
Obesity Subtyping: The Etiology, Prevention, and Management of Acquired versus Inherited Obese Phenotypes

{kind=link}
Abstract
:1. Introduction
2. Etiologic Subtypes of Obesity
3. Nongenetic versus Genetic Inheritance and the Role of Genes in Obesity
4. Acquired Obesity: Its Etiology and Response to Intervention
5. The Prevention and Management of the Acquired Obese Phenotype
6. Inherited Obesity: Its Etiology and Response to Intervention
7. The Prevention and Management of the Inherited Obese Phenotype
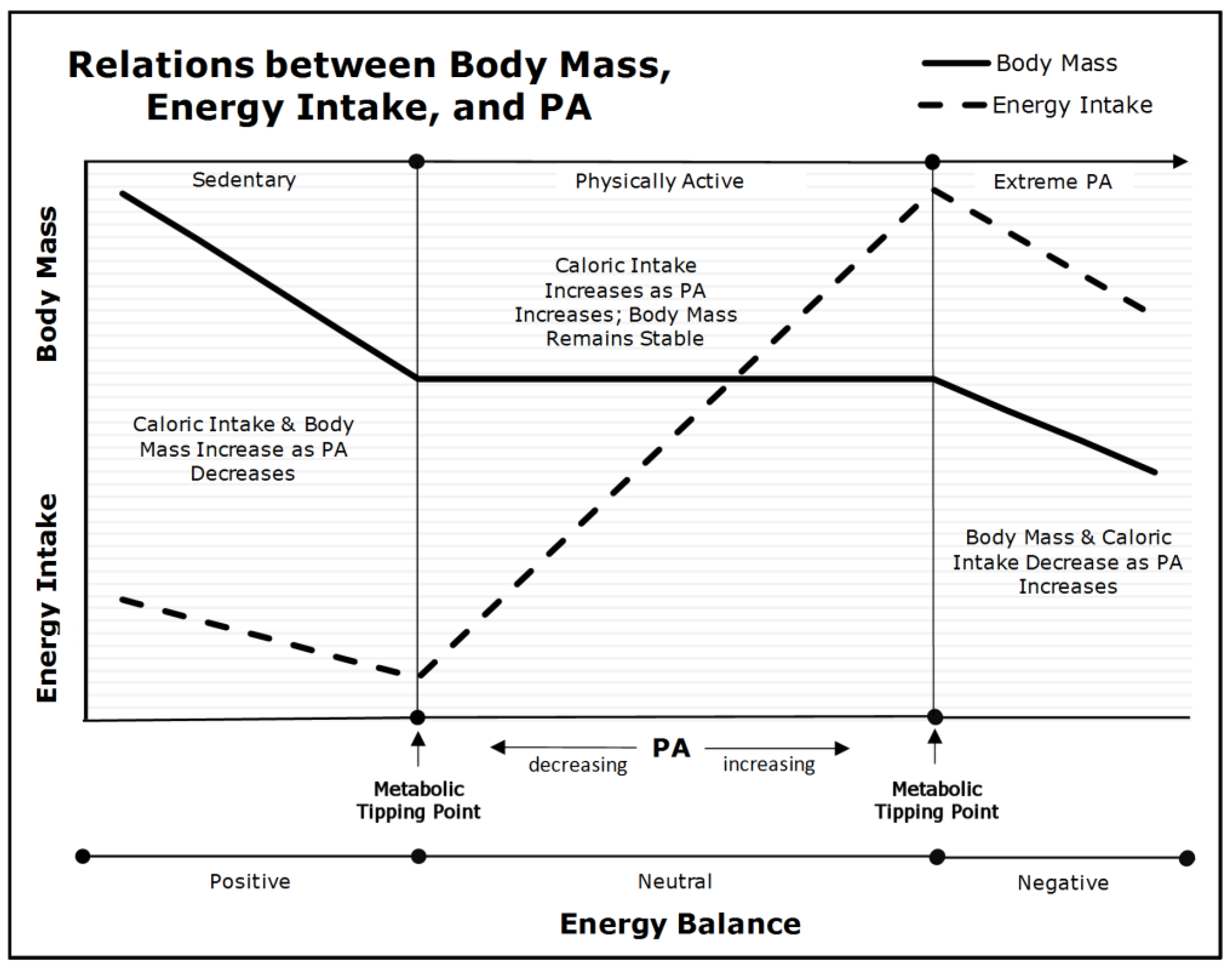
8. The ‘Metabolic Tipping-Point’ and Its Effect on Intervention
9. Assumptions and Limitations
10. Summary and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Archer, E.; Lavie, C.J.; Hill, J.O. The Contributions of ‘Diet’, ‘Genes’, and Physical Activity to the Etiology of Obesity: Contrary Evidence and Consilience. Prog. Cardiovasc. Dis. 2018, 61, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, M.W.; Randy, J.; Seeley, R.J.; Zeltser, L.M.; Drewnowski, A.; Ravussin, E.; Redman, L.M.; Leibel, R.L. Obesity Pathogenesis: An Endocrine Society Scientific Statement. Endocr. Rev. 2017, 38, 267–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Archer, E. The Childhood Obesity Epidemic as a Result of Nongenetic Evolution: The Maternal Resources Hypothesis. Mayo Clin. Proc. 2015, 90, 77–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heymsfield, S.B.; Wadden, T.A. Mechanisms, Pathophysiology, and Management of Obesity. N. Engl. J. Med. 2017, 376, 254–266. [Google Scholar] [CrossRef] [PubMed]
- Blair, S.N.; Sallis, R.E.; Hutber, A.; Archer, E. Exercise therapy—The public health message. Scand. J. Med. Sci. Sports 2012, 22, e24–e28. [Google Scholar] [CrossRef]
- Fryar, C.D.; Carroll, M.D.; Ogden, C.L. Prevalence of Overweight, Obesity, and Severe Obesity among Children and Adolescents Aged 2–19 Years: United States, 1963–1965 through 2015–2016; Centers for Disease Control and Prevention, 2018. Available online: https://www.cdc.gov/nchs/data/hestat/obesity_adult_15_16/obesity_adult_15_16.htm (accessed on 28 May 2022).
- Lavie, C.J.; Parto, P.; Archer, E. Obesity, fitness, hypertension, and prognosis: Is physical activity the common denominator? JAMA Intern. Med. 2016, 176, 217–218. [Google Scholar] [CrossRef]
- Lavie, C.J.; De Schutter, A.; Parto, P.; Jahangir, E.; Kokkinos, P.; Ortega, F.B.; Arena, R.; Milani, R.V. Obesity and Prevalence of Cardiovascular Diseases and Prognosis-The Obesity Paradox Updated. Prog. Cardiovasc. Dis. 2016, 58, 537–547. [Google Scholar] [CrossRef] [Green Version]
- Gadde, K.M.; Martin, C.K.; Berthoud, H.-R.; Heymsfield, S.B. Obesity. J. Am. Coll. Cardiol. 2018, 71, 69–84. [Google Scholar] [CrossRef]
- Field, A.E.; Camargo, C.A.; Ogino, S. The merits of subtyping obesity: One size does not fit all. JAMA 2013, 310, 2147–2148. [Google Scholar] [CrossRef]
- Archer, E. The Mother of All Problems. New Sci. 2015, 225, 32–33. [Google Scholar] [CrossRef]
