
Liver Metastatic Breast Cancer: Epidemiology, Dietary Interventions, and Related Metabolism


Abstract
:1. Introduction
2. Breast Cancer Metastasis
3. Breast Cancer Liver Metastasis Diagnosis, Therapies, and Potential Treatments

{kind=link}
| Therapy | Administration | Target | Combination | Status | Year |
|---|---|---|---|---|---|
| Everolimus [42] | Oral | mTOR | Not noted | FDA approved | 2020 |
| Alpelisib [28,42] | Oral | PI3K-alpha | Combination with fulvestrantor letrozole | FDA approved | 2020 |
| Elacestrant [43] | Oral | Estrogen receptor | Low-fat diet combination | Phase Ib | 2020 |
| Giredestrant [44] | Oral | Estrogen receptor | Not noted | Phase III | 2021 |
| AZD9833 [45] | Oral | Estrogen receptor | Not noted | Phase I | 2020 |
4. Link between Diets and Metastatic Breast Cancer
4.1. Western Diet
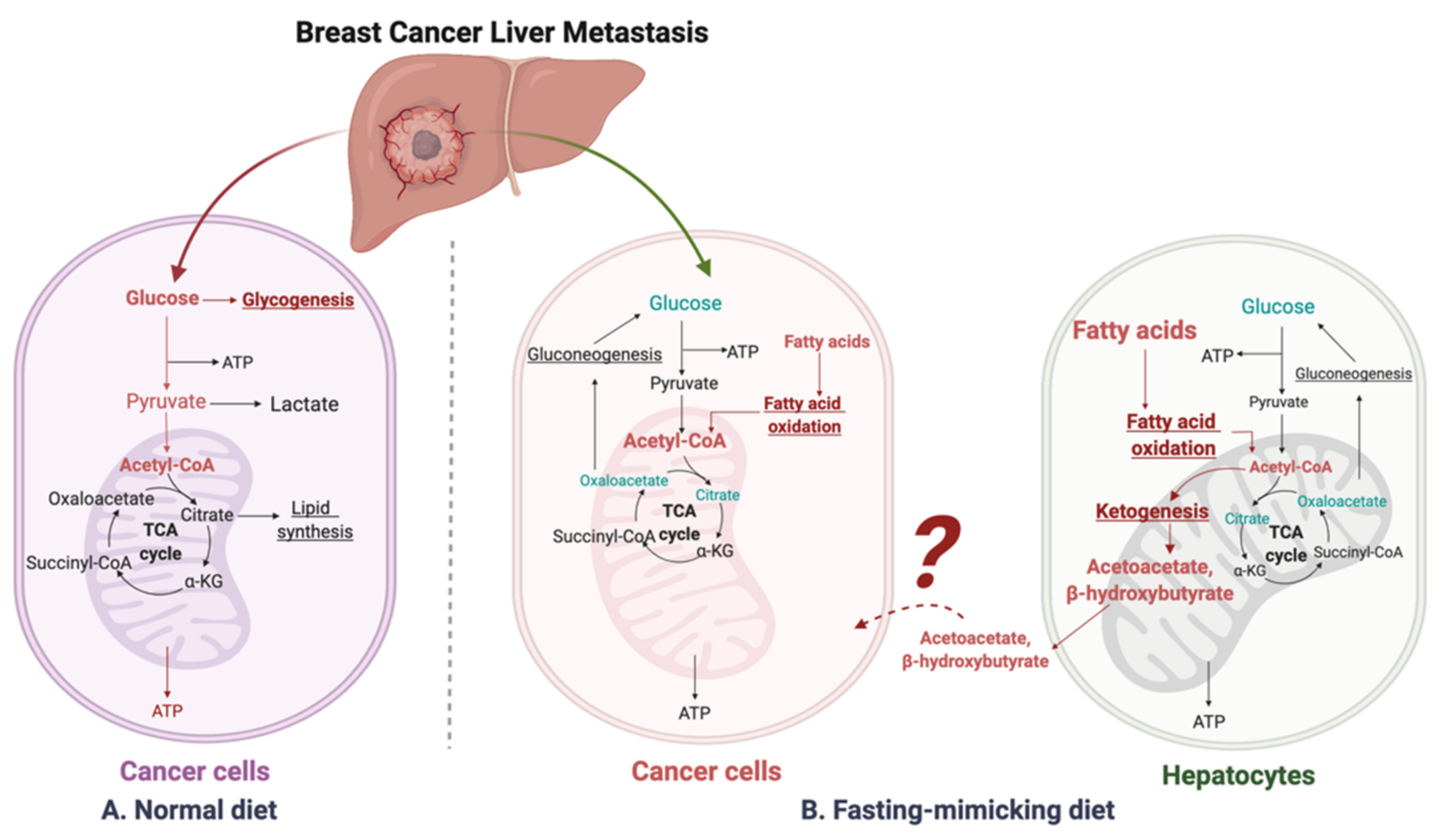
4.2. Fasting-Mimicking Diet
4.3. β-Hydroxybutyrate Paradox
5. Conclusions and Future Perspective
Author Contributions
Funding
Conflicts of Interest
References
- Tarver, T. Breast Cancer Facts & Figures 2021; American Cancer Society: Atlanta, GA, USA, 2021. [Google Scholar]
- National Cancer Institute. Cancer Stat Facts: Female Breast Cancer; National Cancer Institute: Bethesda, MD, USA, 2020.
- Azamjah, N.; Soltan-Zadeh, Y.; Zayeri, F. Global Trend of Breast Cancer Mortality Rate: A 25-Year Study. Asian Pac. J. Cancer Prev. 2019, 20, 2015–2020. [Google Scholar] [CrossRef] [PubMed]
- Lévesque, S.; Pol, J.G.; Ferrere, G.; Galluzzi, L.; Zitvogel, L.; Kroemer, G. Trial watch: Dietary interventions for cancer therapy. OncoImmunology 2019, 8, e1591878. [Google Scholar] [CrossRef]
- Mariotto, A.B.; Etzioni, R.; Hurlbert, M.; Penberthy, L.; Mayer, M. Estimation of the Number of Women Living with Metastatic Breast Cancer in the United States. Cancer Epidemiol. Prev. Biomark. 2017, 26, 809–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, G.P.; Massagué, J. Cancer metastasis: Building a framework. Cell 2006, 127, 679–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Eng, L.G.; Dawood, S.; Sopik, V.; Haaland, B.; Tan, P.S.; Bhoo-Pathy, N.; Warner, E.; Iqbal, J.; Narod, S.A.; Dent, R. Ten-year survival in women with primary stage IV breast cancer. Breast Cancer Res. Treat. 2016, 160, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Howlader, N.; Noone, A.M.; Krapcho, M.; Miller, D.; Brest, A.; Yu, M.; Ruhl, J.; Tatalovich, Z.; Mariotto, A.; Lewis, D.R.; et al. SEER Cancer Statistics Review, 1975–2017; National Cancer Institute: Bethesda, MD, USA, 2020. Available online: https://seer.cancer.gov/csr/1975_2017/ (accessed on 5 April 2022).
