
High Iron Exposure from the Fetal Stage to Adulthood in Mice Alters Lipid Metabolism


Abstract
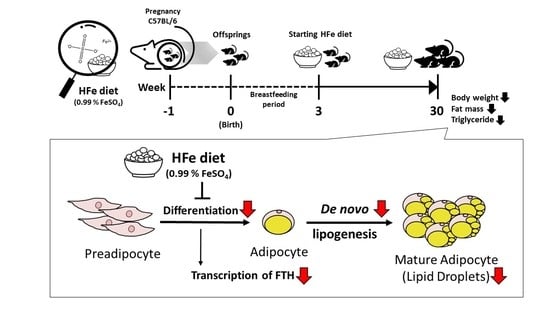
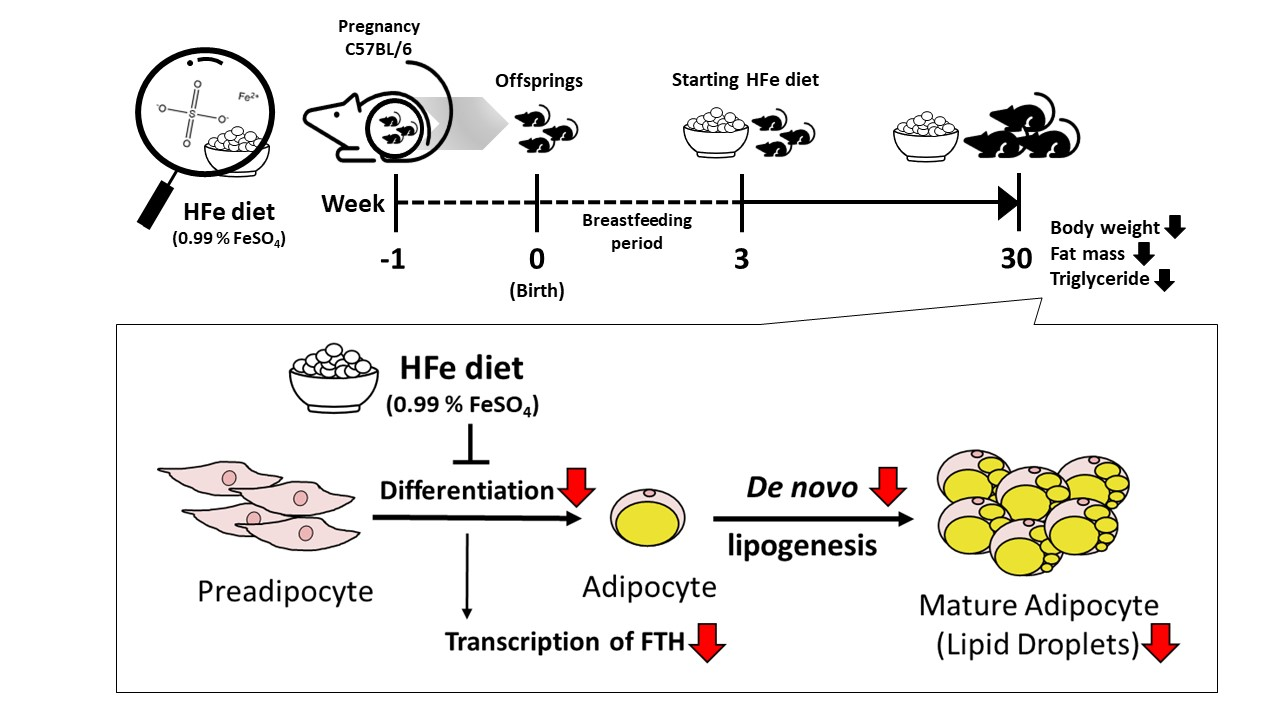
1. Introduction
2. Materials and Methods
2.1. Animal Experimental Protocol
2.2. Biochemistry Measurement in Blood Plasma and Tissue
2.3. Oral Glucose Tolerance Test (OGTT)
2.4. Histological Examination
2.5. Determination of Malondialdehyde (MDA) Level in Mouse Tissues
2.6. Measurement of Total Glutathione (tGSH) Content
2.7. Ferrozine Essay
2.8. cDNA Extraction and Quantitative Real-Time PCR
2.9. Western Blot
2.10. Methylation Specific Polymerase Chain Reaction (MSP)
2.11. Prediction of Transcription Factor (TF) Binding Sites by Bioinformatics Analysis
2.12. Statistical Analysis
3. Results
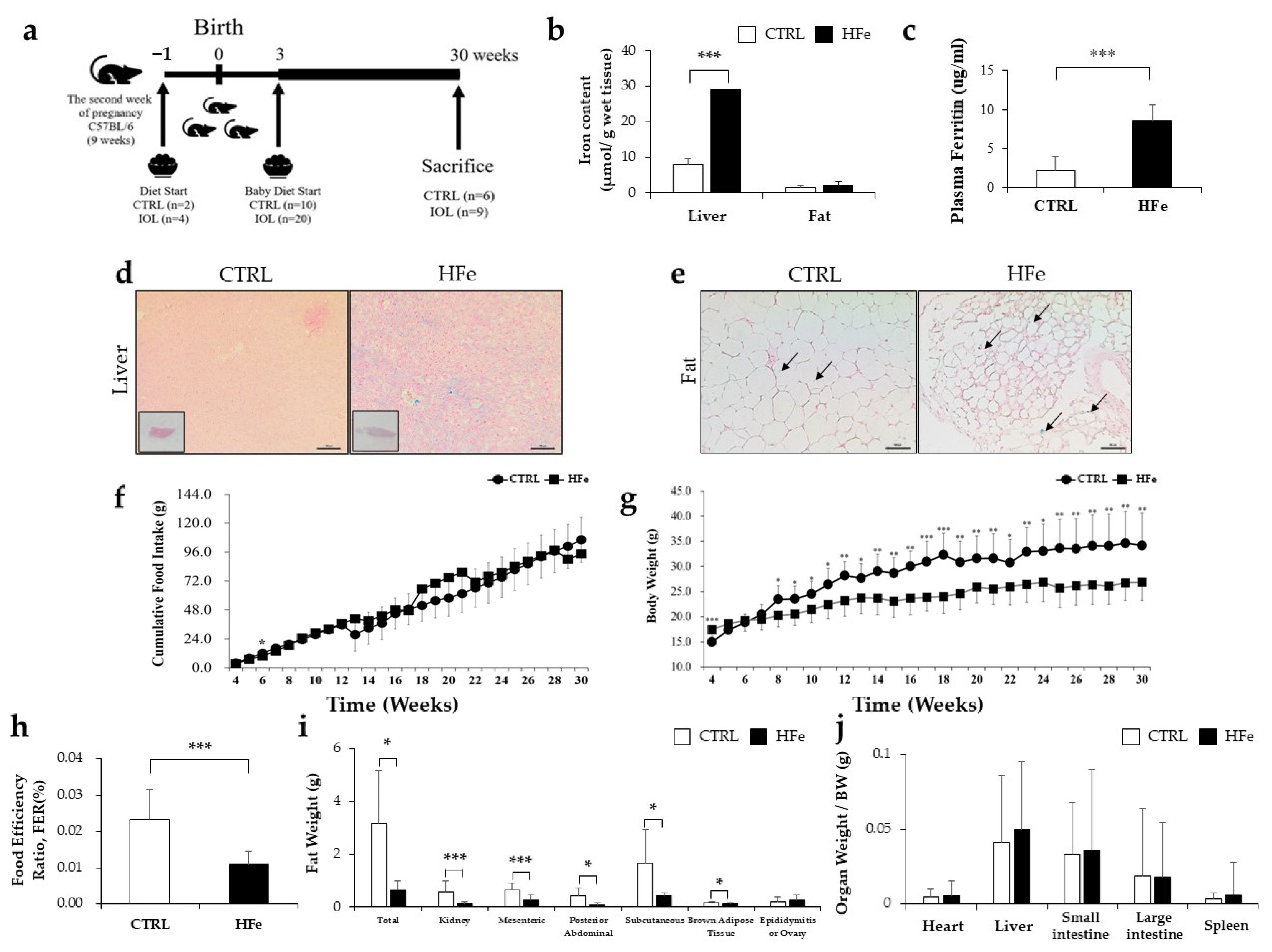
3.1. High Iron Intake in the Hfe Decreased Body Weights and Fat Mass
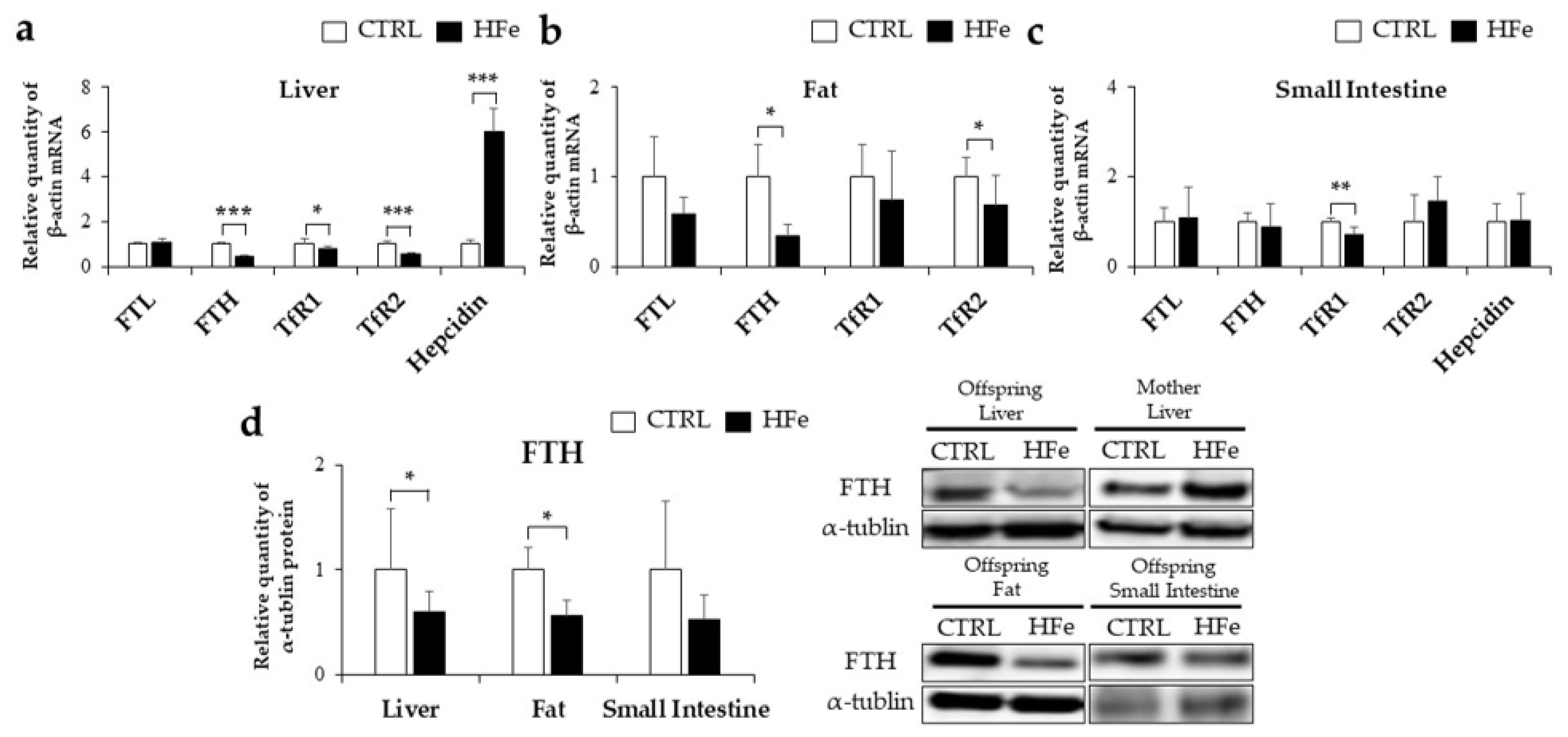
3.2. Iron-Metabolism Gene Expressions Were Affected by HFe Diet
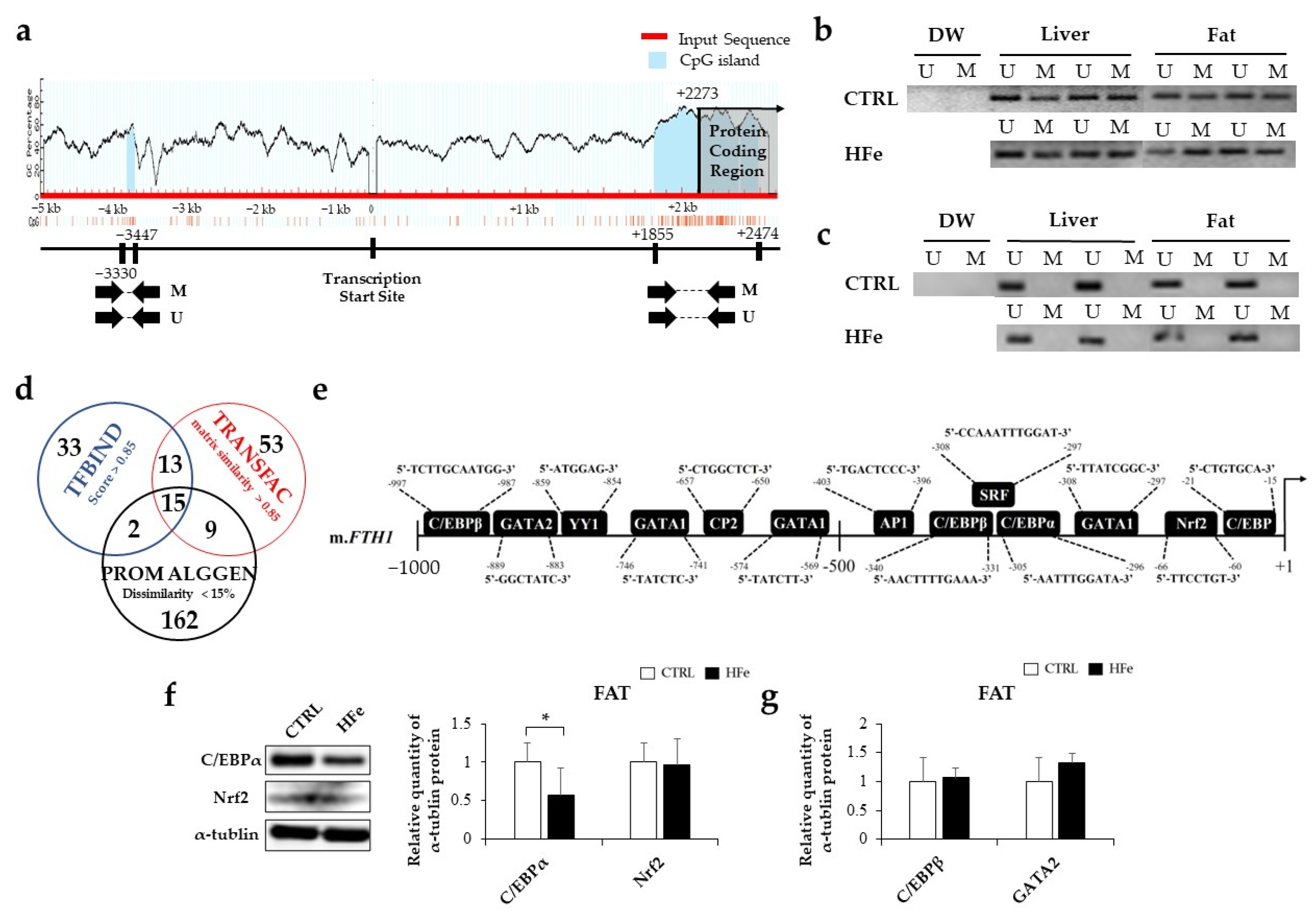
3.3. Low Gene Expression of FTH in Pups Was Not Associated with DNA Methylation in CpG Island
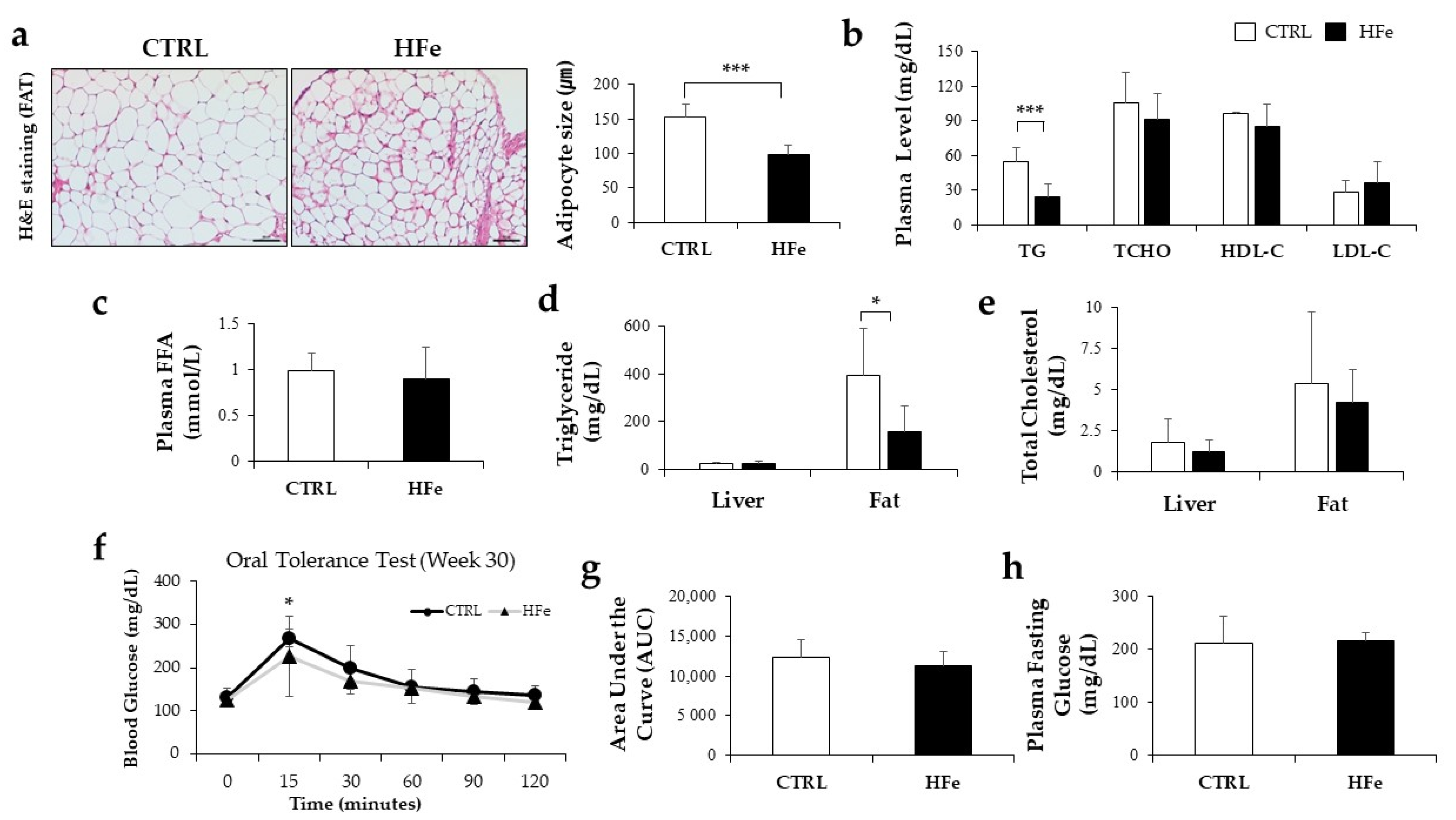
3.4. The HFe Decreased the Adipocyte Size and TG Levels in Plasma and Fat Tissues
3.5. HFe Diet Affected Differentiation of Adipocyte, FA Biosynthesis, and FA β-Oxidation
3.6. Prenatal to Postnatal Exposure of HFe Diet Enhanced the Antioxidant System and Induced Depletion of GSH
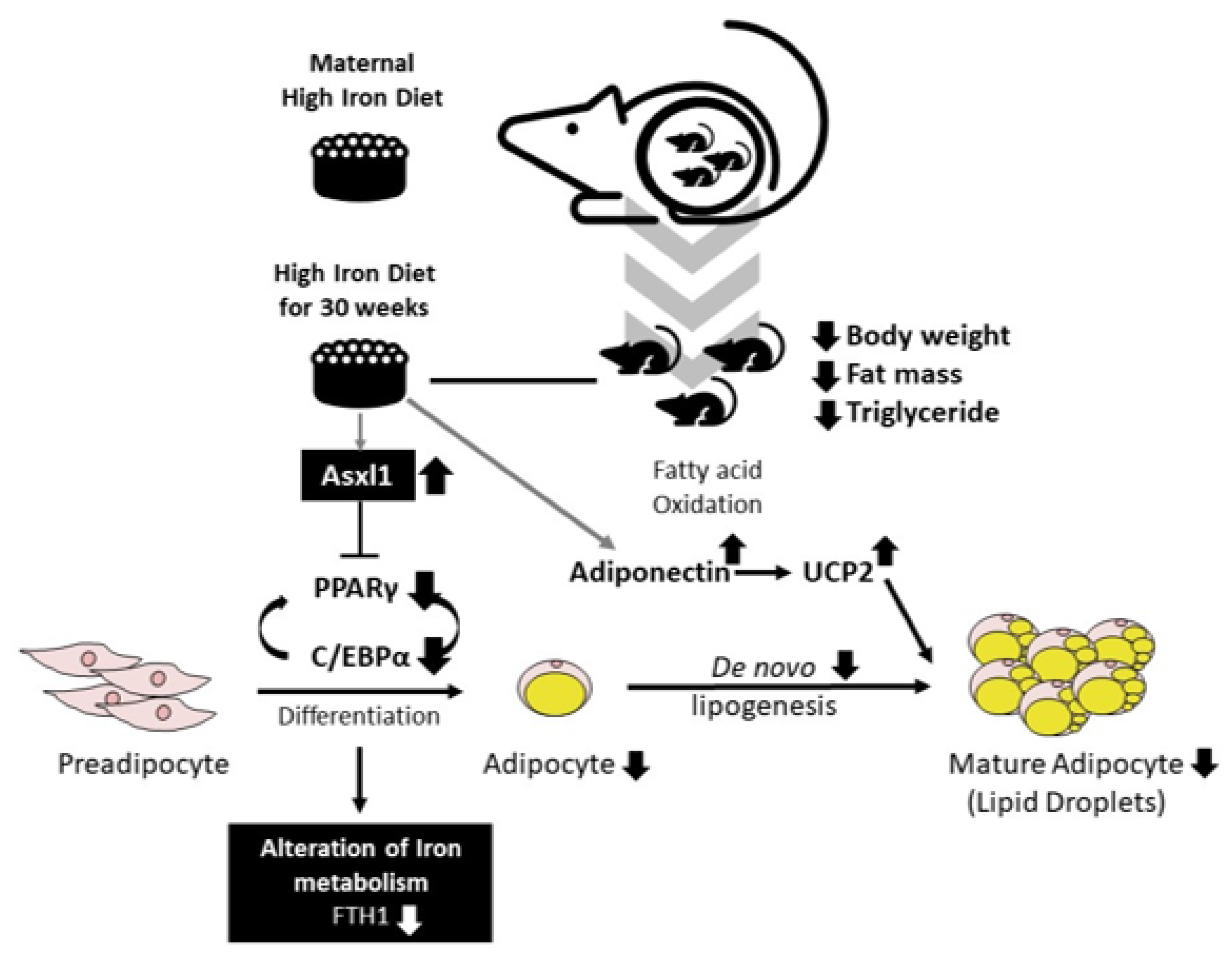
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fisher, A.L.; Nemeth, E. Iron homeostasis during pregnancy. Am. J. Clin. Nutr. 2017, 106, 1567S–1574S. [Google Scholar] [CrossRef] [PubMed]
- Bothwell, T.H. Iron requirements in pregnancy and strategies to meet them. Am. J. Clin. Nutr. 2000, 72, 257S–264S. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Bernal, C.; Rebagliato, M.; Ballester, F. Maternal nutrition and fetal growth: The role of iron status and intake during pregnancy. Nutr. Diet. Suppl. 2012, 4, 25–37. [Google Scholar] [CrossRef]
- Alwan, N.A.; Greenwood, D.C.; Simpson, N.A.; McArdle, H.J.; Godfrey, K.M.; Cade, J.E. Dietary iron intake during early pregnancy and birth outcomes in a cohort of British women. Mol. Hum. Reprod. 2011, 26, 911–919. [Google Scholar] [CrossRef]
- Ronnenberg, A.G.; Wood, R.J.; Wang, X.; Xing, H.; Chen, C.; Chen, D.; Guang, W.; Huang, A.; Wang, L.; Xu, X. Preconception hemoglobin and ferritin concentrations are associated with pregnancy outcome in a prospective cohort of Chinese women. J. Nutr. 2004, 134, 2586–2591. [Google Scholar] [CrossRef]
- Koenig, M.D.; Tussing-Humphreys, L.; Day, J.; Cadwell, B.; Nemeth, E. Hepcidin and iron homeostasis during pregnancy. Nutrients 2014, 6, 3062–3083. [Google Scholar] [CrossRef]
- World Health Organization. Guideline: Daily Iron and Folic Acid Supplementation in Pregnant Women; World Health Organization: Geneva, Switzerland, 2012; p. 27. [Google Scholar]
- The Korean Nutrition Society. Dietary Reference Intakes for Koreans 2020; Ministry of Health and Welfare: Sejong, Korea, 2020. [Google Scholar]
- Institute of Medicine. Iron: Tolerable Upper Intake Levels. Nutr. Clin. Care 2002, 5, 236–250. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. Recommendations to prevent and control iron deficiency in the United States. Morb. Mortal. Wkly. Rep. 1998, 47, 1–29. [Google Scholar]
- Rybo, G.; Sölvell, L. Side-effect studies on a new sustained release iron preparation. Scand. J. Haematol. Suppl. 1971, 8, 257–264. [Google Scholar] [CrossRef]
- Ribot, B.; Aranda, N.; Giralt, M.; Romeu, M.; Balaguer, A.; Arija, V. Effect of different doses of iron supplementation during pregnancy on maternal and infant health. Ann. Hematol. 2013, 92, 221–229. [Google Scholar] [CrossRef]