- Archer, E.; Pavela, G.; McDonald, S.; Lavie, C.J.; Hill, J.O. Cell-Specific “Competition for Calories” Drives Asymmetric Nutrient-Energy Partitioning, Obesity, and Metabolic Diseases in Human and Non-human Animals. Front. Physiol. 2018, 9, 1053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Archer, E.; McDonald, S.M. The Maternal Resources Hypothesis and Childhood Obesity. In Fetal and Early Postnatal Programming and Its Influence on Adult Health; Patel, M.S., Nielsen, J.S., Eds.; CRC Press: Boca Raton, FL, USA; Taylor and Francis Group: New York, NY, USA, 2017; pp. 17–32. [Google Scholar]
- Archer, E. In reply-Maternal, paternal, and societal efforts are needed to “cure” childhood obesity. Mayo Clin. Proc. 2015, 90, 555–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Archer, E. In reply—epigenetics and childhood obesity. Mayo Clin. Proc. 2015, 90, 693–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Archer, E.; Hill, J.O. Body and Fat Mass are not Regulated, Controlled, or Defended: An Introduction to the ‘Invisible Hand’ and ‘Competition Model of Metabolism’. Prog. Cardiovasc. Dis. 2022, in press. [Google Scholar]
- Bonduriansky, R.; Day, T. Nongenetic Inheritance and Its Evolutionary Implications. Annu. Rev. Ecol. Evol. Syst. 2009, 40, 103–125. [Google Scholar] [CrossRef] [Green Version]
- Bonduriansky, R.; Day, T. Extended Heredity: A New Understanding of Inheritance and Evolution; Princeton University Press: Princeton, NJ, USA, 2018. [Google Scholar]
- Day, T.; Bonduriansky, R. A unified approach to the evolutionary consequences of genetic and nongenetic inheritance. Am. Nat. 2011, 178, E18–E36. [Google Scholar] [CrossRef]
- Feldman, M.W.; Lewontin, R.C. The heritability hang-up. Science 1975, 190, 1163–1168. [Google Scholar] [CrossRef]
- Lewontin, R.C. The analysis of variance and the analysis of causes. Am. J. Hum. Genet. 1974, 26, 400–411. [Google Scholar] [CrossRef] [Green Version]
- Weinsier, R.L.; Hunter, G.R.; Heini, A.F.; Goran, M.I.; Sell, S.M. The etiology of obesity: Relative contribution of metabolic factors, diet, and physical activity. Am. J. Med. 1998, 105, 145–150. [Google Scholar] [CrossRef]
- Jebb, S.A.; Moore, M.S. Contribution of a sedentary lifestyle and inactivity to the etiology of overweight and obesity: Current evidence and research issues. Med. Sci. Sports Exerc. 1999, 31, S534–S541. [Google Scholar] [CrossRef]
- LaMonte, M.J.; Blair, S.N.; Church, T.S. Physical activity and diabetes prevention. J. Appl. Physiol. 2005, 99, 1205–1213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blair, S.N.; Church, T.S. The fitness, obesity, and health equation: Is physical activity the common denominator? JAMA 2004, 292, 1232–1234. [Google Scholar] [CrossRef] [PubMed]
