Alisol B Alleviates Hepatocyte Lipid Accumulation and Lipotoxicity via Regulating RARα-PPARγ-CD36 Cascade and Attenuates Non-Alcoholic Steatohepatitis in Mice

Abstract
:
1. Introduction
2. Materials and Methods
2.1. Extraction of Alisol B from Alisma orientalis (Sam.)
2.2. Animal Experiments
2.3. Biochemical Assays
2.4. Histological Analysis and Definition of Scoring System
2.5. RNA-Seq Analysis
2.6. Cell Culture and Treatment
2.7. Oil Red O Staining and Fatty Acid Uptake
2.8. Reactive Oxygen Species (ROS) Determination
2.9. RNA Interference
2.10. Transient Plasmid Transfection
2.11. Luciferase Reporter Activity Assay
2.12. Quantitative RT-PCR
2.13. Western Blot Analysis
2.14. Statistical Analysis
3. Results
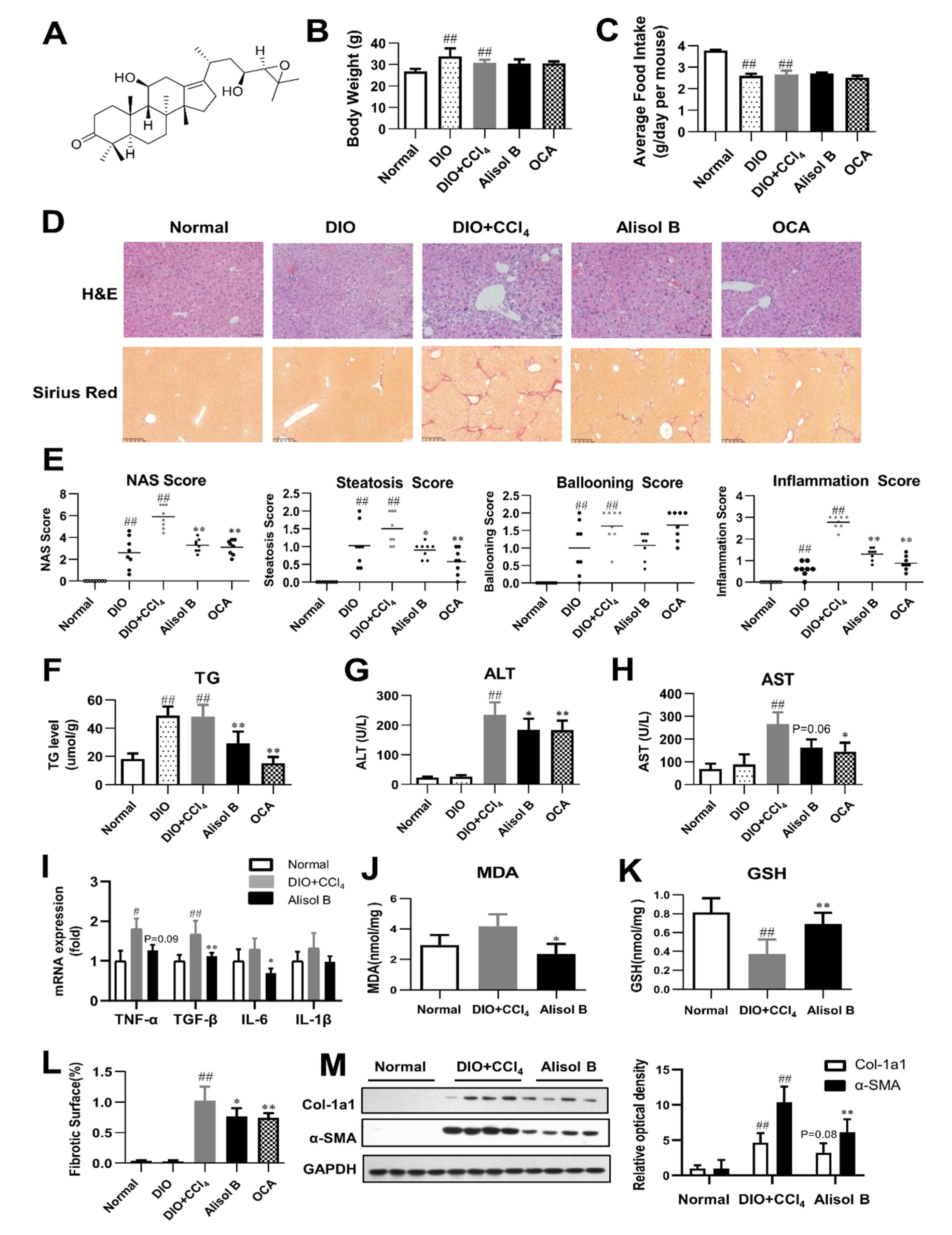
3.1. Alisol B ameliorated NASH in a DIO+CCl4-Induced Murine Model
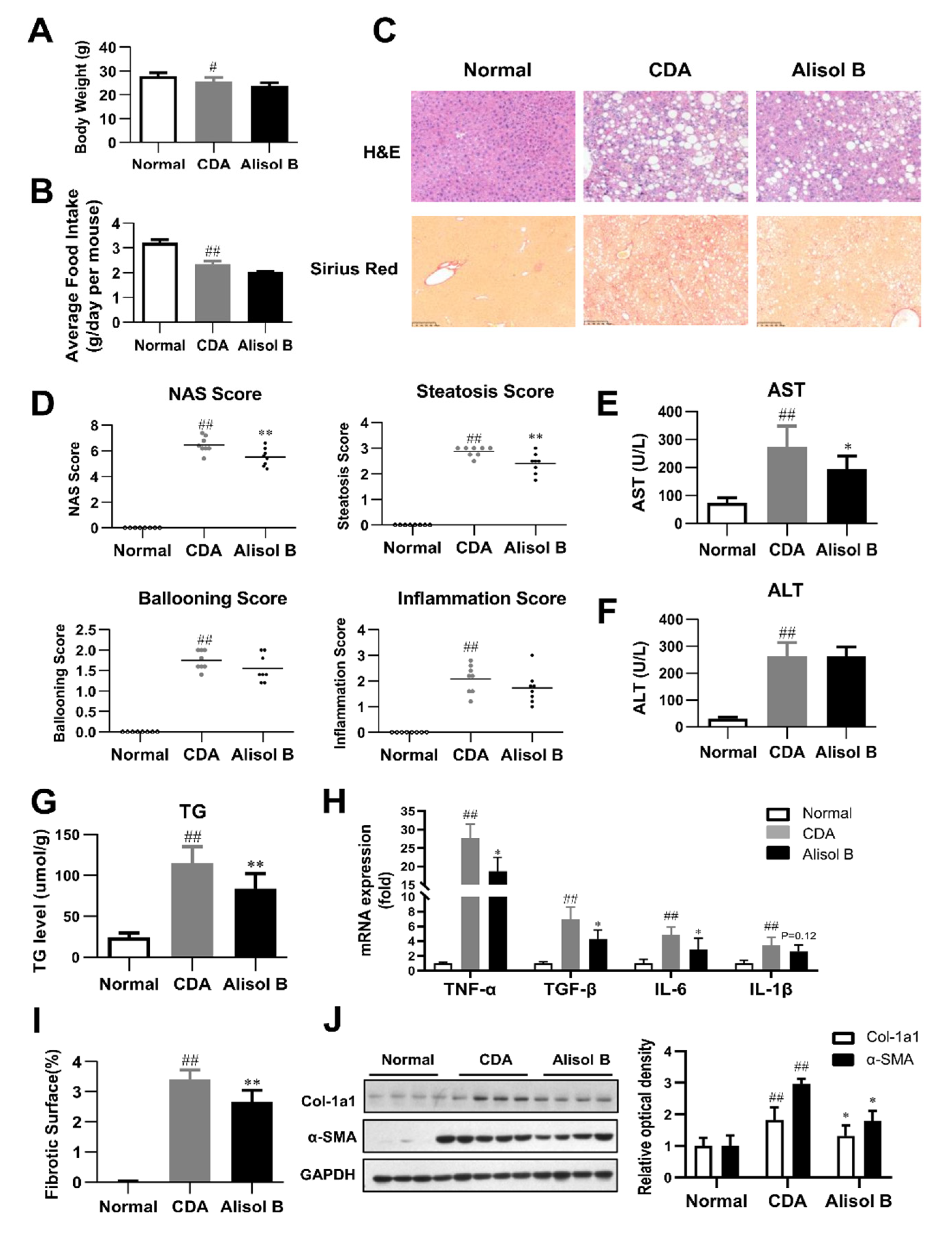
3.2. Alisol B Ameliorated NASH in a CDA Diet-Induced Murine Model
3.3. RNA-Seq Analysis
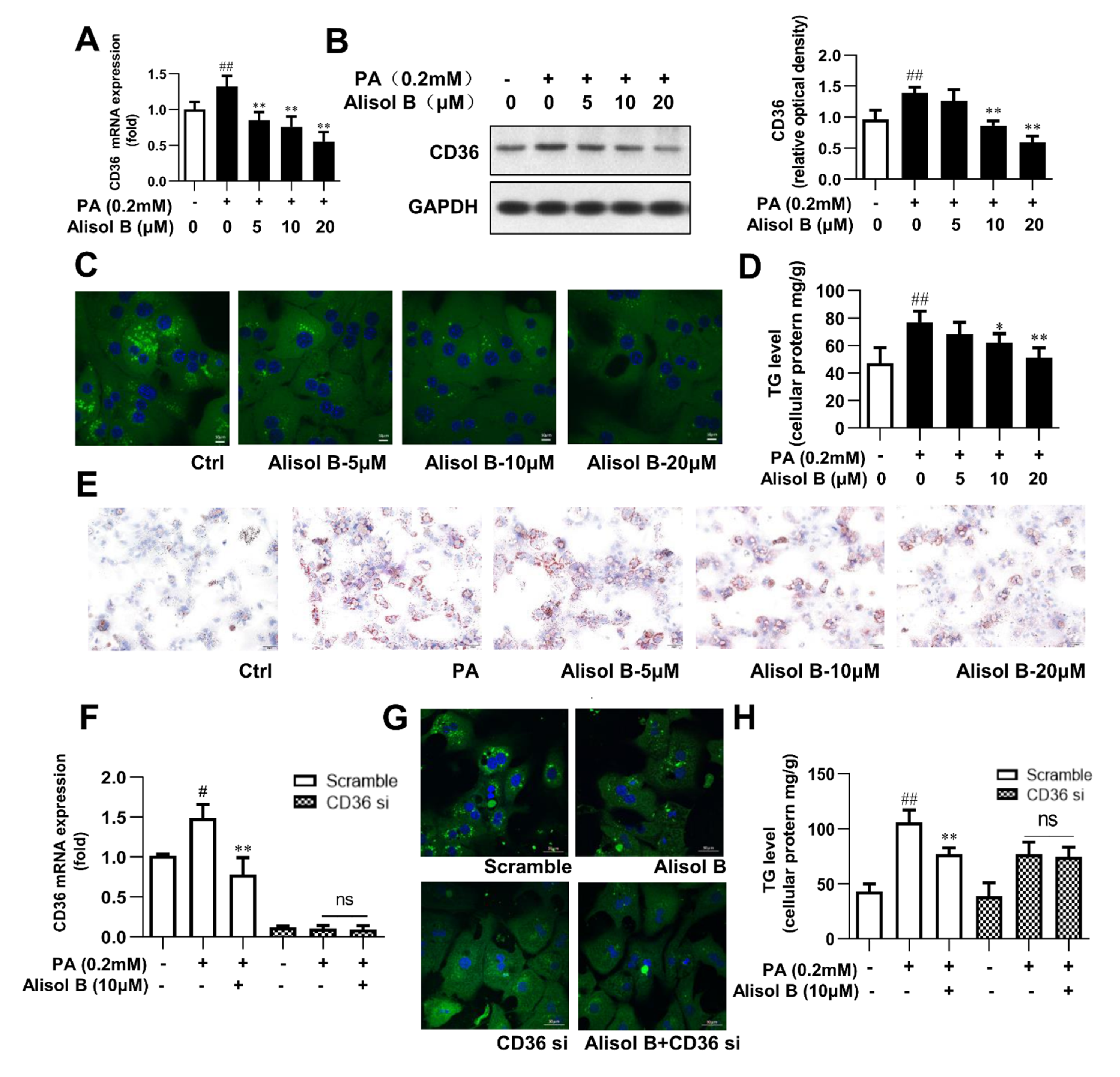
3.4. Alisol B Ameliorated Cellular TG Accumulation in Primary Hepatocytes by Inhibiting FFA Uptake in a CD36-Dependent Manner
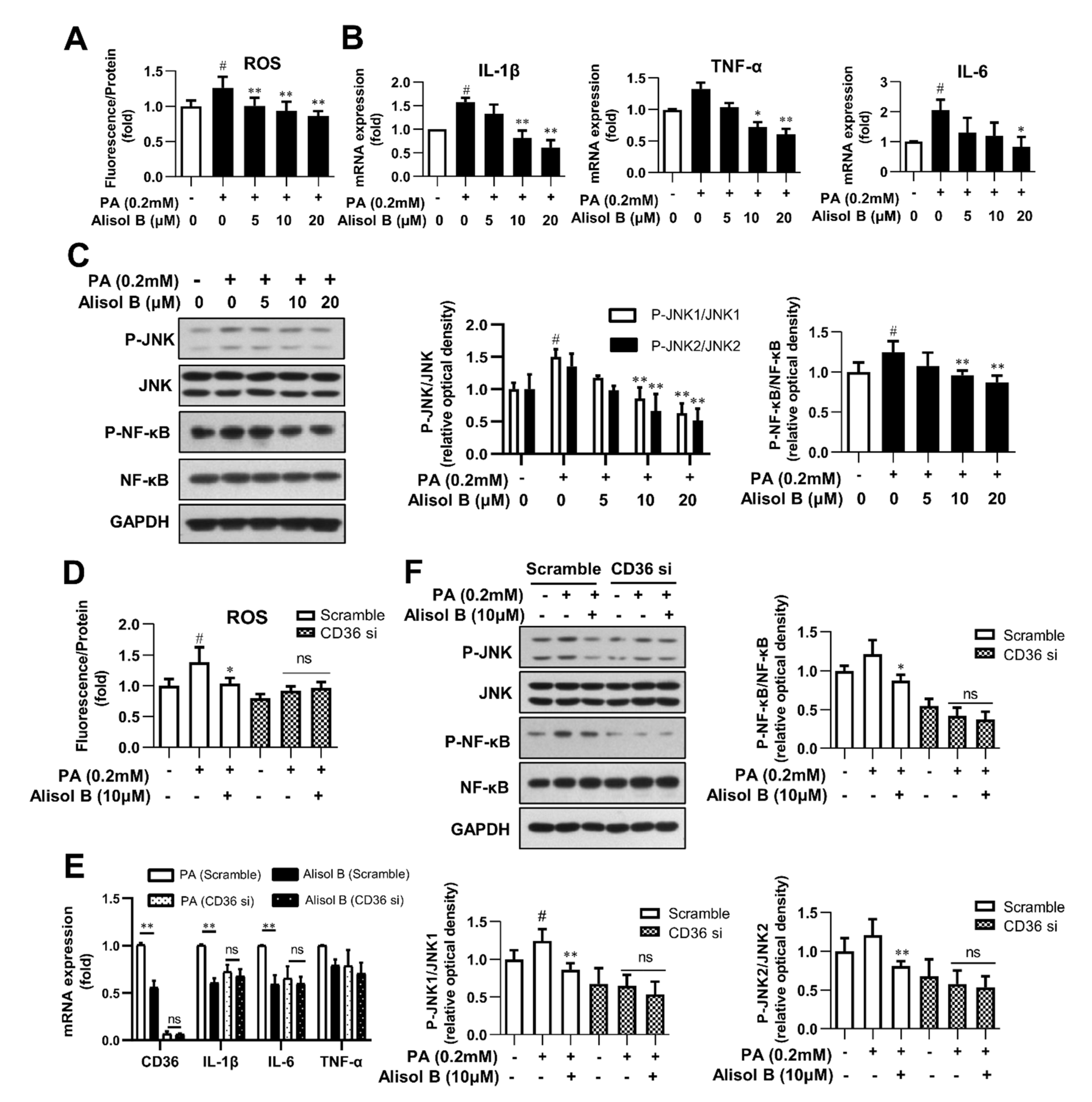
3.5. Alisol B Inhibited Oxidative Stress and Inflammation in Primary Hepatocytes in a CD36-Dependent Manner
3.6. Alisol B Decreased CD36 Expression through Downregulating PPARγ, and This Effect Was Independent of FXR
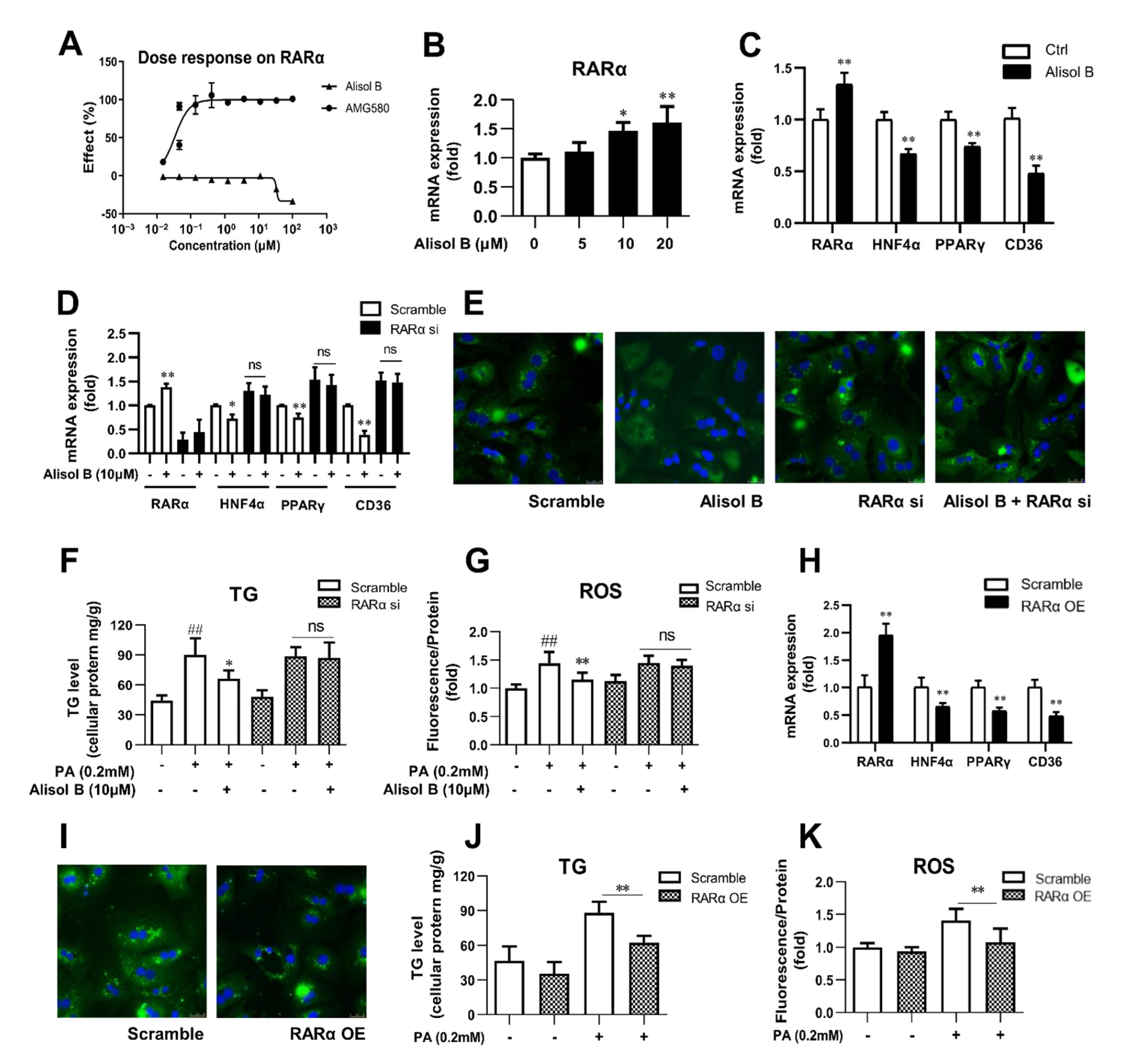
3.7. Alisol B Decreased CD36 Expression and Attenuated Hepatocyte Lipid Accumulation and Lipotoxicity via RARα-HNF4α-PPARγ Transcriptional Cascade
3.8. Chronic Treatment of Alisol B Regulated RARα-PPARγ-CD36 Transcriptional Cascade and Inhibited JNK/NF-κB Signaling Pathway in NASH Mice