- Thame, M.; Wilks, R.J.; McFarlane-Anderson, N.; Bennett, F.I.; Forrester, T.E. Relationship between maternal nutritional status and infant’s weight and body proportions at birth. Eur. J. Clin. Nutr. 1997, 51, 134–138. [Google Scholar] [CrossRef] [PubMed]
- Shastri, L.; Mishra, P.; Dwarkanath, P.; Thomas, T.; Duggan, C.; Bosch, R.; McDonald, C.; Thomas, A.; Kurpad, A. Association of oral iron supplementation with birth outcomes in non-anaemic South Indian pregnant women. Eur. J. Clin. Nutr. 2015, 69, 609–613. [Google Scholar] [CrossRef] [PubMed]
- Moretti, D.; Goede, J.S.; Zeder, C.; Jiskra, M.; Chatzinakou, V.; Tjalsma, H.; Melse-Boonstra, A.; Brittenham, G.; Swinkels, D.W.; Zimmermann, M.B. Oral iron supplements increase hepcidin and decrease iron absorption from daily or twice-daily doses in iron-depleted young women. Blood 2015, 126, 1981–1989. [Google Scholar] [CrossRef] [PubMed]
- Lachili, B.; Hininger, I.; Faure, H.; Arnaud, J.; Richard, M.-J.; Favier, A.; Roussel, A.-M. Increased lipid peroxidation in pregnant women after iron and vitamin C supplementation. Biol. Trace Elem. Res. 2001, 83, 103–110. [Google Scholar] [CrossRef]
- Amin, S.B.; Myers, G.; Wang, H. Association between neonatal iron overload and early human brain development in premature infants. Early Hum. Dev. 2012, 88, 583–587. [Google Scholar] [CrossRef] [PubMed]
- Yip, R. Iron supplementation during pregnancy: Is it effective? Am. J. Clin. Nutr. 1996, 63, 853–855. [Google Scholar] [CrossRef] [PubMed]
- Volani, C.; Paglia, G.; Smarason, S.V.; Pramstaller, P.P.; Demetz, E.; Pfeifhofer-Obermair, C.; Weiss, G. Metabolic Signature of Dietary Iron Overload in a Mouse Model. Cells 2018, 7, 264. [Google Scholar] [CrossRef]
- Silva, M.; da Costa Guerra, J.F.; Sampaio, A.F.S.; Lima, W.G.d.; Silva, M.E.; Pedrosa, M.L. Iron dextran increases hepatic oxidative stress and alters expression of genes related to lipid metabolism contributing to hyperlipidaemia in murine model. BioMed Res. Int. 2015, 2015, 272617. [Google Scholar] [CrossRef]
- Choi, J.S.; Koh, I.-U.; Lee, H.J.; Kim, W.H.; Song, J. Effects of excess dietary iron and fat on glucose and lipid metabolism. J. Nutr. Biochem. 2013, 24, 1634–1644. [Google Scholar] [CrossRef]
- Fernández-Real, J.M.; López-Bermejo, A.; Ricart, W. Cross-talk between iron metabolism and diabetes. Diabetes 2002, 51, 2348–2354. [Google Scholar] [CrossRef]
- Rawal, S.; Hinkle, S.N.; Bao, W.; Zhu, Y.; Grewal, J.; Albert, P.S.; Weir, N.L.; Tsai, M.Y.; Zhang, C. A longitudinal study of iron status during pregnancy and the risk of gestational diabetes: Findings from a prospective, multiracial cohort. Diabetologia 2017, 60, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Meroño, T.; Gómez Rosso, L.; Sorroche, P.; Boero, L.; Arbelbide, J.; Brites, F. High risk of cardiovascular disease in iron overload patients. Eur. J. Clin. Investig. 2011, 41, 479–486. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Wang, D.; Zhang, H.; Zhang, Y.; Wang, J.; Qi, R.; Yang, J.; Shen, H.; Xu, Y.; Li, M. Rapid responses of adipocytes to iron overload increase serum TG level by decreasing adiponectin. J. Cell. Physiol. 2021, 236, 544–7553. [Google Scholar] [CrossRef] [PubMed]
- Tsay, J.; Yang, Z.; Ross, F.P.; Cunningham-Rundles, S.; Lin, H.; Coleman, R.; Mayer-Kuckuk, P.; Doty, S.B.; Grady, R.W.; Giardina, P.J. Bone loss caused by iron overload in a murine model: Importance of oxidative stress. Blood Am. J. Hematol. 2010, 116, 2582–2589. [Google Scholar] [CrossRef] [PubMed]
- Andreelli, F.; Foretz, M.; Knauf, C.; Cani, P.D.; Perrin, C.; Iglesias, M.A.; Pillot, B.; Bado, A.; Tronche, F.; Mithieux, G. Liver adenosine monophosphate-activated kinase-α2 catalytic subunit is a key target for the control of hepatic glucose production by adiponectin and leptin but not insulin. Endocrinology 2006, 147, 2432–2441. [Google Scholar] [CrossRef]