- Blair, S.N.; Archer, E.; Hand, G.A. Commentary: Luke and Cooper are wrong: Physical activity has a crucial role in weight management and determinants of obesity. Int. J. Epidemiol. 2013, 42, 1836–1838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayer, J.; Marshall, N.B.; Vitale, J.J.; Christensen, J.H.; Mashayekhi, M.B.; Stare, F.J. Exercise, food intake and body weight in normal rats and genetically obese adult mice. Am. J. Physiol. 1954, 177, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Mayer, J. Decreased activity and energy balance in the hereditary obesity-diabetes syndrome of mice. Science 1953, 117, 504–505. [Google Scholar] [CrossRef]
- Chirico, A.M.; Stunkard, A.J. Physical activity and human obesity. N. Engl. J. Med. 1960, 263, 935–940. [Google Scholar] [CrossRef]
- Archer, E.; Blair, S.N. Physical activity and the prevention of cardiovascular disease: From evolution to epidemiology. Prog. Cardiovasc. Dis. 2011, 53, 387–396. [Google Scholar] [CrossRef]
- Melzer, K.; Kayser, B.; Saris, W.H.; Pichard, C. Effects of physical activity on food intake. Clin. Nutr. 2005, 24, 885–895. [Google Scholar] [CrossRef] [Green Version]
- Lavie, C.J.; Ozemek, C.; Carbone, S.; Katzmarzyk, P.T.; Blair, S.N. Sedentary behavior, exercise, and cardiovascular health. Circ. Res. 2019, 124, 799–815. [Google Scholar] [CrossRef]
- Archer, E.; Artero, E.G.; Blair, S.N. Sedentary Behavior and Cardiovascular Disease. In Sedentary Behavior and Health: Concepts, Assessment & Intervention—Human Kinetics; Zhu, W., Owen, N., Eds.; Human Kinetics: Champaign, IL, USA, 2017; pp. 203–225. [Google Scholar]
- Stubbs, R.J.; Hughes, D.A.; Johnstone, A.M.; Horgan, G.W.; King, N.; Blundell, J.E. A decrease in physical activity affects appetite, energy, and nutrient balance in lean men feeding ad libitum. Am. J. Clin. Nutr. 2004, 79, 62–69. [Google Scholar] [CrossRef]
- Bosy-Westphal, A.; Hägele, F.A.; Müller, M.J. Impact of Energy Turnover on the Regulation of Energy and Macronutrient Balance. Obesity 2021, 29, 1114–1119. [Google Scholar] [CrossRef] [PubMed]
- Shook, R.P.; Hand, G.A.; Drenowatz, C.; Hebert, J.R.; Paluch, A.E.; Blundell, J.E.; Hill, J.O.; Katzmarzyk, P.T.; Church, T.S.; Blair, S.N. Low levels of physical activity are associated with dysregulation of energy intake and fat mass gain over 1 year. Am. J. Clin. Nutr. 2015, 102, 1332–1338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westerterp, K.R.; Meijer, G.A.; Janssen, E.M.; Saris, W.H.; Ten Hoor, F. Long-term effect of physical activity on energy balance and body composition. Br. J. Nutr. 1992, 68, 21–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeFronzo, R.A.; Tripathy, D. Skeletal Muscle Insulin Resistance Is the Primary Defect in Type 2 Diabetes. Diabetes Care 2009, 32, S157–S163. [Google Scholar] [CrossRef] [Green Version]
- DeFronzo, R.A. Lilly lecture 1987. The triumvirate: Beta-cell, muscle, liver. A collusion responsible for NIDDM. Diabetes 1988, 37, 667–687. [Google Scholar] [CrossRef] [PubMed]
- DeFronzo, R.A.; Bonadonna, R.C.; Ferrannini, E. Pathogenesis of NIDDM. A balanced overview. Diabetes Care 1992, 15, 318–368. [Google Scholar] [CrossRef]
