Abstract
To date, the tumor microenvironment (TME) has gained considerable attention in various areas of cancer research due to its role in driving a loss of immune surveillance and enabling rapid advanced tumor development and progression. The TME plays an integral role in driving advanced aggressive breast cancers, including triple-negative breast cancer (TNBC), a pivotal mediator for tumor cells to communicate with the surrounding cells via lymphatic and circulatory systems. Furthermore, the TME plays a significant role in all steps and stages of carcinogenesis by promoting and stimulating uncontrolled cell proliferation and protecting tumor cells from the immune system. Various cellular components of the TME work together to drive cancer processes, some of which include tumor-associated adipocytes, fibroblasts, macrophages, and neutrophils which sustain perpetual amplification and release of pro-inflammatory molecules such as cytokines. Thymoquinone (TQ), a natural chemical component from black cumin seed, is widely used traditionally and now in clinical trials for the treatment/prevention of multiple types of cancer, showing a potential to mitigate components of TME at various stages by various pathways. In this review, we focus on the role of TME in TNBC cancer progression and the effect of TQ on the TME, emphasizing their anticipated role in the prevention and treatment of TNBC. It was concluded from this review that the multiple components of the TME serve as a critical part of TNBC tumor promotion and stimulation of uncontrolled cell proliferation. Meanwhile, TQ could be a crucial compound in the prevention and progression of TNBC therapy through the modulation of the TME.
1. Introduction
Breast cancer (BC) is one of the most life-threatening diseases with the highest number of human cancer subtypes and is the second leading cause of death in women [1]. In 2020, there were approximately 2.3 million new breast cancer cases and 685,000 breast cancer deaths globally [2], with 1,898,160 new cases and 608,570 deaths projected in the United States by the end of 2021 [3]. Triple-negative breast cancer (TNBC) is characterized by a lack of expressed progesterone receptors, estrogen receptors, and human epidermal growth factor 2 (HER2) [4]. It is one of the major metastatic, drug-resistant, and aggressive sub-classes of BC [5], which comprises around 20% of total BC globally [6], and 83% of deaths compared with other BC subtypes [7]. There are various factors involved in the initiation and progression of TNBC. The most indispensable is the TME, directly linked with angiogenesis, rapid cell proliferation, immune system suppression, apoptosis inhibition, and eventual evasion of immune surveillance in the tumor area [8]. Because of these activities, TME can be noted as a critical hallmark in cancer pathogenicity and a potential target for TNBC [9].
Even though combination chemotherapy with taxanes and anthracyclines is the main treatment of TNBC [10], mortality rates are associated with a heterogeneity of oncogenic drivers, chemoresistance, systemic toxicity, and poor selectivity, establishing challenging limiting factors [11]. Side effects of conventional therapy for BC patients involve impaired fertility, osteoporosis, premature menopause, congestive heart failure, cardiomyopathy, arthralgia, and myalgia [12]. Given that TNBC is one of the most common life-threatening and complex kinds of BC, substantial research has been devoted to discovering novel biomarkers and biologically targeted treatment sites to enhance patient prognosis and clinical outcomes compared to other subtypes [13].
2. Tumor Microenvironment (TME) and TNBC
The tumor microenvironment is crucial for the induction and development of TNBC. The initiation of angiogenesis, proliferation, apoptosis inhibition, immune system suppression, and evasion of immune surveillance is inherently linked to TME [8]. Naturally, TME is heterogeneous and highly complex [14], enabling cancer cells’ rapid proliferation, subsequent development of hypoxia, and concomitant reprogramming of cancer cells in the TME to acclimatize to changes within TME. The TME generates a dwelling place for interacting cancer cells with their neighboring immune and endothelial cells and fibroblasts and can provide potential targets for TNBC [9].