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef] [PubMed]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.; Cabrera, D.; Arrese, M.; Feldstein, A.E. Triggering and resolution of inflammation in NASH. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Geisler, C.E.; Renquist, B.J. Hepatic lipid accumulation: Cause and consequence of dysregulated glucoregulatory hormones. J. Endocrinol. 2017, 234, R1–R21. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Investig. 2005, 115, 1343–1351. [Google Scholar] [CrossRef] [Green Version]
- Miquilena-Colina, M.E.; Lima-Cabello, E.; Sanchez-Campos, S.; Garcia-Mediavilla, M.V.; Fernandez-Bermejo, M.; Lozano-Rodriguez, T.; Vargas-Castrillon, J.; Buque, X.; Ochoa, B.; Aspichueta, P.; et al. Hepatic fatty acid translocase CD36 upregulation is associated with insulin resistance, hyperinsulinaemia and increased steatosis in non-alcoholic steatohepatitis and chronic hepatitis C. Gut 2011, 60, 1394–1402. [Google Scholar] [CrossRef]
- Wilson, C.G.; Tran, J.L.; Erion, D.M.; Vera, N.B.; Febbraio, M.; Weiss, E.J. Hepatocyte-Specific Disruption of CD36 Attenuates Fatty Liver and Improves Insulin Sensitivity in HFD-Fed Mice. Endocrinology 2016, 157, 570–585. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Febbraio, M.; Wada, T.; Zhai, Y.; Kuruba, R.; He, J.; Lee, J.H.; Khadem, S.; Ren, S.; Li, S.; et al. Hepatic fatty acid transporter Cd36 is a common target of LXR, PXR, and PPARgamma in promoting steatosis. Gastroenterology 2008, 134, 556–567. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.; Wang, C.; Zhang, X.; Peng, L.; Liu, W.; Zhang, X.; Liu, Y.; He, J.; Jiang, C.; Ai, D.; et al. Hyperhomocysteinemia activates the aryl hydrocarbon receptor/CD36 pathway to promote hepatic steatosis in mice. Hepatology 2016, 64, 92–105. [Google Scholar] [CrossRef]