- Soni, D.A.; Ren, Z.; Hameed, O.; Chanda, D.; Morgan, C.J.; Siegal, G.P.; Wei, S. Breast Cancer Subtypes Predispose the Site of Distant Metastases. Am. J. Clin. Pathol. 2015, 143, 471–478. [Google Scholar] [CrossRef]
- Gong, Y.; Liu, Y.-R.; Ji, P.; Hu, X.; Shao, Z.-M. Impact of molecular subtypes on metastatic breast cancer patients: A SEER population-based study. Sci. Rep. 2017, 7, 45411. [Google Scholar] [CrossRef] [Green Version]
- Horn, S.R.; Stoltzfus, K.C.; Lehrer, E.J.; Dawson, L.A.; Tchelebi, L.; Gusani, N.J.; Sharma, N.K.; Chen, H.; Trifiletti, D.M.; Zaorsky, N.G. Epidemiology of liver metastases. Cancer Epidemiol. 2020, 67, 101760. [Google Scholar] [CrossRef] [PubMed]
- Rashid, N.S.; Grible, J.M.; Clevenger, C.V.; Harrell, J.C. Breast cancer liver metastasis: Current and future treatment approaches. Clin. Exp. Metastasis 2021, 38, 263–277. [Google Scholar] [CrossRef] [PubMed]
- Adam, R.; Aloia, T.; Krissat, J.; Bralet, M.P.; Paule, B.; Giacchetti, S.; Delvart, V.; Azoulay, D.; Bishmuth, H.; Castaing, D. Is liver resection justified for patients with hepatic metastases from breast cancer? Ann. Surg. 2006, 244, 897. [Google Scholar] [CrossRef] [PubMed]
- De Ridder, J.; De Wilt, J.H.W.; Simmer, F.; Overbeek, L.; Lemmens, V.; Nagtegaal, I. Incidence and origin of histologically confirmed liver metastases: An explorative case-study of 23,154 patients. Oncotarget 2016, 7, 55368–55376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cummings, M.C.; Simpson, P.T.; Reid, L.E.; Jayanthan, J.; Skerman, J.; Song, S.; Reed, A.E.M.; Kutasovic, J.R.; Morey, A.L.; Marquart, L.; et al. Metastatic progression of breast cancer: Insights from 50 years of autopsies. J. Pathol. 2014, 232, 23–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, L.; Cheng, L.; Zhu, X.; Gao, Y.; Fan, L.; Wang, Z. Risk and prognostic factors of breast cancer with liver metastases. BMC Cancer 2021, 21, 1–15. [Google Scholar] [CrossRef]
- Xie, J.Z.; Xu, A. Population-Based Study on Liver Metastases in Women with Newly Diagnosed Breast Cancer. Cancer Epidemiol Biomark. Prev. 2019, 28, 283–292. [Google Scholar] [CrossRef] [Green Version]
- Diamond, J.R.; Finlayson, C.A.; Borges, V.F. Hepatic complications of breast cancer. Lancet Oncol. 2009, 10, 615–621. [Google Scholar] [CrossRef]
- Patanaphan, V.; Salazar, O.M.; Risco, R. Breast cancer: Metastatic patterns and their prognosis. South. Med. J. 1988, 81, 1109–1112. [Google Scholar] [CrossRef]
- Cao, R.; Wang, L.-P. Serological Diagnosis of Liver Metastasis in Patients with Breast Cancer. Cancer Biol. Med. 2012, 9, 57–62. [Google Scholar] [CrossRef]
- Bale, R.; Putzer, D.; Schullian, P. Local Treatment of Breast Cancer Liver Metastasis. Cancers 2019, 11, 1341. [Google Scholar] [CrossRef] [Green Version]
- Higgins, M.J.; Baselga, J. Targeted therapies for breast cancer. J. Clin. Investig. 2011, 121, 3797–3803. [Google Scholar] [CrossRef]
- Carrick, S.; Parker, S.; Thornton, C.E.; Ghersi, D.; Simes, J.; Wilcken, N. Single agent versus combination chemotherapy for metastatic breast cancer. Cochrane Database Syst. Rev. 2009, 2021, CD003372. [Google Scholar] [CrossRef]
- Osborne, C.K. Tamoxifen in the treatment of breast cancer. N. Engl. J. Med. 1998, 339, 1609–1618. [Google Scholar] [CrossRef] [PubMed]
- Smith, I.E.; Dowsett, M. Aromatase inhibitors in breast cancer. N. Engl. J. Med. 2003, 348, 2431–2442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spoerke, J.M.; Gendreau, S.; Walter, K.; Qiu, J.; Wilson, T.R.; Savage, H.; Aimi, J.; Derynck, M.K.; Chen, M.; Chan, I.T.; et al. Heterogeneity and clinical significance of ESR1 mutations in ER-positive metastatic breast cancer patients receiving fulvestrant. Nat. Commun. 2016, 7, 1–10. [Google Scholar] [CrossRef]
- Mayer, I.A.; Abramson, V.G.; Formisano, L.; Balko, J.M.; Estrada, M.V.; Sanders, M.E. A phase Ib study of alpelisib (BYL719), a PI3Kα-specific inhibitor, with letrozole in ER+/HER2− metastatic breast cancer. Clin. Cancer Res. 2017, 23, 26–34. [Google Scholar] [CrossRef] [Green Version]
- Coleman, R.L.; Brady, M.F.; Herzog, T.J.; Sabbatini, P.; Armstrong, D.K.; Walker, J.L.; Kim, B.G.; Fujiwara, K.; Tewari, K.S.; O’Malley, D.M.; et al. Bevacizumab and paclitaxel–carboplatin chemotherapy and secondary cytoreduction in recurrent, platinum-sensitive ovarian cancer (NRG Oncology/Gynecologic Oncology Group study GOG-0213): A multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2017, 18, 779–791. [Google Scholar] [CrossRef] [Green Version]
- Delaloge, S.; Pérol, D.; Courtinard, C.; Brain, E.; Asselain, B.; Bachelot, T.; Debled, M.; Dieras, V.; Campone, M.; Levy, C.; et al. Paclitaxel plus bevacizumab or paclitaxel as first-line treatment for HER2-negative metastatic breast cancer in a multicenter national observational study. Ann. Oncol. 2016, 27, 1725–1732. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.S.; Martin, M.; Rugo, H.S.; Jones, S.; Im, S.-A.; Gelmon, K.; Harbeck, N.; Lipatov, O.N.; Walshe, J.M.; Moulder, S.; et al. Palbociclib and Letrozole in Advanced Breast Cancer. N. Engl. J. Med. 2016, 375, 1925–1936. [Google Scholar] [CrossRef]