The reciprocal communication between stromal cells and cancer cells produces changes in the cellular elements of TME, which predisposes tumor cells to metastasis [15]. Cancer cells generally gain a supportive microenvironment by activating the wound healing response of the host [16]. Morphologically, the TME resembles a wound healing site [17], composed of different histological changes, including inflammatory cell infiltration, extravascular clotting, accumulation of activated fibroblast, angiogenesis, and synthesis of extracellular matrix [18]. Likewise, the stromal cells, such as tumor-associated macrophages or cancer-associated fibroblasts, promote tumor progression by generating growth factors, chemokines, and promigratory extracellular matrix components [19]. Additionally, TME stimulates the transition of epithelial cells to TNBC stem cells [20]. A mounting number of studies have focused on the TME to discover the complex mechanisms underlying tumorigenesis, elucidate new biomarkers and drug targets that may predict clinical outcomes, and guide therapy in TNBCs [21]. The various components in the microenvironment promote tumorigenesis, protect tumor cells from host immunity, and reduce chemotherapeutic response effectiveness [22] (Figure 1).
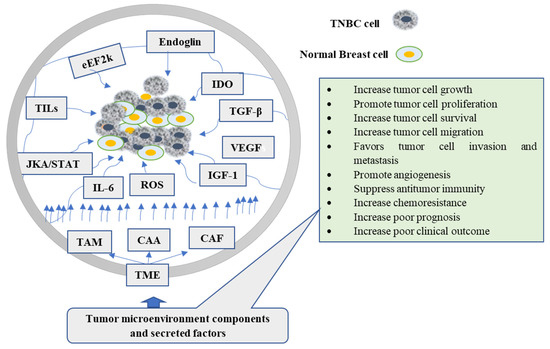
Figure 1.
The role of tumor microenvironment components and their secreted factors in the carcinogenesis of TNBC cells. TAM, CAA, and CAF are the major cellular components of TME, while also shown in the figure are secreted factors and signals. CAA: Cancer-associated adipocytes; CAF: Cancer-associated fibroblast; eEF2k: eukaryotic elongation facto 2k; TAMs: tumor-associated macrophages; TGF-β: transforming growth factor-β; IGF: insulin-like growth factor; ROS: reactive oxygen species; IL-6: Interleukin-6; TNBC: triple-negative breast cancer; VEGF: Vascular endothelial growth factor; IDO: Indoleamine 2,3-dioxygenase.
3. Natural Products Targeting TME
Natural products have been utilized for centuries in traditional medicines, ubiquitously distributed throughout plants and marine organisms [23]. The majority of bioactive natural products are secondary metabolites that have been isolated and experimentally validated for their anti-bacterial, anti-viral, anti-fungal, anti-inflammatory, and anti-cancer properties [24,25]. For many decades there has been growing interest in natural compounds with anti-cancer activities since they are less/nontoxic, inexpensive, and easily accessible by most people, which is preferable for the prevention and treatment of various ailments [26]. Around 45% of all anti-cancer drugs in clinical use originate from natural compound secondary metabolites [27,28]. Natural compounds exhibit a significant effect on the tumor cells via the modulation of TME [29] and research directions have now shifted toward natural compounds and specific TME-modulating properties [30,31,32]. Considering the data emerging from these efforts, it has become advisable that we revisit TQ to understand its broader significance in the modulation of TME and associated mechanisms. The expectation is that the current review would encourage future translational advancements of this compound in clinical use to prevent and treat TNBC.
4. Thymoquinone (TQ)
The Nigella sativa seed, also known as black seed, is commonly used in different areas globally to prevent and treat diseases [33]. It contains various bioactive compounds, such as thymoquinone, p-cymene, α-piene, thymo-hydroquinone, dithymo-quinone, and t-anethol. These compounds possess several pharmacological properties, including anti-neoplastic, anti-inflammatory, antioxidant, anti-asthmatic, analgesic, antipyretic, antihypertensive, and antimicrobial [34,35]. Among these constituents, TQ has shown a promising role against various diseases, including cancer, such as breast cancer, prostate cancer, bone cancer, gastric cancer, bladder cancer, colon cancer, and lung cancer [26,36].