- Han, J.; Hajjar, D.P.; Febbraio, M.; Nicholson, A.C. Native and modified low density lipoproteins increase the functional expression of the macrophage class B scavenger receptor, CD36. J. Biol. Chem. 1997, 272, 21654–21659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goncalves, M.B.; Wu, Y.; Trigo, D.; Clarke, E.; Malmqvist, T.; Grist, J.; Hobbs, C.; Carlstedt, T.P.; Corcoran, J.P.T. Retinoic acid synthesis by NG2 expressing cells promotes a permissive environment for axonal outgrowth. Neurobiol. Dis. 2018, 111, 70–79. [Google Scholar] [CrossRef]
- Koterazawa, Y.; Koyanagi-Aoi, M.; Uehara, K.; Kakeji, Y.; Aoi, T. Retinoic acid receptor gamma activation promotes differentiation of human induced pluripotent stem cells into esophageal epithelium. J. Gastroenterol. 2020, 55, 763–774. [Google Scholar] [CrossRef]
- Blaner, W.S. Vitamin A signaling and homeostasis in obesity, diabetes, and metabolic disorders. Pharmacol. Ther. 2019, 197, 153–178. [Google Scholar] [CrossRef]
- Yanagitani, A.; Yamada, S.; Yasui, S.; Shimomura, T.; Murai, R.; Murawaki, Y.; Hashiguchi, K.; Kanbe, T.; Saeki, T.; Ichiba, M.; et al. Retinoic acid receptor alpha dominant negative form causes steatohepatitis and liver tumors in transgenic mice. Hepatology 2004, 40, 366–375. [Google Scholar] [CrossRef]
- Zarei, L.; Farhad, N.; Abbasi, A. All-Trans Retinoic Acid (atRA) effectively improves liver steatosis in a rabbit model of high fat induced liver steatosis. Arch. Physiol. Biochem. 2020, 23, 1–6. [Google Scholar] [CrossRef]
- Tsuchiya, H.; Ikeda, Y.; Ebata, Y.; Kojima, C.; Katsuma, R.; Tsuruyama, T.; Sakabe, T.; Shomori, K.; Komeda, N.; Oshiro, S.; et al. Retinoids ameliorate insulin resistance in a leptin-dependent manner in mice. Hepatology 2012, 56, 1319–1330. [Google Scholar] [CrossRef] [Green Version]