- Ramey, G.; Faye, A.; Durel, B.; Viollet, B.; Vaulont, S. Iron overload in Hepc1−/− mice is not impairing glucose homeostasis. FEBS Lett. 2007, 581, 1053–1057. [Google Scholar] [CrossRef]
- Giustarini, D.; Dalle-Donne, I.; Milzani, A.; Fanti, P.; Rossi, R. Analysis of GSH and GSSG after derivatization with N-ethylmaleimide. Nat. Protoc. 2013, 8, 1660. [Google Scholar] [CrossRef]
- Sönmez Aydın, F.; Hukkamlı, B.; Budak, H. Coaction of hepatic thioredoxin and glutathione systems in iron overload-induced oxidative stress. J. Biochem. Mol. Toxicol. 2021, 35, e22704. [Google Scholar] [CrossRef]
- Sheikh, N.; Dudas, J.; Ramadori, G. Changes of gene expression of iron regulatory proteins during turpentine oil-induced acute-phase response in the rat. Lab. Investig. 2007, 87, 713–725. [Google Scholar] [CrossRef][Green Version]
- Siah, C.W.; Trinder, D.; Olynyk, J.K. Iron overload. Clin. Chim. Acta 2005, 358, 24–36. [Google Scholar] [CrossRef]
- Laker, R.C.; Lillard, T.S.; Okutsu, M.; Zhang, M.; Hoehn, K.L.; Connelly, J.J.; Yan, Z. Exercise prevents maternal high-fat diet–induced hypermethylation of the Pgc-1α gene and age-dependent metabolic dysfunction in the offspring. Diabetes 2014, 63, 1605–1611. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-H.; Yang, C.-M.; Chang, S.-P.; Hu, M.-L. C/EBP beta and C/EBP delta expression is elevated in the early phase of ethanol-induced hepatosteatosis in mice. Acta Pharmacol. Sin. 2009, 30, 1138–1143. [Google Scholar] [CrossRef] [PubMed]
- Kerins, M.J.; Ooi, A. The roles of NRF2 in modulating cellular iron homeostasis. Antioxid. Redox Signal. 2018, 29, 1756–1773. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Kulkarni, S.R.; Donepudi, A.C.; More, V.R.; Slitt, A.L. Enhanced Nrf2 activity worsens insulin resistance, impairs lipid accumulation in adipose tissue, and increases hepatic steatosis in leptin-deficient mice. Diabetes 2012, 61, 3208–3218. [Google Scholar] [CrossRef]
- Polat, A.; Dinulescu, M.; Fraitag, S.; Nimubona, S.; Toutain, F.; Jouneau, S.; Poullot, E.; Droitcourt, C.; Dupuy, A. Skin manifestations among GATA 2-deficient patients. Br. J. Dermatol. Suppl. 2018, 178, 781–785. [Google Scholar] [CrossRef]
- Somparn, N.; Prawan, A.; Senggunprai, L.; Kukongviriyapan, U.; Jetsrisuparb, A.; Lee, M.-H.; Kim, D.-H.; Kukongviriyapan, V.; Surh, Y.-J. Cellular adaptation mediated through Nrf2-induced glutamate cysteine ligase up-regulation against oxidative stress caused by iron overload in β-thalassemia/HbE patients. Free Radic. Res. 2019, 53, 791–799. [Google Scholar] [CrossRef]
- Park, U.-H.; Yoon, S.K.; Park, T.; Kim, E.-J.; Um, S.-J. Additional sex comb-like (ASXL) proteins 1 and 2 play opposite roles in adipogenesis via reciprocal regulation of peroxisome proliferator-activated receptor γ. J. Biol. Chem. 2011, 286, 1354–1363. [Google Scholar] [CrossRef]
- Eberle, D.; Hegarty, B.; Bossard, P.; Ferré, P.; Foufelle, F. SREBP transcription factors: Master regulators of lipid homeostasis. Biochimie 2004, 86, 839–848. [Google Scholar] [CrossRef]
- Sanchez, J.; Palou, A.; Pico, C. Response to carbohydrate and fat refeeding in the expression of genes involved in nutrient partitioning and metabolism: Striking effects on fibroblast growth factor-21 induction. Endocrinology 2009, 150, 5341–5350. [Google Scholar] [CrossRef]
- Gamberi, T.; Magherini, F.; Modesti, A.; Fiaschi, T. Adiponectin signaling pathways in liver diseases. Biomedicines 2018, 6, 52. [Google Scholar] [CrossRef]
- Wu, Z.; Rosen, E.D.; Brun, R.; Hauser, S.; Adelmant, G.; Troy, A.E.; McKeon, C.; Darlington, G.J.; Spiegelman, B.M. Cross-regulation of C/EBPα and PPARγ controls the transcriptional pathway of adipogenesis and insulin sensitivity. Mol. Cell 1999, 3, 151–158. [Google Scholar] [CrossRef]
- Farmer, S. Regulation of PPAR γ activity during adipogenesis. Int. J. Obes. 2005, 29, S13–S16. [Google Scholar] [CrossRef] [PubMed]