- Taylor, R. Pathogenesis of type 2 diabetes: Tracing the reverse route from cure to cause. Diabetologia 2008, 51, 1781–1789. [Google Scholar] [CrossRef] [Green Version]
- Saris, W.; Blair, S.; Van Baak, M.; Eaton, S.; Davies, P.; Di Pietro, L.; Fogelholm, M.; Rissanen, A.; Schoeller, D.; Swinburn, B. How much physical activity is enough to prevent unhealthy weight gain? Outcome of the IASO 1st Stock Conference and consensus statement. Obes. Rev. 2003, 4, 101–114. [Google Scholar] [CrossRef]
- Piercy, K.L.; Troiano, R.P.; Ballard, R.M.; Carlson, S.A.; Fulton, J.E.; Galuska, D.A.; George, S.M.; Olson, R.D. The physical activity guidelines for Americans. JAMA 2018, 320, 2020–2028. [Google Scholar] [CrossRef]
- Boulé, N.G.; Haddad, E.; Kenny, G.P.; Wells, G.A.; Sigal, R.J. Effects of Exercise on Glycemic Control and Body Mass in Type 2 Diabetes MellitusA Meta-analysis of Controlled Clinical Trials. JAMA 2001, 286, 1218–1227. [Google Scholar] [CrossRef]
- Umpierre, D.; Ribeiro, P.A.B.; Kramer, C.K.; Leitão, C.B.; Zucatti, A.T.N.; Azevedo, M.J.; Gross, J.L.; Ribeiro, J.P.; Schaan, B.D. Physical Activity Advice Only or Structured Exercise Training and Association with HbA1c Levels in Type 2 Diabetes: A Systematic Review and Meta-analysis. JAMA 2011, 305, 1790–1799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavie, C.J. Sugar Wars-Commentary from the Editor. Prog. Cardiovasc. Dis. 2018, 61, 382–383. [Google Scholar] [CrossRef] [PubMed]
- Lavie, C.J.; Laddu, D.; Arena, R.; Ortega, F.B.; Alpert, M.A.; Kushner, R.F. Healthy weight and obesity prevention: JACC health promotion series. J. Am. Coll. Cardiol. 2018, 72, 1506–1531. [Google Scholar] [CrossRef] [PubMed]
- Lavie, C.J.; Milani, R.V.; O’Keefe, J.H. Dyslipidemia intervention in metabolic syndrome: Emphasis on improving lipids and clinical event reduction. Am. J. Med. Sci. 2011, 341, 388–393. [Google Scholar] [CrossRef]
- Hales, C.N.; Barker, D.J.; Clark, P.M.; Cox, L.J.; Fall, C.; Osmond, C.; Winter, P.D. Fetal and infant growth and impaired glucose tolerance at age 64. BMJ 1991, 303, 1019–1022. [Google Scholar] [CrossRef] [Green Version]
- Barker, D.J.P.; Osmond, C. Infant Mortality, Childhood Nutrition, And Ischaemic Heart Disease in England And Wales. Lancet 1986, 327, 1077–1081. [Google Scholar] [CrossRef]
- Hales, C.N.; Barker, D.J. Type 2 (non-insulin-dependent) diabetes mellitus: The thrifty phenotype hypothesis. Diabetologia 1992, 35, 595–601. [Google Scholar] [CrossRef]
- Archer, E.; Lavie, C.J.; McDonald, S.M.; Thomas, D.M.; Hebert, J.R.; Taverno Ross, S.E.; McIver, K.L.; Malina, R.M.; Blair, S.N. Maternal inactivity: 45-year trends in mothers’ use of time. Mayo Clin. Proc. 2013, 88, 1368–1377. [Google Scholar] [CrossRef] [Green Version]
- Archer, E.; Shook, R.P.; Thomas, D.M.; Church, T.S.; Katzmarzyk, P.T.; Hebert, J.R.; McIver, K.L.; Hand, G.A.; Lavie, C.J.; Blair, S.N. 45-Year trends in women’s use of time and household management energy expenditure. PLoS ONE 2013, 8, e56620. [Google Scholar] [CrossRef] [Green Version]