- Turner, N.C.; Ro, J.; André, F.; Loi, S.; Verma, S.; Iwata, H.; Harbeck, N.; Loibl, S.; Bartlett, C.H.; Zhang, K.; et al. Palbociclib in Hormone-Receptor–Positive Advanced Breast Cancer. N. Engl. J. Med. 2015, 373, 209–219. [Google Scholar] [CrossRef] [Green Version]
- Hortobagyi, G.N.; Stemmer, S.M.; Burris, H.A.; Yap, Y.-S.; Sonke, G.S.; Paluch-Shimon, S.; Campone, M.; Blackwell, K.L.; André, F.; Winer, E.P.; et al. Ribociclib as First-Line Therapy for HR-Positive, Advanced Breast Cancer. N. Engl. J. Med. 2016, 375, 1738–1748. [Google Scholar] [CrossRef]
- Senkus, E.; Łacko, A. Over-treatment in metastatic breast cancer. Breast 2017, 31, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, F.; Harbeck, N.; Fallowfield, L.; Kyriakides, S.; Senkus, E. ESMO Guidelines Working Group. Locally recurrent or metastatic breast cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2012, 23, vii11–vii19. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.; Lee, L.; Werfel, T.; Joly, M.M.M.; Hicks, D.J.; Rahman, B.; Elion, D.; McKernan, C.; Sanchez, V.; Estrada, M.V.; et al. Intrinsic apoptotic pathway activation increases response to anti-estrogens in luminal breast cancers. Cell Death Dis. 2018, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Richman, J.; Dowsett, M. Beyond 5 years: Enduring risk of recurrence in oestrogen receptor-positive breast cancer. Nat. Rev. Clin. Oncol. 2019, 16, 296–311. [Google Scholar] [CrossRef]
- Razavi, P.; Chang, M.T.; Xu, G.; Bandlamudi, C.; Ross, D.S.; Vasan, N.; Cai, Y.; Bielski, C.M.; Donoghue, M.T.A.; Jonsson, P.; et al. The Genomic Landscape of Endocrine-Resistant Advanced Breast Cancers. Cancer Cell 2018, 34, 427–438.e6. [Google Scholar] [CrossRef] [Green Version]
- Boudreau, M.W.; Duraki, D.; Wang, L.; Mao, C.; Kim, J.E.; Henn, M.A.; Tang, B.; Fanning, S.W.; Kiefer, J.; Tarasow, T.M.; et al. A small-molecule activator of the unfolded protein response eradicates human breast tumors in mice. Sci. Transl. Med. 2021, 13, eabf1383. [Google Scholar] [CrossRef]
- Holland, S.J.; Pan, A.; Franci, C.; Hu, Y.; Chang, B.; Li, W.; Duan, M.; Torneros, A.; Yu, J.; Heckrodt, T.J.; et al. R428, a Selective Small Molecule Inhibitor of Axl Kinase, Blocks Tumor Spread and Prolongs Survival in Models of Metastatic Breast Cancer. Cancer Res. 2010, 70, 1544–1554. [Google Scholar] [CrossRef] [Green Version]
- Deng, S.; Krutilina, R.I.; Wang, Q.; Lin, Z.; Parke, D.N.; Playa, H.C.; Chen, H.; Miller, D.D.; Seagroves, T.N.; Li, W. An Orally Available Tubulin Inhibitor, VERU-111, Suppresses Triple-Negative Breast Cancer Tumor Growth and Metastasis and Bypasses Taxane Resistance. Mol. Cancer Ther. 2020, 19, 348–363. [Google Scholar] [CrossRef] [Green Version]
- Vernieri, C.; Corti, F.; Nichetti, F.; Ligorio, F.; Manglaviti, S.; Zattarin, E.; Rea, C.G.; Capri, G.; Bianchi, G.V.; De Braud, F. Everolimus versus alpelisib in advanced hormone receptor-positive HER2-negative breast cancer: Targeting different nodes of the PI3K/AKT/mTORC1 pathway with different clinical implications. Breast Cancer Res. 2020, 22, 1–13. [Google Scholar] [CrossRef]
- Jager, A.; De Vries, E.G.E.; Oordt, C.W.M.-V.D.H.V.; Neven, P.; Venema, C.M.; Glaudemans, A.W.J.M.; Wang, Y.; Bagley, R.G.; Conlan, M.G.; Aftimos, P. A phase 1b study evaluating the effect of elacestrant treatment on estrogen receptor availability and estradiol binding to the estrogen receptor in metastatic breast cancer lesions using 18F-FES PET/CT imaging. Breast Cancer Res. 2020, 22, 1–11. [Google Scholar] [CrossRef]
- Liang, J.; Zbieg, J.R.; Blake, R.A.; Chang, J.H.; Daly, S.; DiPasquale, A.G.; Friedman, L.S.; Gelzleichter, T.; Gill, M.; Giltnane, J.M.; et al. GDC-9545 (Giredestrant): A Potent and Orally Bioavailable Selective Estrogen Receptor Antagonist and Degrader with an Exceptional Preclinical Profile for ER+ Breast Cancer. J. Med. Chem. 2021, 64, 11841–11856. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.S.; Moss, T.A.; Balazs, A.; Barlaam, B.; Breed, J.; Carbajo, R.J.; Chiarparin, E.; Davey, P.R.J.; Delpuech, O.; Fawell, S.; et al. Discovery of AZD9833, a Potent and Orally Bioavailable Selective Estrogen Receptor Degrader and Antagonist. J. Med. Chem. 2020, 63, 14530–14559. [Google Scholar] [CrossRef] [PubMed]
- Doll, R.; Peto, R. The Causes of Cancer: Quantitative Estimates of Avoidable Risks of Cancer in the United States Today. J. Natl. Cancer Inst. 1981, 66, 1192–1308. [Google Scholar] [CrossRef]
- Nicodemus, K.K.; Jacobsr, D.R., Jr.; Folsom, A.R. Whole and refined grain intake and risk of incident postmenopausal breast cancer (United States). Cancer Causes Control 2001, 12, 917–925. [Google Scholar] [CrossRef] [PubMed]
- Dydjow-Bendek, D.; Zagozdzon, P. Total Dietary Fats, Fatty Acids, and Omega-3/Omega-6 Ratio as Risk Factors of Breast Cancer in the Polish Population-a Case-Control Study. In Vivo 2020, 34, 423–431. [Google Scholar] [CrossRef]