- Le Lay, S.; Simard, G.; Martinez, M.C.; Andriantsitohaina, R. Oxidative stress and metabolic pathologies: From an adipocentric point of view. Oxid. Med. Cell. Longev. 2014, 2014, 908539. [Google Scholar] [CrossRef] [PubMed]
- Del Rio, D.; Stewart, A.J.; Pellegrini, N. A review of recent studies on malondialdehyde as toxic molecule and biological marker of oxidative stress. Nutr. Metab. Cardiovasc. 2005, 15, 316–328. [Google Scholar] [CrossRef] [PubMed]
- Stolwijk, J.M.; Falls-Hubert, K.C.; Searby, C.C.; Wagner, B.A.; Buettner, G.R. Simultaneous detection of the enzyme activities of GPx1 and GPx4 guide optimization of selenium in cell biological experiments. Redox Biol. 2020, 32, 101518. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Lv, X.; Song, E.; Song, Y. Fostered Nrf2 expression antagonizes iron overload and glutathione depletion to promote resistance of neuron-like cells to ferroptosis. Toxicol. Appl. Pharmacol. 2020, 407, 115241. [Google Scholar] [CrossRef]
- Helal, M.G.; El-Kashef, D.H. Krill oil alleviates oxidative stress, iron accumulation and fibrosis in the liver and spleen of iron-overload rats. Environ. Sci. Pollut. Res. 2020, 27, 3950–3961. [Google Scholar] [CrossRef]
- Zheng, R.; Heck, D.E.; Mishin, V.; Black, A.T.; Shakarjian, M.P.; Kong, A.-N.T.; Laskin, D.L.; Laskin, J.D. Modulation of keratinocyte expression of antioxidants by 4-hydroxynonenal, a lipid peroxidation end product. Toxicol. Appl. Pharmacol. 2014, 275, 113–121. [Google Scholar] [CrossRef]
- Onyenwoke, R.U.; Wiegel, J. Iron (III) reduction: A novel activity of the human NAD (P) H: Oxidoreductase. Biochem. Biophys. Res. Commun 2007, 353, 389–393. [Google Scholar] [CrossRef]
- Lushchak, V.I. Glutathione homeostasis and functions: Potential targets for medical interventions. J. Amino Acids 2012, 2012, 736837. [Google Scholar] [CrossRef]
- Lisle, S.; Lewis, R.; Petry, C.; Ozanne, S.; Hales, C.; Forhead, A. Effect of maternal iron restriction during pregnancy on renal morphology in the adult rat offspring. Br. J. Nutr. 2003, 90, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.; Petry, C.; Ozanne, S.; Hales, C. Effects of maternal iron restriction in the rat on blood pressure, glucose tolerance, and serum lipids in the 3-month-old offspring. Metab. Clin. Exp. 2001, 50, 562–567. [Google Scholar] [CrossRef] [PubMed]
- Samuelsson, A.-M.; Matthews, P.A.; Argenton, M.; Christie, M.R.; McConnell, J.M.; Jansen, E.H.; Piersma, A.H.; Ozanne, S.E.; Twinn, D.F.; Remacle, C. Diet-induced obesity in female mice leads to offspring hyperphagia, adiposity, hypertension, and insulin resistance: A novel murine model of developmental programming. Hypertension 2008, 51, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Dahri, S.; Snoeck, A.; Reusens-Billen, B.; Remacle, C.; Hote, J.J. Islet function in offspring of mothers on low-protein diet during gestation. Diabetes 1991, 40, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Størdal, K.; McArdle, H.J.; Hayes, H.; Tapia, G.; Viken, M.K.; Lund-Blix, N.A.; Haugen, M.; Joner, G.; Skrivarhaug, T.; Mårild, K. Prenatal iron exposure and childhood type 1 diabetes. Sci. Rep. 2018, 8, 9067. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Zhang, D.; Liu, S.; Dong, Z.; Zhou, J.; Yin, Y.; Wan, D. Maternal Iron Supplementation During Pregnancy Affects Placental Function and Iron Status in Offspring. J. Trace Elem. Med. Biol. 2022, 71, 126950. [Google Scholar] [CrossRef]
- Lakhal-Littleton, S. Advances in understanding the crosstalk between mother and fetus on iron utilization. Semin. Hematol. 2021, 58, 153–160. [Google Scholar] [CrossRef]
- Kakhlon, O.; Gruenbaum, Y.; Cabantchik, Z.I. Repression of ferritin expression increases the labile iron pool, oxidative stress, and short-term growth of human erythroleukemia cells. Am. J. Hematol. 2001, 97, 2863–2871. [Google Scholar] [CrossRef]