- Church, T.S.; Thomas, D.M.; Tudor-Locke, C.; Katzmarzyk, P.T.; Earnest, C.P.; Rodarte, R.Q.; Martin, C.K.; Blair, S.N.; Bouchard, C. Trends over 5 decades in U.S. occupation-related physical activity and their associations with obesity. PLoS ONE 2011, 6, e19657. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, A.; Lavie, C.J.; Blair, S.N. Fitness or fatness: Which is more important? JAMA 2018, 319, 231–232. [Google Scholar] [CrossRef]
- Ortega, F.B.; Ruiz, J.R.; Labayen, I.; Lavie, C.J.; Blair, S.N. The Fat but Fit paradox: What we know and don’t know about it. Br. J. Sports Med. 2018, 52, 151–153. [Google Scholar] [CrossRef] [PubMed]
- Ortega, F.B.; Lavie, C.J.; Blair, S.N. Obesity and cardiovascular disease. Circ. Res. 2016, 118, 1752–1770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elagizi, A.; Kachur, S.; Lavie, C.J.; Carbone, S.; Pandey, A.; Ortega, F.B.; Milani, R.V. An overview and update on obesity and the obesity paradox in cardiovascular diseases. Prog. Cardiovasc. Dis. 2018, 61, 142–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myers, J.; McAuley, P.; Lavie, C.J.; Despres, J.-P.; Arena, R.; Kokkinos, P. Physical activity and cardiorespiratory fitness as major markers of cardiovascular risk: Their independent and interwoven importance to health status. Prog. Cardiovasc. Dis. 2015, 57, 306–314. [Google Scholar] [CrossRef]
- Blundell, J.E.; Caudwell, P.; Gibbons, C.; Hopkins, M.; Naslund, E.; King, N.; Finlayson, G. Role of resting metabolic rate and energy expenditure in hunger and appetite control: A new formulation. Dis. Models Mech. 2012, 5, 608–613. [Google Scholar] [CrossRef] [Green Version]
- Blundell, J.E.; Caudwell, P.; Gibbons, C.; Hopkins, M.; Naslund, E.; King, N.A.; Finlayson, G. Body composition and appetite: Fat-free mass (but not fat mass or BMI) is positively associated with self-determined meal size and daily energy intake in humans. Br. J. Nutr. 2012, 107, 445–449. [Google Scholar] [CrossRef] [Green Version]
- Caudwell, P.; Finlayson, G.; Gibbons, C.; Hopkins, M.; King, N.; Naslund, E.; Blundell, J.E. Resting metabolic rate is associated with hunger, self-determined meal size, and daily energy intake and may represent a marker for appetite. Am. J. Clin. Nutr. 2013, 97, 7–14. [Google Scholar] [CrossRef]
- Blundell, J.E.; Finlayson, G.; Gibbons, C.; Caudwell, P.; Hopkins, M. The biology of appetite control: Do resting metabolic rate and fat-free mass drive energy intake? Physiol. Behav. 2015, 152, 473–478. [Google Scholar] [CrossRef]
- Blundell, J.E.; Gibbons, C.; Beaulieu, K.; Casanova, N.; Duarte, C.; Finlayson, G.; Stubbs, R.J.; Hopkins, M. The drive to eat in homo sapiens: Energy expenditure drives energy intake. Physiol. Behav. 2020, 219, 112846. [Google Scholar] [CrossRef]
- Mayer, J.; Vitale, J.; Bates, M.W. Mechanism of the regulation of food intake. Nature 1951, 167, 562–563. [Google Scholar] [CrossRef] [PubMed]
- Beaudoin, R.; Mayer, J. Food intakes of obese and non-obese women. J. Am. Diet. Assoc. 1953, 29, 29–33. [Google Scholar] [CrossRef]