- Tsilidis, K.K.; Travis, R.C.; Appleby, P.N.; Allen, N.E.; Lindström, S.; Albanes, D.; Ziegler, R.G.; McCullough, M.L.; Siddiq, A.; Barricarte, A.; et al. Insulin-like growth factor pathway genes and blood concentrations, dietary protein and risk of prostate cancer in the NCI Breast and Prostate Cancer Cohort Consortium (BPC3). Int. J. Cancer 2013, 133, 495–504. [Google Scholar] [CrossRef] [Green Version]
- Williams, C.M.; Dickerson, J.W. Dietary fat, hormones and breast cancer: The cell membrane as a possible site of interaction of these two risk factors. Eur. J. Surg. Oncol. 1987, 13, 89–104. [Google Scholar]
- Levi, F.; La Vecchia, C.; Gulie, C.; Negri, E. Dietary Factors and Breast-Cancer Risk in Vaud, Switzerland. Nutr. Cancer Int. J. 1993, 19, 327–335. [Google Scholar] [CrossRef]
- Mauvais-Jarvis, F.; Clegg, D.J.; Hevener, A.L. The Role of Estrogens in Control of Energy Balance and Glucose Homeostasis. Endocr. Rev. 2013, 34, 309–338. [Google Scholar] [CrossRef] [Green Version]
- Madak-Erdogan, Z.; Band, S.; Zhao, Y.C.; Smith, B.P.; Kulkoyluoglu-Cotul, E.; Zuo, Q.; Casiano, A.S.; Wrobel, K.; Rossi, G.; Smith, R.L.; et al. Free fatty acids rewire cancer metabolism in obesity-associated breast cancer via estrogen receptor and mTOR signaling. Cancer Res. 2019, 79, 2494–2510. [Google Scholar] [CrossRef] [Green Version]
- Zuo, Q.; Band, S.; Kesavadas, M.; Erdogan, Z.M. Obesity and Postmenopausal Hormone Receptor-positive Breast Cancer: Epidemiology and Mechanisms. Endocrinology 2021, 162, bqab195. [Google Scholar] [CrossRef] [PubMed]
- Jiralerspong, S.; Goodwin, P.J. Obesity and Breast Cancer Prognosis: Evidence, Challenges, and Opportunities. J. Clin. Oncol. 2016, 34, 4203–4216. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, R.; Orsini, N.; Saji, S.; Key, T.J.; Wolk, A. Body weight and incidence of breast cancer defined by estrogen and progesterone receptor status-A meta-analysis. Int. J. Cancer 2009, 124, 698–712. [Google Scholar] [CrossRef] [PubMed]
- Iyengar, N.M.; Arthur, R.; Manson, J.E.; Chlebowski, R.T.; Kroenke, C.H.; Peterson, L.; Cheng, T.D.; Feliciano, E.; Lane, D.; Luo, J.; et al. Association of Body Fat and Risk of Breast Cancer in Postmenopausal Women with Normal Body Mass Index A Secondary Analysis of a Randomized Clinical Trial and Observational Study. JAMA Oncol. 2019, 5, 155–163. [Google Scholar] [CrossRef]
- Picon-Ruiz, M.; Morata-Tarifa, C.; Valle-Goffin, J.J.; Friedman, E.R.; Slingerland, J.M. Obesity and adverse breast cancer risk and outcome: Mechanistic insights and strategies for intervention. CA Cancer J. Clin. 2017, 67, 378–397. [Google Scholar] [CrossRef]
- Keum, N.; Greenwood, D.C.; Lee, D.H.; Kim, R.; Aune, D.; Ju, W.; Hu, F.B.; Giovannucci, E.L. Adult Weight Gain and Adiposity-Related Cancers: A Dose-Response Meta-Analysis of Prospective Observational Studies. J. Natl. Cancer Inst. 2015, 107, djv088. [Google Scholar]
- Renehan, A.G.; Tyson, M.; Egger, M.; Heller, R.F.; Zwahlen, M. Body-mass index and incidence of cancer: A systematic review and meta-analysis of prospective observational studies. Lancet 2008, 371, 569–578. [Google Scholar] [CrossRef]
- Chan, D.S.M.; Vieira, A.R.; Aune, D.; Bandera, E.V.; Greenwood, D.C.; McTiernan, A.; Rosenblatt, D.N.; Thune, I.; Vieira, R.; Norat, T. Body mass index and survival in women with breast cancer—systematic literature review and meta-analysis of 82 follow-up studies. Ann. Oncol. 2014, 25, 1901–1914. [Google Scholar] [CrossRef]
- Gathirua-Mwangi, W.G.; Palmer, J.R.; Champion, V.; Castro-Webb, N.; Stokes, A.C.; Adams-Campbell, L.; Marley, A.R.; Forman, M.R.; Rosenberg, L.; Bertrand, K.A. Maximum and Time-Dependent Body Mass Index and Breast Cancer Incidence Among Postmenopausal Women in the Black Women’s Health Study. Am. J. Epidemiol. 2022, 191, 646–654. [Google Scholar] [CrossRef]
- Chauhan, R.; Trivedi, V.; Rani, R.; Singh, U. A comparative analysis of body mass index with estrogen receptor, progesterone receptor and human epidermal growth factor receptor 2 status in pre- and postmenopausal breast cancer patients. J. Mid-Life Health 2020, 11, 210–216. [Google Scholar] [CrossRef]
- Statovci, D.; Aguilera, M.; Mac Sharry, J.; Melgar, S. The Impact of Western Diet and Nutrients on the Microbiota and Immune Response at Mucosal Interfaces. Front. Immunol. 2017, 8, 838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Montero, C.; Fraile-Martínez, O.; Gómez-Lahoz, A.M.; Pekarek, L.; Castellanos, A.J.; Noguerales-Fraguas, F.; Coca, S.; Guijarro, L.G.; García-Honduvilla, N.; Asúnsolo, A.; et al. Nutritional Components in Western Diet Versus Mediterranean Diet at the Gut Microbiota-Immune System Interplay. Implications for Health and Disease. Nutrients 2021, 13, 699. [Google Scholar] [CrossRef] [PubMed]
- Zinöcker, M.K.; Lindseth, I.A. The Western Diet–Microbiome-Host Interaction and Its Role in Metabolic Disease. Nutrients 2018, 10, 365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cully, M.; You, H.; Levine, A.J.; Mak, T.W. Beyond PTEN mutations: The PI3K pathway as an integrator of multiple inputs during tumorigenesis. Nat. Rev. Cancer 2006, 6, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Haythorne, E.; Rohm, M.; Van De Bunt, M.; Brereton, M.F.; Tarasov, A.I.; Blacker, T.S.; Sachse, G.; Dos Santos, M.S.; Exposito, R.T.; Davis, S.; et al. Diabetes causes marked inhibition of mitochondrial metabolism in pancreatic β-cells. Nat. Commun. 2019, 10, 1–17. [Google Scholar] [CrossRef]
