., Castellana, S., Bevilacqua, A., Latiano, A., Latiano, T., Panza, A., Fontana, R., Ippolito, A. M., Biscaglia, G., Gentile, A., Gioffreda, D., Decina, I., Tricarico, M., Sinigaglia, M., Corbo, M. R., Mazza, T., Perri, F., & Lamacchia, C. (2022). Adherence to Gluten-Free Diet Restores Alpha Diversity in Celiac People but the Microbiome Composition Is Different to Healthy People. Nutrients, 14(12), 2452. https://doi.org/10.3390/nu14122452