- Zhang, Q.; Lu, X.-M.; Zhang, M.; Yang, C.-Y.; Lv, S.-Y.; Li, S.-F.; Zhong, C.-Y.; Geng, S.-S. Adverse effects of iron deficiency anemia on pregnancy outcome and offspring development and intervention of three iron supplements. Sci. Rep. 2021, 11, 1347. [Google Scholar] [CrossRef]
- Dongiovanni, P.; Ruscica, M.; Rametta, R.; Recalcati, S.; Steffani, L.; Gatti, S.; Girelli, D.; Cairo, G.; Magni, P.; Fargion, S. Dietary iron overload induces visceral adipose tissue insulin resistance. Am. J. Clin. Pathol. 2013, 182, 2254–2263. [Google Scholar] [CrossRef]
- Yusrawati, Y.; Defrin, D.; Karmia, H.R. Neonatal Growth, Neurotrophine, Zinc, and Ferritin Concentration in Normal and Iron Deficience Pregnancy: An Observational Analitic Study. Open Access Maced. J. Med. Sci. 2019, 7, 1114. [Google Scholar] [CrossRef] [PubMed]
- Cunnane, S.; McAdoo, K.; Prohaska, J. Lipid and fatty acid composition of organs from copper-deficient mice. J. Nutr. 1986, 116, 1248–1256. [Google Scholar] [CrossRef] [PubMed]
- Gupta, M.; Solanki, M.H.; Chatterjee, P.K.; Xue, X.; Roman, A.; Desai, N.; Rochelson, B.; Metz, C.N. Maternal magnesium deficiency in mice leads to maternal metabolic dysfunction and altered lipid metabolism with fetal growth restriction. Mol. Med. 2014, 20, 332–340. [Google Scholar] [CrossRef] [PubMed]
- Guthrie, H.A.; Froozani, M.; Sherman, A.R.; Barron, G.P. Hyperlipidemia in offspring of iron-deficient rats. J. Nutr. 1974, 104, 1273–1278. [Google Scholar] [CrossRef] [PubMed]
- LeBlanc, C.P.; Fiset, S.; Surette, M.E.; Turgeon O’Brien, H.; Rioux, F.M. Maternal iron deficiency alters essential fatty acid and eicosanoid metabolism and increases locomotion in adult guinea pig offspring. J. Nutr. 2009, 139, 1653–1659. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Lewis, R.M.; Wang, C.; Hales, N.; Byrne, C.D. Maternal dietary iron restriction modulates hepatic lipid metabolism in the fetuses. Am. J. Physiol. Regul. Integr. Comp. 2005, 288, R104–R111. [Google Scholar] [CrossRef]
- Dillard, C.J.; Tappel, A.L. Volatile hydrocarbon and carbonyl products of lipid peroxidation: A comparison of pentane, ethane, hexanal, and acetone as in vivo indices. Lipids 1979, 14, 989–995. [Google Scholar] [CrossRef]
- Brunet, S.; Thibault, L.; Delvin, E.; Yotov, W.; Bendayan, M.; Levy, E. Dietary iron overload and induced lipid peroxidation are associated with impaired plasma lipid transport and hepatic sterol metabolism in rats. Hepatology 1999, 29, 1809–1817. [Google Scholar] [CrossRef]
- Lefterova, M.I.; Lazar, M.A. New developments in adipogenesis. Trends Endocrinol. Metab. 2009, 20, 107–114. [Google Scholar] [CrossRef]
- Shimomura, I.; Bashmakov, Y.; Ikemoto, S.; Horton, J.D.; Brown, M.S.; Goldstein, J.L. Insulin selectively increases SREBP-1c mRNA in the livers of rats with streptozotocin-induced diabetes. Proc. Natl. Acad. Sci. USA 1999, 96, 13656–13661. [Google Scholar] [CrossRef]
- Barrera, C.; Valenzuela, R.; Rincón, M.Á.; Espinosa, A.; Echeverria, F.; Romero, N.; Gonzalez-Mañan, D.; Videla, L.A. Molecular mechanisms related to the hepatoprotective effects of antioxidant-rich extra virgin olive oil supplementation in rats subjected to short-term iron administration. Free Radic. Biol. Med. 2018, 126, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Bonomo, L.d.F.; Silva, M.; Oliveira, R.D.P.; Silva, M.E.; Pedrosa, M.L. Iron overload potentiates diet-induced hypercholesterolemia and reduces liver ppar-α expression in hamsters. J. Biochem. Mol. Toxicol. 2012, 26, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Furuhashi, M.; Hotamisligil, G.S. Fatty acid-binding proteins: Role in metabolic diseases and potential as drug targets. Nat. Rev. Drug Discov. 2008, 7, 489–503. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Cho, Y.M.; Lee, K.H.; Jeong, S.-W.; Kwon, O.-J. Oleate protects macrophages from palmitate-induced apoptosis through the downregulation of CD36 expression. Biochem. Biophys. Res. Commun. 2017, 488, 477–482. [Google Scholar] [CrossRef]