- Newsholme, P.; Keane, K.N.; Carlessi, R.; Cruzat, V. Oxidative stress pathways in pancreatic β-cells and insulin-sensitive cells and tissues: Importance to cell metabolism, function, and dysfunction. Am. J. Physiol. Cell Physiol. 2019, 317, C420–C433. [Google Scholar] [CrossRef]
- Klement, R.J.; Kammerer, U. Is there a role for carbohydrate restriction in the treatment and prevention of cancer? Nutr. Metab. 2011, 8, 1–16. [Google Scholar] [CrossRef] [Green Version]
- LaPensee, C.R.; Hugo, E.R.; Ben-Jonathan, N. Insulin Stimulates Interleukin-6 Expression and Release in LS14 Human Adipocytes through Multiple Signaling Pathways. Endocrinology 2008, 149, 5415–5422. [Google Scholar] [CrossRef]
- Makino, T.; Noguchi, Y.; Yoshikawa, T.; Doi, C.; Nomura, K. Circulating interleukin 6 concentrations and insulin resistance in patients with cancer. Br. J. Surg. 1998, 85, 1658–1662. [Google Scholar] [CrossRef]
- Mccall, J.L.; Tuckey, J.A.; Parry, B.R. Serum Tumor-Necrosis-Factor-Alpha and Insulin Resistance in Gastrointestinal Cancer. Br. J. Surg. 1992, 79, 1361–1363. [Google Scholar] [CrossRef]
- Tian, M.; Zhang, H.; Nakasone, Y.; Mogi, K.; Endo, K. Expression of Glut-1 and Glut-3 in untreated oral squamous cell carcinoma compared with FDG accumulation in a PET study. Eur. J. Nucl. Med. Mol. Imaging 2004, 31, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.-L.; Wang, M.-D.; Zhou, X.; Qin, C.-J.; Fu, G.-B.; Tang, L.; Wu, H.; Huang, S.; Zhao, L.-H.; Zeng, M.; et al. Blocking preferential glucose uptake sensitizes liver tumor-initiating cells to glucose restriction and sorafenib treatment. Cancer Lett. 2017, 388, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maldonado, R.; Talana, C.A.; Song, C.; Dixon, A.; Uehara, K.; Weichhaus, M. β-hydroxybutyrate does not alter the effects of glucose deprivation on breast cancer cells. Oncol. Lett. 2021, 21, 1. [Google Scholar] [CrossRef] [PubMed]
- Varghese, S.; Samuel, S.M.; Varghese, E.; Kubatka, P.; Büsselberg, D. High Glucose Represses the Anti-Proliferative and Pro-Apoptotic Effect of Metformin in Triple Negative Breast Cancer Cells. Biomolecules 2019, 9, 16. [Google Scholar] [CrossRef] [Green Version]
- Zuo, Q.; Mogol, A.N.; Liu, Y.-J.; Casiano, A.S.; Chien, C.; Drnevich, J.; Imir, O.B.; Kulkoyluoglu-Cotul, E.; Park, N.H.; Shapiro, D.J.; et al. Targeting metabolic adaptations in the breast cancer-liver metastatic niche using dietary approaches to improve endocrine therapy efficacy. Mol. Cancer Res. 2022, 20, 923–937. [Google Scholar] [CrossRef]
- Healy, M.E.; Chow, J.D.; Byrne, F.L.; Breen, D.S.; Leitinger, N.; Li, C.; Lackner, C.; Caldwell, S.H.; Hoehn, K.L. Dietary effects on liver tumor burden in mice treated with the hepatocellular carcinogen diethylnitrosamine. J. Hepatol. 2015, 62, 599–606. [Google Scholar] [CrossRef] [Green Version]
- Bechmann, L.P.; Hannivoort, R.A.; Gerken, G.; Hotamisligil, G.S.; Trauner, M.; Canbay, A. The interaction of hepatic lipid and glucose metabolism in liver diseases. J. Hepatol. 2012, 56, 952–964. [Google Scholar] [CrossRef] [Green Version]
- Zhong, X.-Y.; Yuan, X.-M.; Xu, Y.-Y.; Yin, M.; Yan, W.-W.; Zou, S.-W.; Wei, L.-M.; Lu, H.-J.; Wang, Y.-P.; Lei, Q.-Y. CARM1 Methylates GAPDH to Regulate Glucose Metabolism and Is Suppressed in Liver Cancer. Cell Rep. 2018, 24, 3207–3223. [Google Scholar] [CrossRef] [Green Version]
- Krstic, J.; Reinisch, I.; Schindlmaier, K.; Galhuber, M.; Riahi, Z.; Berger, N.; Kupper, N.; Moyschewitz, E.; Auer, M.; Michenthaler, H.; et al. Fasting improves therapeutic response in hepatocellular carcinoma through p53-dependent metabolic synergism. Sci. Adv. 2022, 8, eabh2635. [Google Scholar] [CrossRef]
- Wahdan-Alaswad, R.S.; Edgerton, S.M.; Salem, H.S.; Thor, A.D. Metformin Targets Glucose Metabolism in Triple Negative Breast Cancer. J. Oncol. Transl. Res. 2018, 4, 129. [Google Scholar] [CrossRef]
- Roy, R.; Hahm, E.R.; White, A.G.; Anderson, C.J.; Singh, S.V. AKT-dependent sugar addiction by benzyl isothiocyanate in breast cancer cells. Mol. Carcinog. 2019, 58, 996–1007. [Google Scholar] [CrossRef] [PubMed]
- Gluschnaider, U.; Hertz, R.; Ohayon, S.; Smeir, E.; Smets, M.; Pikarsky, E.; Bar-Tana, J. Long-Chain Fatty Acid Analogues Suppress Breast Tumorigenesis and Progression. Cancer Res. 2014, 74, 6991–7002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, M.; Brandhorst, S.; Shelehchi, M.; Mirzaei, H.; Cheng, C.W.; Budniak, J.; Groshen, S.; Mack, W.J.; Guen, E.; Di Biase, S.; et al. Fasting-mimicking diet and markers/risk factors for aging, diabetes, cancer, and cardiovascular disease. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Poff, A.; Ari, C.; Arnold, P.; Seyfried, T.; D’Agostino, D. Ketone supplementation decreases tumor cell viability and prolongs survival of mice with metastatic cancer. Int. J. Cancer 2014, 135, 1711–1720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, B.G.; Bhatia, S.K.; Anderson, C.M.; Eichenberger-Gilmore, J.M.; Sibenaller, Z.A.; Mapuskar, K.A.; Schoenfeld, J.D.; Buatti, J.M.; Spitz, D.R.; Fath, M.A. Ketogenic diets as an adjuvant cancer therapy: History and potential mechanism. Redox Biol. 2014, 2, 963–970. [Google Scholar] [CrossRef] [Green Version]