TQ can protect the healthy cells from oxidative damage and provide long recovery for cells by preventing toxic side effects [37]. TQ exhibits antiproliferative effects on different cancer cell lines of the breast, ovary, larynx, colon, myeloblastic leukemia, osteosarcoma, and lung [38,39]. In BC models, TQ can lower VEGF, enhance serum INF-γ levels, suppress angiogenesis, and shift the immune response toward T helper1 [40]. It has also been reported that by targeting cyclin E, cyclin D1, and p27 proteins, TQ caused cell cycle arrest, suppressing progression from G1 to S phase [41]. TQ can also induce apoptosis by activating caspase-8, cleaving poly (ADP-ribose) polymerase, and lowering AKT phosphorylation [42,43]. Moreover, TQ is effective in xenograft tumor models through multiple mechanisms, including inducing p38 phosphorylation and inhibiting protein expression of anti-apoptotic genes, i.e., Bcl-2, Bcl-Xl XIAP, and survivin. These findings are fairly consistent throughout the literature, where TQ enhances TUNEL, lowers Ki67 [44,45], activates caspase-9, induces apoptotic cell death, and interferes with the survival of cells involving the PI3-K/Akt pathway [46,47]. Figure 2 describes the possible anti-cancer mechanisms of TQ.
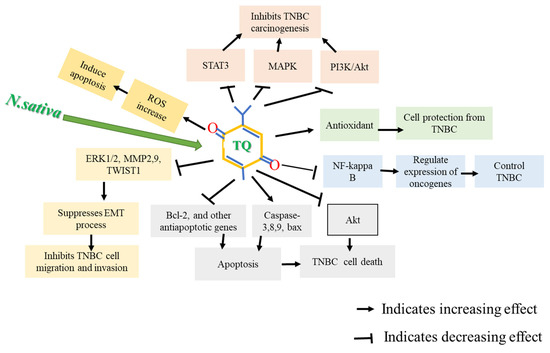
Figure 2.
Possible anti-cancer mechanism of thymoquinone (TQ). TQ caused apoptosis in TNBC cells by inducing apoptotic genes, downregulating anti-apoptotic genes, and generating reactive oxygen species (ROS). TQ also suppresses Akt activation, deactivates Nf-kB, and increases antioxidant enzymes, inhibiting TNBC carcinogenesis.
5. Thymoquinone (TQ) Targeting the Cellular Components of TME
The cellular components and secretary molecules of TME targeted by TQ as discussed here are tumor-associated macrophage(TAM), cancer-associated fibroblast(CAF), cancer-associated adipocytes(CAA), cholesterol synthesis, reactive oxygen species (ROS), eukaryotic elongation factor-2 kinase (eEF-2K), tumor-infiltrating lymphocytes (TILs), indoleamine 2,3-dioxygenase (IDO), vascular endothelial growth factor (VEGF), transforming growth factor-β (TGF-β), insulin-like growth factors (IGF-I), endoglin, IL-6, and JAK/STAT signaling.
5.1. Effect of TQ on Cholesterol Synthesis and Its Metabolites
Cholesterol is a crucial lipid molecule in normal physiological processes, being critical for intracellular signal transduction, an essential component of the plasma membrane, and a precursor for steroid hormone [48]. However, the accumulation of cholesterol is associated with increased cancer cell proliferation in different types of cancer, including TNBC [49]. Because cancer cells proliferate rapidly, they rely heavily on cholesterol to meet their growing demand for substrates for membrane production [50]. As a result, cholesterol is helpful for cancer growth and development because it increases oncogenic signaling, apoptosis evasion, cell motility, and invasion [51,52]. Cancer cells, for example, increase their cholesterol requirement by boosting de novo production or absorption, modifying cholesterol outflow or increasing cholesterol storage [53]. Cholesterol and its metabolites produced by tumor cells have influenced the phenotype and function of cells forming the TME. Increased glucose uptake and de novo steroidogenesis principally satisfy the high nutrient demand of tumor cells. However, increased cholesterol metabolism and de novo lipogenesis are also detrimental for the cells forming the TME, such as immune cells [54].