- Panera, N.; Ceccarelli, S.; De Stefanis, C.; Nobili, V.; Alisi, A. Retinoids counteract insulin resistance and liver steatosis: What’s the potential mechanism? Hepatology 2013, 58, 1185. [Google Scholar] [CrossRef]
- Kim, S.C.; Kim, C.K.; Axe, D.; Cook, A.; Lee, M.; Li, T.; Smallwood, N.; Chiang, J.Y.; Hardwick, J.P.; Moore, D.D.; et al. All-trans-retinoic acid ameliorates hepatic steatosis in mice by a novel transcriptional cascade. Hepatology 2014, 59, 1750–1760. [Google Scholar] [CrossRef] [Green Version]
- Tian, T.; Chen, H.; Zhao, Y.Y. Traditional uses, phytochemistry, pharmacology, toxicology and quality control of Alisma orientale (Sam.) Juzep: A review. J. Ethnopharmacol. 2014, 158 Pt A, 373–387. [Google Scholar] [CrossRef]
- Choi, E.; Jang, E.; Lee, J.H. Pharmacological Activities of Alisma orientale against Nonalcoholic Fatty Liver Disease and Metabolic Syndrome: Literature Review. Evid. Based Complement. Alternat. Med. 2019, 2019, 2943162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeon, S.H.; Jang, E.; Park, G.; Lee, Y.; Jang, Y.P.; Lee, K.T.; Inn, K.S.; Lee, J.K.; Lee, J.H. Beneficial Activities of Alisma orientale Extract in a Western Diet-Induced Murine Non-Alcoholic Steatohepatitis and Related Fibrosis Model via Regulation of the Hepatic Adiponectin and Farnesoid X Receptor Pathways. Nutrients 2022, 14, 695. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.; Gao, Y.; Zheng, D.; Liu, Y.; Shan, S.; Fang, B.; Zhao, Y.; Song, D.; Zhang, Y.; Li, Q. Alisol A attenuates high-fat-diet-induced obesity and metabolic disorders via the AMPK/ACC/SREBP-1c pathway. J. Cell. Mol. Med. 2019, 23, 5108–5118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, L.; Tang, W.; Yin, J.; Feng, L.; Li, Y.; Yao, X.; Zhou, B. Alisol A 24-Acetate Prevents Hepatic Steatosis and Metabolic Disorders in HepG2 Cells. Cell. Physiol. Biochem. 2016, 40, 453–464. [Google Scholar] [CrossRef]