- Xiao, L.; Luo, G.; Li, H.; Yao, P.; Tang, Y. Dietary iron overload mitigates atherosclerosis in high-fat diet-fed apolipoprotein E knockout mice: Role of dysregulated hepatic fatty acid metabolism. Biochim. Biophys. Acta 2021, 1866, 159004. [Google Scholar] [CrossRef]
- Foretz, M.; Even, P.C.; Viollet, B. AMPK activation reduces hepatic lipid content by increasing fat oxidation in vivo. Int. J. Mol. Sci. 2018, 19, 2826. [Google Scholar] [CrossRef]
- Huang, H.; Chen, J.; Lu, H.; Zhou, M.; Chai, Z.; Hu, Y. Iron-induced generation of mitochondrial ROS depends on AMPK activity. Biometals 2017, 30, 623–628. [Google Scholar] [CrossRef]
- Zheng, Q.; Zhao, Y.; Guo, J.; Zhao, S.; Fei, C.; Xiao, C.; Wu, D.; Wu, L.; Li, X.; Chang, C. Iron overload promotes mitochondrial fragmentation in mesenchymal stromal cells from myelodysplastic syndrome patients through activation of the AMPK/MFF/Drp1 pathway. Cell Death Dis. 2018, 9, 515. [Google Scholar] [CrossRef]
- Lee, W.J.; Kim, M.; Park, H.-S.; Kim, H.S.; Jeon, M.J.; Oh, K.S.; Koh, E.H.; Won, J.C.; Kim, M.-S.; Oh, G.T. AMPK activation increases fatty acid oxidation in skeletal muscle by activating PPARα and PGC-1. Biochem. Biophys. Res. Commun. 2006, 340, 291–295. [Google Scholar] [CrossRef]
- Leone, T.C.; Weinheimer, C.J.; Kelly, D.P. A critical role for the peroxisome proliferator-activated receptor α (PPARα) in the cellular fasting response: The PPARα-null mouse as a model of fatty acid oxidation disorders. Proc. Natl. Acad. Sci. USA 1999, 96, 7473–7478. [Google Scholar] [CrossRef]
- Awazawa, M.; Ueki, K.; Inabe, K.; Yamauchi, T.; Kaneko, K.; Okazaki, Y.; Bardeesy, N.; Ohnishi, S.; Nagai, R.; Kadowaki, T. Adiponectin suppresses hepatic SREBP1c expression in an AdipoR1/LKB1/AMPK dependent pathway. Biochem. Biophys. Res. Commun. 2009, 382, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Dahyaleh, K.; Sung, H.K.; Prioriello, M.; Rengasamy, P.; Lam, N.H.; Kim, J.B.; Gross, S.; Sweeney, G. Iron overload reduces adiponectin receptor expression via a ROS/FOXO1-dependent mechanism leading to adiponectin resistance in skeletal muscle cells. J. Cell. Physiol. 2021, 236, 5339–5351. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Xu, A.; Tam, P.K.; Lam, K.S.; Huang, B.; Liang, Y.; Lee, I.-K.; Wu, D.; Wang, Y. Upregulation of UCP2 by adiponectin: The involvement of mitochondrial superoxide and hnRNP K. PLoS ONE 2012, 7, e32349. [Google Scholar] [CrossRef] [PubMed]
- Gabrielsen, J.S.; Gao, Y.; Simcox, J.A.; Huang, J.; Thorup, D.; Jones, D.; Cooksey, R.C.; Gabrielsen, D.; Adams, T.D.; Hunt, S.C. Adipocyte iron regulates adiponectin and insulin sensitivity. J. Clin. Investig. 2012, 122, 3529–3540. [Google Scholar] [CrossRef]
- Ricquier, D. Uncoupling protein-2 (UCP2): Molecular and genetic studies. Int. J. Obes. 1999, 23, S38–S42. [Google Scholar] [CrossRef]
- Vaziri, N.D.; Wang, X.Q.; Oveisi, F.; Rad, B. Induction of oxidative stress by glutathione depletion causes severe hypertension in normal rats. Hypertension 2000, 36, 142–146. [Google Scholar] [CrossRef]
- Vertuani, S.; Angusti, A.; Manfredini, S. The antioxidants and pro-antioxidants network: An overview. Curr. Pharm. Des. 2004, 10, 1677–1694. [Google Scholar] [CrossRef]
- Huang, H.-C.; Nguyen, T.; Pickett, C.B. Regulation of the antioxidant response element by protein kinase C-mediated phosphorylation of NF-E2-related factor 2. Proc. Natl. Acad. Sci. USA 2000, 97, 12475–12480. [Google Scholar] [CrossRef]
- Lim, P.J.; Duarte, T.L.; Arezes, J.; Garcia-Santos, D.; Hamdi, A.; Pasricha, S.-R.; Armitage, A.E.; Mehta, H.; Wideman, S.; Santos, A.G. Nrf2 controls iron homoeostasis in haemochromatosis and thalassaemia via Bmp6 and hepcidin. Nat. Metab. 2019, 1, 519–531. [Google Scholar] [CrossRef]