- Mayer, J.; Roy, P.; Mitra, K.P. Relation between Caloric Intake, Body Weight, and Physical Work: Studies in an Industrial Male Population in West Bengal. Am. J. Clin. Nutr. 1956, 4, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Mayer, J. Correlation between metabolism and feeding behavior and multiple etiology of obesity. Bull. N. Y. Acad. Med. 1957, 33, 744–761. [Google Scholar] [PubMed]
- Archer, E. In Defense of Sugar: A Critique of Diet-Centrism. Prog. Cardiovasc. Dis. 2018, 61, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.C.; Lavie, C.J.; Vedanthan, R. Optimal dose of running for longevity: Is more better or worse? J. Am. Coll. Cardiol. 2015, 65, 420–422. [Google Scholar] [CrossRef] [Green Version]
- Friedman, J.M. Modern science versus the stigma of obesity. Nat. Med. 2004, 10, 563–569. [Google Scholar] [CrossRef]
- Hartlev, M. Stigmatisation as a Public Health Tool against Obesity—A Health and Human Rights Perspective. Eur. J. Health Law 2014, 21, 365–386. [Google Scholar] [CrossRef] [Green Version]
- Archer, E.; Lavie, C.J.; Hill, J.O. The Failure to Measure Dietary Intake Engendered a Fictional Discourse on Diet-Disease Relations. Front. Nutr. 2018, 5, 105. [Google Scholar] [CrossRef] [Green Version]
- Archer, E. The Demonization of ‘Diet’ Is Nothing New. Prog. Cardiovasc. Dis. 2018, 61, 386–387. [Google Scholar] [CrossRef]
- Archer, E.; Arjmandi, B. Falsehoods and facts about dietary sugars: A call for evidence-based policy. Crit. Rev. Food Sci. Nutr. 2020, 61, 3725–3739. [Google Scholar] [CrossRef] [PubMed]
- Dobersek, U.; Wy, G.; Adkins, J.; Altmeyer, S.; Krout, K.; Lavie, C.J.; Archer, E. Meat and mental health: A systematic review of meat abstention and depression, anxiety, and related phenomena. Crit. Rev. Food Sci. Nutr. 2020, 61, 622–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fletcher, G.F.; Landolfo, C.; Niebauer, J.; Ozemek, C.; Arena, R.; Lavie, C.J. Promoting physical activity and exercise: JACC health promotion series. J. Am. Coll. Cardiol. 2018, 72, 1622–1639. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Archer, E.; Lavie, C.J. Obesity Subtyping: The Etiology, Prevention, and Management of Acquired versus Inherited Obese Phenotypes. Nutrients 2022, 14, 2286. https://doi.org/10.3390/nu14112286
Archer E, Lavie CJ. Obesity Subtyping: The Etiology, Prevention, and Management of Acquired versus Inherited Obese Phenotypes. Nutrients. 2022; 14(11):2286. https://doi.org/10.3390/nu14112286
Chicago/Turabian StyleArcher, Edward, and Carl J. Lavie. 2022. "Obesity Subtyping: The Etiology, Prevention, and Management of Acquired versus Inherited Obese Phenotypes" Nutrients 14, no. 11: 2286. https://doi.org/10.3390/nu14112286
APA StyleArcher, E., & Lavie, C. J. (2022). Obesity Subtyping: The Etiology, Prevention, and Management of Acquired versus Inherited Obese Phenotypes. Nutrients, 14(11), 2286. https://doi.org/10.3390/nu14112286