- Luan, Z.L.; Huo, X.K.; Dong, P.P.; Tian, X.G.; Sun, C.P.; Lv, X.; Feng, L.; Ning, J.; Wang, C.; Zhang, B.J.; et al. Highly potent non-steroidal FXR agonists protostane-type triterpenoids: Structure-activity relationship and mechanism. Eur. J. Med. Chem. 2019, 182, 111652. [Google Scholar] [CrossRef]
- Kanno, Y.; Yatsu, T.; Yamashita, N.; Zhao, S.; Li, W.; Imai, M.; Kashima, M.; Inouye, Y.; Nemoto, K.; Koike, K. Alisol B 23-acetate from the rhizomes of Alisma orientale is a natural agonist of the human pregnane X receptor. Phytomedicine 2017, 26, 22–27. [Google Scholar] [CrossRef]
- Meng, Q.; Duan, X.P.; Wang, C.Y.; Liu, Z.H.; Sun, P.Y.; Huo, X.K.; Sun, H.J.; Peng, J.Y.; Liu, K.X. Alisol B 23-acetate protects against non-alcoholic steatohepatitis in mice via farnesoid X receptor activation. Acta Pharmacol. Sin. 2017, 38, 69–79. [Google Scholar] [CrossRef] [Green Version]
- Xu, F.; Lu, C.; Wu, Q.; Gu, W.; Chen, J.; Fang, F.; Zhao, B.; Du, W.; You, M. Studies on the lipid-regulating mechanism of alisol-based compounds on lipoprotein lipase. Bioorg. Chem. 2018, 80, 347–360. [Google Scholar] [CrossRef]
- Li, C.; Yan, W.; Cui, E.; Zheng, C. Anti-bacterial effect of phytoconstituents isolated from Alimatis rhizoma. Appl. Biol. Chem. 2021, 64, 9. [Google Scholar] [CrossRef]
- Kleiner, D.E.; Brunt, E.M.; Van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.C.; Torbenson, M.S.; Unalp-Arida, A.; et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef]
- Liu, S.; Huang, S.; Wu, X.; Feng, Y.; Shen, Y.; Zhao, Q.S.; Leng, Y. Activation of SIK1 by phanginin A inhibits hepatic gluconeogenesis by increasing PDE4 activity and suppressing the cAMP signaling pathway. Mol. Metab. 2020, 41, 101045. [Google Scholar] [CrossRef]