- Rigo, P.; Paulus, P.; Kaschten, B.J.; Hustinx, R.; Bury, T.; Jerusalem, G.; Benoit, T.; Foidart-Willems, J. Oncological applications of positron emission tomography with fluorine-18 fluorodeoxyglucose. Eur. J. Nucl. Med. 1996, 23, 1641–1674. [Google Scholar] [CrossRef]
- Kaji, K.; Nishimura, N.; Seki, K.; Sato, S.; Saikawa, S.; Nakanishi, K.; Furukawa, M.; Kawaratani, H.; Kitade, M.; Moriya, K.; et al. Sodium glucose cotransporter 2 inhibitor canagliflozin attenuates liver cancer cell growth and angiogenic activity by inhibiting glucose uptake. Int. J. Cancer 2018, 142, 1712–1722. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Qiao, Y.; Wu, Q.; Chen, Y.; Zou, S.; Liu, X.; Zhu, G.; Zhao, Y.; Chen, Y.; Yu, Y.; et al. The essential role of YAP O-GlcNAcylation in high-glucose-stimulated liver tumorigenesis. Nat. Commun. 2017, 8, 1–15. [Google Scholar] [CrossRef]
- Bu, P.; Chen, K.-Y.; Xiang, K.; Johnson, C.; Crown, S.B.; Rakhilin, N.; Ai, Y.; Wang, L.; Xi, R.; Astapova, I.; et al. Aldolase B-Mediated Fructose Metabolism Drives Metabolic Reprogramming of Colon Cancer Liver Metastasis. Cell Metab. 2018, 27, 1249–1262.e4. [Google Scholar] [CrossRef] [Green Version]
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Anykin-Burns, N.; Ahmad, I.A.; Zhu, Y.; Oberley, L.W.; Spitz, D.R. Increased levels of superoxide and hydrogen peroxide mediate the differential susceptibility of cancer cells vs. Normal cells to glucose deprivation. Biochem. J. 2009, 418, 29–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boros, L.G.; Lee, P.W.N.; Brandesa, J.L.; Cascante, M.; Muscarellaa, P.; Schirmera, W.J.; Melvina, W.S.; Ellisona, E.C. Nonoxidative pentose phosphate pathways and their direct role in ribose synthesis in tumors: Is cancer a disease of cellular glucose metabolism? Med. Hypotheses 1998, 50, 55–59. [Google Scholar] [CrossRef]
- Weber, D.D.; Aminazdeh-Gohari, S.; Kofler, B. Ketogenic diet in cancer therapy. Aging 2018, 10, 164–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallace, D.C. Mitochondria and cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buettner, G.R. Superoxide dismutase in redox biology: The roles of superoxide and hydrogen peroxide. Anti-Cancer Agents Med. Chem. 2011, 11, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Veech, R.L. Ketone ester effects on metabolism and transcription. J. Lipid Res. 2014, 55, 2004–2006. [Google Scholar] [CrossRef] [Green Version]
- Hopkins, B.D.; Pauli, C.; Du, X.; Wang, D.G.; Li, X.; Wu, D.; Amadiume, S.C.; Goncalves, M.D.; Hodakoski, C.; Lundquist, M.R.; et al. Suppression of insulin feedback enhances the efficacy of PI3K inhibitors. Nature 2018, 560, 499–503, Erratum in Nature 2018, 563, E24. [Google Scholar] [CrossRef] [Green Version]
- Ho, V.W.; Leung, K.; Hsu, A.; Luk, B.; Lai, J.; Shen, S.Y.; Minchinton, A.I.; Waterhouse, D.; Bally, M.; Lin, W.; et al. A Low Carbohydrate, High Protein Diet Slows Tumor Growth and Prevents Cancer Initiation. Cancer Res. 2011, 71, 4484–4493. [Google Scholar] [CrossRef] [Green Version]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- Byrne, F.L.; Hargett, S.R.; Lahiri, S.; Roy, R.J.; Berr, S.S.; Caldwell, S.H.; Hoehn, K.L. Serial MRI Imaging Reveals Minimal Impact of Ketogenic Diet on Established Liver Tumor Growth. Cancers 2018, 10, 312. [Google Scholar] [CrossRef] [Green Version]
- Tan-Shalaby, J. Ketogenic diets and cancer: Emerging evidence. Fed. Pract. 2017, 34, 37S. [Google Scholar] [PubMed]
- Allen, B.G.; Bhatia, S.K.; Buatti, J.; Brandt, K.E.; Lindholm, K.E.; Button, A.; Szweda, L.I.; Smith, B.J.; Spitz, D.R.; Fath, M.A. Ketogenic Diets Enhance Oxidative Stress and Radio-Chemo-Therapy Responses in Lung Cancer Xenografts. Clin. Cancer Res. 2013, 19, 3905–3913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdelwahab, M.G.; Fenton, K.E.; Preul, M.C.; Rho, J.M.; Lynch, A.; Stafford, P.; Scheck, A.C. The Ketogenic Diet Is an Effective Adjuvant to Radiation Therapy for the Treatment of Malignant Glioma. PLoS ONE 2012, 7, e36197. [Google Scholar] [CrossRef] [PubMed]
- Fokidis, H.B.; Chin, M.Y.; Ho, V.W.; Adomat, H.H.; Soma, K.K.; Fazli, L.; Nip, K.M.; Cox, M.; Krystal, G.; Zoubeidi, A.; et al. A low carbohydrate, high protein diet suppresses intratumoral androgen synthesis and slows castration-resistant prostate tumor growth in mice. J. Steroid Biochem. Mol. Biol. 2015, 150, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Ho, V.W.; Hamilton, M.J.; Dang, N.-H.T.; Hsu, B.E.; Adomat, H.H.; Guns, E.S.; Weljie, A.; Samudio, I.; Bennewith, K.L.; Krystal, G. A low carbohydrate, high protein diet combined with celecoxib markedly reduces metastasis. Carcinogenesis 2014, 35, 2291–2299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martuscello, R.T.; Vedam-Mai, V.; McCarthy, D.J.; Schmoll, M.E.; Jundi, M.A.; Louviere, C.D.; Griffith, B.G.; Skinner, C.L.; Suslov, O.; Deleyrolle, L.P.; et al. A Supplemented High-Fat Low-Carbohydrate Diet for the Treatment of Glioblastoma. Clin. Cancer Res. 2016, 22, 2482–2495. [Google Scholar] [CrossRef] [Green Version]