- Brandsch, C.; Schmidt, T.; Behn, D.; Weiße, K.; Mueller, A.S.; Stangl, G.I. Glutathione deficiency down-regulates hepatic lipogenesis in rats. Lipids Health Dis. 2010, 9, 1–8. [Google Scholar] [CrossRef]
- Lee, A.; Choi, K.-M.; Jung, W.-B.; Jeong, H.; Kim, G.-Y.; Lee, J.H.; Lee, M.K.; Hong, J.T.; Roh, Y.-S.; Sung, S.-H. Enhancement of glucose uptake by meso-dihydroguaiaretic acid through GLUT4 up-regulation in 3T3-L1 adipocytes. Molecules 2017, 22, 1423. [Google Scholar] [CrossRef] [PubMed]
- Kaestner, K.H.; Christy, R.J.; Lane, M.D. Mouse insulin-responsive glucose transporter gene: Characterization of the gene and trans-activation by the CCAAT/enhancer binding protein. Proc. Natl. Acad. Sci. USA 1990, 87, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, K.; Araki, E.; Yoshimura, R.; Tsuruzoe, K.; Furukawa, N.; Kaneko, K.; Motoshima, H.; Yoshizato, K.; Hideki, K.; Shichiri, M. Cell-specific regulation of IRS-1 gene expression: Role of E box and C/EBP binding site in HepG2 cells and CHO cells. Diabetes 1997, 46, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Yan, K.; Sun, B.; Gao, S.; Yang, X.; Ni, Y.; Ma, W.; Zhao, R. Long-term high-fat diet decreases hepatic iron storage associated with suppressing TFR2 and ZIP14 expression in rats. J. Agric. Food Chem. 2018, 66, 11612–11621. [Google Scholar] [CrossRef]
- Sie, K.K.; Li, J.; Ly, A.; Sohn, K.J.; Croxford, R.; Kim, Y.I. Effect of maternal and postweaning folic acid supplementation on global and gene-specific DNA methylation in the liver of the rat offspring. Mol. Nutr. Food Res. 2013, 57, 677–685. [Google Scholar] [CrossRef]
- Perera, F.; Herbstman, J. Prenatal environmental exposures, epigenetics, and disease. Reprod. Toxicol. 2011, 31, 363–373. [Google Scholar] [CrossRef]
- Bachman, K.E.; Park, B.H.; Rhee, I.; Rajagopalan, H.; Herman, J.G.; Baylin, S.B.; Kinzler, K.W.; Vogelstein, B. Histone modifications and silencing prior to DNA methylation of a tumor suppressor gene. Cancer Cell 2003, 3, 89–95. [Google Scholar] [CrossRef]
- Masuyama, H.; Hiramatsu, Y. Effects of a High-Fat Diet Exposure in Utero on the Metabolic Syndrome-Like Phenomenon in Mouse Offspring through Epigenetic Changes in Adipocytokine Gene Expression. Endocrinology 2012, 153, 2823–2830. [Google Scholar] [CrossRef]
- Strakovsky, R.S.; Zhang, X.; Zhou, D.; Pan, Y.X. Gestational high fat diet programs hepatic phosphoenolpyruvate carboxykinase gene expression and histone modification in neonatal offspring rats. Physiol. J. 2011, 589, 2707–2717. [Google Scholar] [CrossRef]
- IV, A.N.; Nair, A.S. Bioinformatics screening of ETV4 transcription factor oncogenes and identifying small-molecular anticancer drugs. Chem. Biol. Drug Des. 2022, 99, 277–285. [Google Scholar] [CrossRef]
- Singha, B.; Gatla, H.R.; Vancurova, I. Transcriptional regulation of chemokine expression in ovarian cancer. Biomolecules 2015, 5, 223–243. [Google Scholar] [CrossRef] [PubMed]
- Doty, R.T.; Yan, X.; Lausted, C.; Munday, A.D.; Yang, Z.; Yi, D.; Jabbari, N.; Liu, L.; Keel, S.B.; Tian, Q. Single-cell analyses demonstrate that a heme–GATA1 feedback loop regulates red cell differentiation. Am. J. Hematol. 2019, 133, 457–469. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.; Truksa, J.; Beutler, E. Association of SMAD3/4, COUP-Tf, HNF4α, C/EBPα, GATA2 and STAT5 to the Hamp1 Promoter. Blood 2008, 112, 3837. [Google Scholar] [CrossRef]
- Lee, S.-E.; Hwang, S.-L.; Jang, W.-G.; Chang, H.W.; Kim, Y.D. B-cell translocation gene 2 promotes hepatic hepcidin production via induction of Yin Yang 1. Biochem. Biophys. Res. Commun. 2015, 460, 996–1001. [Google Scholar] [CrossRef]