- Ma, S.Y.; Ning, M.M.; Zou, Q.A.; Feng, Y.; Ye, Y.L.; Shen, J.H.; Leng, Y. OL3, a novel low-absorbed TGR5 agonist with reduced side effects, lowered blood glucose via dual actions on TGR5 activation and DPP-4 inhibition. Acta Pharmacol. Sin. 2016, 37, 1359–1369. [Google Scholar] [CrossRef] [Green Version]
- Musso, G.; Cassader, M.; Gambino, R. Non-alcoholic steatohepatitis: Emerging molecular targets and therapeutic strategies. Nat. Rev. Drug Discov. 2016, 15, 249–274. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Jing, M.; Yang, L.; Jin, L.; Ding, Y.; Lu, J.; Cao, Q.; Jiang, Y. Alisol A 24-acetate ameliorates nonalcoholic steatohepatitis by inhibiting oxidative stress and stimulating autophagy through the AMPK/mTOR pathway. Chem. Biol. Interact 2018, 291, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, T.; Lee, Y.A.; Fujiwara, N.; Ybanez, M.; Allen, B.; Martins, S.; Fiel, M.I.; Goossens, N.; Chou, H.I.; Hoshida, Y.; et al. A simple diet- and chemical-induced murine NASH model with rapid progression of steatohepatitis, fibrosis and liver cancer. J. Hepatol. 2018, 69, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Caballero, F.; Fernandez, A.; Matias, N.; Martinez, L.; Fucho, R.; Elena, M.; Caballeria, J.; Morales, A.; Fernandez-Checa, J.C.; Garcia-Ruiz, C. Specific contribution of methionine and choline in nutritional nonalcoholic steatohepatitis: Impact on mitochondrial S-adenosyl-L-methionine and glutathione. J. Biol. Chem. 2010, 285, 18528–18536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, J.; Lee, J.H.; Febbraio, M.; Xie, W. The emerging roles of fatty acid translocase/CD36 and the aryl hydrocarbon receptor in fatty liver disease. Exp. Biol. Med. 2011, 236, 1116–1121. [Google Scholar] [CrossRef]