- Caffa, I.; Spagnolo, V.; Vernieri, C.; Valdemarin, F.; Becherini, P.; Wei, M.; Brandhorst, S.; Zucal, C.; Driehuis, E.; Ferrando, L.; et al. Fasting-mimicking diet and hormone therapy induce breast cancer regression. Nature 2020, 583, 620–624. [Google Scholar] [CrossRef]
- Klement, R.J.; Champ, C.E.; Otto, C.; Kämmerer, U. Anti-Tumor Effects of Ketogenic Diets in Mice: A Meta-Analysis. PLoS ONE 2016, 11, e0155050. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; TeSlaa, T.; Ng, S.; Nofal, M.; Wang, L.; Lan, T.; Zeng, X.; Cowan, A.; McBride, M.; Lu, W.; et al. Ketogenic diet and chemotherapy combine to disrupt pancreatic cancer metabolism and growth. Med 2022, 3, 119–136.e8. [Google Scholar] [CrossRef] [PubMed]
- Di Biase, S.; Shim, H.S.; Kim, K.H.; Vinciguerra, M.; Rappa, F.; Wei, M.; Brandhorst, S.; Cappello, F.; Mirzaei, H.; Lee, C.; et al. Fasting regulates EGR1 and protects from glucose-and dexamethasone-dependent sensitization to chemotherapy. PLoS Biol. 2017, 15, e2001951. [Google Scholar] [CrossRef] [Green Version]
- Salvadori, G.; Zanardi, F.; Iannelli, F.; Lobefaro, R.; Vernieri, C.; Longo, V.D. Fasting-mimicking diet blocks triple-negative breast cancer and cancer stem cell escape. Cell Metab. 2021, 33, 2247–2259.e6. [Google Scholar] [CrossRef] [PubMed]
- Elgendy, M.; Cirò, M.; Hosseini, A.; Weiszmann, J.; Mazzarella, L.; Ferrari, E.; Cazzoli, R.; Curigliano, G.; DeCensi, A.; Bonanni, B.; et al. Combination of Hypoglycemia and Metformin Impairs Tumor Metabolic Plasticity and Growth by Modulating the PP2A-GSK3β-MCL-1 Axis. Cancer Cell 2019, 35, 798–815.e5. [Google Scholar] [CrossRef] [PubMed]
- Puchalska, P.; Crawford, P.A. Multi-dimensional Roles of Ketone Bodies in Fuel Metabolism, Signaling, and Therapeutics. Cell Metab. 2017, 25, 262–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laffel, L. Ketone bodies: A review of physiology, pathophysiology and application of monitoring to diabetes. Diabetes/Metab. Res. Rev. 1999, 15, 412–426. [Google Scholar] [CrossRef]
- Newman, J.C.; Verdin, E. β-Hydroxybutyrate: A Signaling Metabolite. Annu. Rev. Nutr. 2017, 37, 51–76. [Google Scholar] [CrossRef]
- Dhillon, K.K.; Gupta, S. Biochemistry, Ketogenesis. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK493179/ (accessed on 10 February 2022).
- Kim, D.Y.; Rho, J.M. The ketogenic diet and epilepsy. Curr. Opin. Clin. Nutr. Metab. Care 2008, 11, 113–120. [Google Scholar] [CrossRef]
- Duan, Y.; Zhang, Y.; Dong, H.; Wang, Y.; Zhang, J. Effects of dietary poly-beta-hydroxybutyrate (PHB) on microbiota composition and the mTOR signaling pathway in the intestines of litopenaeus vannamei. J. Microbiol. 2017, 55, 946–954. [Google Scholar] [CrossRef]
- Huang, C.; Wang, P.; Xu, X.; Zhang, Y.; Gong, Y.; Hu, W.; Gao, M.; Wu, Y.; Ling, Y.; Zhao, X.; et al. The ketone body metabolite beta-hydroxybutyrate induces an antidepression-associated ramification of microglia via HDACs inhibition-triggered Akt-small RhoGTPase activation. Glia 2018, 66, 256–278. [Google Scholar] [CrossRef]
- Vernieri, C.; Fucà, G.; Ligorio, F.; Huber, V.; Vingiani, A.; Iannelli, F.; Raimondi, A.; Rinchai, D.; Frigè, G.; Belfiore, A.; et al. Fasting-Mimicking Diet Is Safe and Reshapes Metabolism and Antitumor Immunity in Patients with Cancer. Cancer Discov. 2022, 12, 90–107. [Google Scholar] [CrossRef]
- Han, Y.-M.; Ramprasath, T.; Zou, M.-H. β-hydroxybutyrate and its metabolic effects on age-associated pathology. Exp. Mol. Med. 2020, 52, 548–555. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.H.; Park, M.H.; Ha, S.; Bang, E.J.; Lee, Y.; Lee, A.K.; Lee, J.; Yu, B.P.; Chung, H.Y. Anti-inflammatory action of beta-hydroxybutyrate via modulation of PGC-1alpha and FoxO1, mimicking calorie restriction. Aging 2019, 11, 1283–1304. [Google Scholar] [CrossRef]
- Shimazu, T.; Hirschey, M.D.; Newman, J.; He, W.; Shirakawa, K.; Le Moan, N.; Grueter, C.A.; Lim, H.; Saunders, L.R.; Stevens, R.D.; et al. Suppression of oxidative stress by beta-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science 2013, 339, 211–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pant, K.; Peixoto, E.; Richard, S.; Gradilone, S.A. Role of Histone Deacetylases in Carcinogenesis: Potential Role in Cholangiocarcinoma. Cells 2020, 9, 780. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Outschoorn, U.E.; Prisco, M.; Ertel, A.; Tsirigos, A.; Lin, Z.; Pavlides, S.; Wang, C.; Flomenberg, N.; Knudsen, E.S.; Howell, A. Ketones and lactate increase cancer cell “stemness,” driving recurrence, metastasis and poor clinical outcome in breast cancer: Achieving personalized medicine via Metabolo-Genomics. Cell Cycle 2011, 10, 1271–1286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonuccelli, G.; Tsirigos, A.; Whitaker-Menezes, D.; Pavlides, S.; Pestell, R.G.; Chiavarina, B.; Frank, P.G.; Flomenberg, N.; Howell, A.; Martinez-Outschoorn, U.E.; et al. Ketones and lactate “fuel” tumor growth and metastasis: Evidence that epithelial cancer cells use oxidative mitochondrial metabolism. Cell Cycle 2010, 9, 3506–3514. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Chang, P.; Kuo, W.; Chen, C.; Jeng, Y.; Chang, K.; Shew, J.; Hu, C.; Lee, W. Adipocytes promote malignant growth of breast tumours with monocarboxylate transporter 2 expression via beta-hydroxybutyrate. Nat. Commun. 2017, 8, 14706. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, L.M.; Uribe-Lewis, S.; Madhu, B.; Honess, D.J.; Stubbs, M.; Griffiths, J.R. The action of β-hydroxybutyrate on the growth, metabolism and global histone H3 acetylation of spontaneous mouse mammary tumours: Evidence of a β-hydroxybutyrate paradox. Cancer Metab. 2017, 5, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Shukla, S.K.; Gebregiworgis, T.; Purohit, V.; Chaika, N.V.; Gunda, V.; Radhakrishnan, P.; Mehla, K.; Pipinos, I.I.; Powers, R.; Yu, F.; et al. Metabolic reprogramming induced by ketone bodies diminishes pancreatic cancer cachexia. Cancer Metab. 2014, 2, 18. [Google Scholar] [CrossRef] [Green Version]
- Mikami, D.; Kobayashi, M.; Uwada, J.; Yazawa, T.; Kamiyama, K.; Nishimori, K.; Nishikawa, Y.; Nishikawa, S.; Yokoi, S.; Taniguchi, T.; et al. β-Hydroxybutyrate enhances the cytotoxic effect of cisplatin via the inhibition of HDAC/survivin axis in human hepatocellular carcinoma cells. J. Pharmacol. Sci. 2020, 142, 1–8. [Google Scholar] [CrossRef]
- Schmidt, M.; Pfetzer, N.; Schwab, M.; Strauss, I.; Kämmerer, U. Effects of a ketogenic diet on the quality of life in 16 patients with advanced cancer: A pilot trial. Nutr. Metab. 2011, 8, 54. [Google Scholar] [CrossRef] [Green Version]
- Bartmann, C.; Raman, S.R.J.; Flöter, J.; Schulze, A.; Bahlke, K.; Willingstorfer, J.; Strunz, M.; Wöckel, A.; Klement, R.J.; Kapp, M.; et al. Beta-hydroxybutyrate (3-OHB) can influence the energetic phenotype of breast cancer cells, but does not impact their proliferation and the response to chemotherapy or radiation. Cancer Metab. 2018, 6, 8. [Google Scholar] [CrossRef] [PubMed]
- Donohoe, D.R.; Collins, L.B.; Wali, A.; Bigler, R.; Sun, W.; Bultman, S.J. The Warburg Effect Dictates the Mechanism of Butyrate-Mediated Histone Acetylation and Cell Proliferation. Mol. Cell 2012, 48, 612–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lupton, J.R. Microbial Degradation Products Influence Colon Cancer Risk: The Butyrate Controversy. J. Nutr. 2004, 134, 479–482. [Google Scholar] [CrossRef] [Green Version]
- Luo, S.; Li, Z.; Mao, L.; Chen, S.; Sun, S. Sodium butyrate induces autophagy in colorectal cancer cells through LKB1/AMPK signaling. J. Physiol. Biochem. 2019, 75, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Salimi, V.; Shahsavari, Z.; Safizadeh, B.; Hosseini, A.; Khademian, N.; Tavakoli-Yaraki, M. Sodium butyrate promotes apoptosis in breast cancer cells through reactive oxygen species (ROS) formation and mitochondrial impairment. Lipids Health Dis. 2017, 16, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vernieri, C.; Ligorio, F.; Zattarin, E.; Rivoltini, L.; De Braud, F. Fasting-mimicking diet plus chemotherapy in breast cancer treatment. Nat. Commun. 2020, 11, 1–4. [Google Scholar] [CrossRef]
- De Groot, S.; Lugtenberg, R.T.; Cohen, D.; Welters, M.J.P.; Ehsan, I.; Vreeswijk, M.P.G.; Smit, V.T.H.B.M.; de Graaf, H.; Heijns, J.B.; Portielje, J.E.A.; et al. Fasting mimicking diet as an adjunct to neoadjuvant chemotherapy for breast cancer in the multicentre randomized phase 2 DIRECT trial. Nat. Commun. 2020, 11, 1–9. [Google Scholar] [CrossRef]
- Di Biase, S.; Lee, C.; Brandhorst, S.; Manes, B.; Buono, R.; Cheng, C.-W.; Cacciottolo, M.; Martin-Montalvo, A.; De Cabo, R.; Wei, M.; et al. Fasting-Mimicking Diet Reduces HO-1 to Promote T Cell-Mediated Tumor Cytotoxicity. Cancer Cell 2016, 30, 136–146. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zuo, Q.; Park, N.H.; Lee, J.K.; Madak Erdogan, Z. Liver Metastatic Breast Cancer: Epidemiology, Dietary Interventions, and Related Metabolism. Nutrients 2022, 14, 2376. https://doi.org/10.3390/nu14122376
Zuo Q, Park NH, Lee JK, Madak Erdogan Z. Liver Metastatic Breast Cancer: Epidemiology, Dietary Interventions, and Related Metabolism. Nutrients. 2022; 14(12):2376. https://doi.org/10.3390/nu14122376
Chicago/Turabian StyleZuo, Qianying, Nicole Hwajin Park, Jenna Kathryn Lee, and Zeynep Madak Erdogan. 2022. "Liver Metastatic Breast Cancer: Epidemiology, Dietary Interventions, and Related Metabolism" Nutrients 14, no. 12: 2376. https://doi.org/10.3390/nu14122376
APA StyleZuo, Q., Park, N. H., Lee, J. K., & Madak Erdogan, Z. (2022). Liver Metastatic Breast Cancer: Epidemiology, Dietary Interventions, and Related Metabolism. Nutrients, 14(12), 2376. https://doi.org/10.3390/nu14122376