- Courselaud, B.; Pigeon, C.; Inoue, Y.; Inoue, J.; Gonzalez, F.J.; Leroyer, P.; Gilot, D.; Boudjema, K.; Guguen-Guillouzo, C.; Brissot, P. C/EBPα regulates hepatic transcription of hepcidin, an antimicrobial peptide and regulator of iron metabolism: Cross-talk between C/EBP pathway and iron metabolism. J. Biol. Chem. 2002, 277, 41163–41170. [Google Scholar] [CrossRef]
- Swanson, C.A. Iron intake and regulation: Implications for iron deficiency and iron overload. Alcohol 2003, 30, 99–102. [Google Scholar] [CrossRef]
- O’Brien, K.O.; Zavaleta, N.; Caulfield, L.E.; Wen, J.; Abrams, S.A. Prenatal iron supplements impair zinc absorption in pregnant Peruvian women. J. Nutr. 2000, 130, 2251–2255. [Google Scholar] [CrossRef]
- Ntambi, J.M.; Miyazaki, M.; Stoehr, J.P.; Lan, H.; Kendziorski, C.M.; Yandell, B.S.; Song, Y.; Cohen, P.; Friedman, J.M.; Attie, A.D. Loss of stearoyl–CoA desaturase-1 function protects mice against adiposity. Proc. Natl. Acad. Sci. USA 2002, 99, 11482–11486. [Google Scholar] [CrossRef]
- Sekiya, M.; Yahagi, N.; Matsuzaka, T.; Takeuchi, Y.; Nakagawa, Y.; Takahashi, H.; Okazaki, H.; Iizuka, Y.; Ohashi, K.; Gotoda, T. SREBP-1-independent regulation of lipogenic gene expression in adipocytes. J. Lipid Res. 2007, 48, 1581–1591. [Google Scholar] [CrossRef]
- Bowman, T.A.; O’Keeffe, K.R.; D’Aquila, T.; Yan, Q.W.; Griffin, J.D.; Killion, E.A.; Salter, D.M.; Mashek, D.G.; Buhman, K.K.; Greenberg, A.S. Acyl CoA synthetase 5 (ACSL5) ablation in mice increases energy expenditure and insulin sensitivity and delays fat absorption. Mol. Metab. 2016, 5, 210–220. [Google Scholar] [CrossRef]
- Zhang, M.; Liu, Y.; Shi, H.; Guo, M.; Chai, M.; He, Q.; Yan, M.; Cao, D.; Zhao, L.; Cai, H. Evolutionary and expression analyses of soybean basic Leucine zipper transcription factor family. BMC Genom. 2018, 19, 159. [Google Scholar] [CrossRef] [PubMed]
- Lage, R.; Diéguez, C.; Vidal-Puig, A.; López, M. AMPK: A metabolic gauge regulating whole-body energy homeostasis. Trends Mol. Med. 2008, 14, 539–549. [Google Scholar] [CrossRef] [PubMed]
- Schupp, M.; Cristancho, A.G.; Lefterova, M.I.; Hanniman, E.A.; Briggs, E.R.; Steger, D.J.; Qatanani, M.; Curtin, J.C.; Schug, J.; Ochsner, S.A. Re-expression of GATA2 cooperates with peroxisome proliferator-activated receptor-γ depletion to revert the adipocyte phenotype. J. Biol. Chem. 2009, 284, 9458–9464. [Google Scholar] [CrossRef]
- McLachlan, S.; Lee, S.-M.; Steele, T.M.; Hawthorne, P.L.; Zapala, M.A.; Eskin, E.; Schork, N.J.; Anderson, G.J.; Vulpe, C.D. In silico QTL mapping of basal liver iron levels in inbred mouse strains. Physiol. Genom. 2011, 43, 136–147. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ingredient (g/1000 g Diet) | AIN-76A Diet | |
|---|---|---|
| CTRL | HFe | |
| Casein | 200 | 200 |
| Corn starch | 150 | 150 |
| Sucrose | 499.99 | 490.09 |
| Corn oil | 50 | 50 |
| Cellulose | 50 | 50 |
| Vitamin mixture | 10 | 10 |
| AIN 76a mineral mix | 35 | 35 |
| Choline bitartrate | 2 | 2 |
| DL-Methionine | 3 | 3 |
| Butylated hydroxytoluene | 0.01 | 0.01 |
| FeSO4, 7H2O | - | 9.9 |
| Total (g) | 1000 | 1000 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, M.; Kim, Y.-h.; Min, S.; Lee, S.-M. High Iron Exposure from the Fetal Stage to Adulthood in Mice Alters Lipid Metabolism. Nutrients 2022, 14, 2451. https://doi.org/10.3390/nu14122451
Kim M, Kim Y-h, Min S, Lee S-M. High Iron Exposure from the Fetal Stage to Adulthood in Mice Alters Lipid Metabolism. Nutrients. 2022; 14(12):2451. https://doi.org/10.3390/nu14122451
Chicago/Turabian StyleKim, Minju, Yeon-hee Kim, Sohyun Min, and Seung-Min Lee. 2022. "High Iron Exposure from the Fetal Stage to Adulthood in Mice Alters Lipid Metabolism" Nutrients 14, no. 12: 2451. https://doi.org/10.3390/nu14122451
APA StyleKim, M., Kim, Y.-h., Min, S., & Lee, S.-M. (2022). High Iron Exposure from the Fetal Stage to Adulthood in Mice Alters Lipid Metabolism. Nutrients, 14(12), 2451. https://doi.org/10.3390/nu14122451