- Xu, C.; Zhang, C.; Ji, J.; Wang, C.; Yang, J.; Geng, B.; Zhao, T.; Zhou, H.; Mu, X.; Pan, J.; et al. CD36 deficiency attenuates immune-mediated hepatitis in mice by modulating the proapoptotic effects of CXC chemokine ligand 10. Hepatology 2018, 67, 1943–1955. [Google Scholar] [CrossRef] [Green Version]
- Puengel, T.; Liu, H.; Guillot, A.; Heymann, F.; Tacke, F.; Peiseler, M. Nuclear Receptors Linking Metabolism, Inflammation, and Fibrosis in Nonalcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2022, 23, 2668. [Google Scholar] [CrossRef]
- Tang, X.H.; Melis, M.; Lu, C.; Rappa, A.; Zhang, T.; Jessurun, J.; Gross, S.S.; Gudas, L.J. A retinoic acid receptor beta2 agonist attenuates transcriptome and metabolome changes underlying nonalcohol-associated fatty liver disease. J. Biol. Chem. 2021, 297, 101331. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Sequence (5′-3′) |
|---|---|
| ABCG5-F | ATCCAACACCTCTATGCTAAATCAC |
| ABCG5-R | TACATTATTGGACCAGTTCAGTCAC |
| ABCG8-F | GAGAGCTTCACAGCCCACAA |
| ABCG8-R | GCCTGAAGATGTCAGAGCGA |
| AHR-F | TTCTTAGGCTCAGCGTCAGCTA |
| AHR-R | GCAAATCCTGCCAGTCTCTGAT |
| CD36-F | ATGGGCTGTGATCGGAACTG |
| CD36-R | GTCTTCCCAATAAGCATGTCTCC |
| Col-1a1-F | ACCTGTGTGTTCCCTACTCA |
| Col-1a1-R | GACTGTTGCCTTCGCCTCTG |
| CPT1α-F | TGGCATCATCACTGGTGTGTT |
| CPT1α-R | GTCTAGGGTCCGATTGATCTTTG |
| CYP1A1-F | CCTCATGTACCTGGTAACCA |
| CYP1A1-R | AAGGATGAATGCCGGAAGGT |
| CYP4A10-F | TCCAGCAGTTCCCATCACCT |
| CYP4A10-R | TTGCTTCCCCAGAACCATCT |
| CYP4A14-F | CCCAAAGGTATCACAGCCACAA |
| CYP4A14-R | CAGCAATTCAAAGCGGAGCAG |
| GAPDH-F | GGATTTGGCCGTATTGGGCG |
| GAPDH-R | CAGTAGAGGCAGGGATGATG |
| HNF4α-F | GGTTTAGCCGACAATGTGTGG |
| HNF4α-R | TCCCGCTCATTTTGGACAGC |
| IL-1β-F | ACCCTGCAGCTGGAGAGTGT |
| IL-1β-R | TTGACTTCTATCTTGTTGAAGACAAACC |
| IL-6-F | CCACTTCACAAGTCGGAGGCTTA |
| IL-6-R | GCAAGTGCATCATCGTTGTTCATAC |
| LXR-F | TCTGGAGACATCTCGGAGGTA |
| LXR-R | GGCTCACCAGTTTCATTAGCA |
| PPAR-γ-F | GGGGCCTGGACCTCTGCTGGGGATCT |
| PPAR-γ-R | GGCCAGAATGGCATCTCTGTGTCAA |
| PXR-F | CCCATCAACGTAGAGGAGGA |
| PXR-R | GGGGGTTGGTAGTTCCAGAT |
| RARα-F | GGCATCAACAAGCAAGAGTTTGGC |
| RARα-R | CTTTTTGGTGAGGTGATCTGTCCC |
| TGF-β-F | GTGTGGAGCAACATGTGGAACTCTA |
| TGF-β-R | TTGGTTCAGCCACTGCCGTA |
| TNF-α-F | TATGGCCCAGACCCTCACA |
| TNF-α-R | GGAGTAGACAAGGTACAACCCATC |
| UGT1a1-F | AGATTACCCCAGGCCCATC |
| UGT1a1-R | ATGGCTTTCTTCTCCGGAAT |
| α-SMA-F | GACGCTGAAGTATCCGATAGAACACG |
| α-SMA-R | CACCATCTCCAGAGTCCAGCACAAT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, Z.; Deng, Z.-T.; Huang, S.; Ning, M.; Feng, Y.; Shen, Y.; Zhao, Q.-S.; Leng, Y. Alisol B Alleviates Hepatocyte Lipid Accumulation and Lipotoxicity via Regulating RARα-PPARγ-CD36 Cascade and Attenuates Non-Alcoholic Steatohepatitis in Mice. Nutrients 2022, 14, 2411. https://doi.org/10.3390/nu14122411
Zhao Z, Deng Z-T, Huang S, Ning M, Feng Y, Shen Y, Zhao Q-S, Leng Y. Alisol B Alleviates Hepatocyte Lipid Accumulation and Lipotoxicity via Regulating RARα-PPARγ-CD36 Cascade and Attenuates Non-Alcoholic Steatohepatitis in Mice. Nutrients. 2022; 14(12):2411. https://doi.org/10.3390/nu14122411
Chicago/Turabian StyleZhao, Zhuohui, Zhen-Tao Deng, Suling Huang, Mengmeng Ning, Ying Feng, Yu Shen, Qin-Shi Zhao, and Ying Leng. 2022. "Alisol B Alleviates Hepatocyte Lipid Accumulation and Lipotoxicity via Regulating RARα-PPARγ-CD36 Cascade and Attenuates Non-Alcoholic Steatohepatitis in Mice" Nutrients 14, no. 12: 2411. https://doi.org/10.3390/nu14122411
APA StyleZhao, Z., Deng, Z.-T., Huang, S., Ning, M., Feng, Y., Shen, Y., Zhao, Q.-S., & Leng, Y. (2022). Alisol B Alleviates Hepatocyte Lipid Accumulation and Lipotoxicity via Regulating RARα-PPARγ-CD36 Cascade and Attenuates Non-Alcoholic Steatohepatitis in Mice. Nutrients, 14(12), 2411. https://doi.org/10.3390/nu14122411