Several oncogenic signals include steroidogenic acute regulatory protein (STAR), STAR-related lipid transfer domain containing 3 (STARD3), RTK/RAS, PI3K/AKT/mTOR, and TP53, have been shown to modulate cholesterol synthesis in cancer cells. Overexpression of STAR and STARD3 increases cholesterol biosynthesis by increasing the expression of cholesterol synthesis enzymes [55]. These proteins are elevated in cancer cells and resistant to apoptotic signals [56]. While STAR and STARD3 are upregulated in BC cells, including TNBC, genetic knockdown of these genes increases sensitivity to chemotherapeutic agents, reduced cell proliferation, and increases apoptosis [55,56,57]. Constitutive upregulation of PI3K/AKT signaling increases intracellular cholesterol levels by activating sterol responsive element binding protein (SREBP), which enables induction of the LDL (low-density lipoprotein) receptor that mediates cholesterol import. It decreases ABCA1, which mediates cholesterol export in a mTORC1-dependent manner [58]. Evidence indicates that the PI3K/AKT/mTOR pathway contributes to cancer cell growth and progression [59]. Targeting these genes either by inducing p53 or inhibited STAR and STARD3, PI3K/AKT/mTOR, RTK/RAS or targeting the mevalonate–cholesterol biosynthesis pathway to regulate cholesterol synthesis and its metabolites using TQ will be an alternative option in the prevention of TNBC, either alone or in combination with other chemotherapeutic agents.
Moreover, TP53, the most frequently mutated gene in TNBC cells, is another gene deregulating the cholesterol synthesis pathway. Through upregulation of the mevalonate pathway, an essential pathway in the synthesis of cholesterol in cancer cells, tumor cells continuously increase activation of cholesterol synthesis. Any agents that inhibited the mevalonate pathway or downregulated mutant P53 are a potential target for preventing TNBC via modulation of cholesterol synthesis [60].
TNBC cells display distinct adipogenic characteristics in the mevalonate–cholesterol biosynthesis pathway and metabolic drug responses. In a mouse MDAMB-468 xenograft tumor model, co-administration of 2-DG plus gefitinib significantly reduced tumor size. G28, a fatty acid synthase (FASN) inhibitor, had a substantial antiproliferative effect on the TNBC cell line MDA-MB-231) and its doxorubicin (231DXR) and paclitaxel-resistant variants (231PTR) [61]. EGCG, an anti-FASN compound, in combination with cetuximab, showed potent anticancer efficacy in doxorubicin-resistant TNBC cell lines (231DXR and HCCDXR) [62].
The cholesteryl ester-producing enzyme acetyltransferase (ACAT-1) is overexpressed in two TNBC cell lines of human breast cancer, MDA-MB-231, and MDA-MB-436. A faster cell growth rate could be connected to higher enzyme levels. This study was confirmed when TNBC cells were treated with CP-113818, an ACAT-1 inhibitor, which inhibited cell proliferation and migration by modulating cholesterol metabolism [63].
According to in vivo and in vitro studies, phosphatase and tensin homolog (PTEN) loss promotes tumorigenesis and metastatic potential in pancreatic cancer by activating the downstream PI3K/Akt/mTOR/SREBP signaling pathway; higher levels of esterified cholesterol boost carcinogenesis and metastatic potential [64]. Several studies show that TQ induces cell death by inhibiting NF-kβ/Akt activation via upregulation of PTEN. This effect leads to phosphorylation of Akt and induction of p53 protein and its transcriptional target p21, thus evoking G2/M phase arrest and apoptosis in drug-resistant hormone-positive BC cells [46]. Another study looked at the role of TQ in different cells, finding that it regulated several target proteins involved in the PI3K/AKT signaling network. TQ lowered PTEN phosphorylation at Ser380 (PTEN inactivation) in the MDA-MB-468 TNBC cell line, resulting in PTEN activation [65]. In the MCF-7 breast cancer cell line, TQ promotes apoptosis by upregulating P53 expression in a time-dependent manner [66].
TQ will be an alternative option in the prevention of TNBC, either alone or in combination with other chemotherapeutic agents, by inducing p53 or upregulation of PTEN or inhibiting STAR and STARD3, PI3K/AKT/mTOR, RTK/RAS, or targeting the mevalonate–cholesterol biosynthesis pathway to regulate cholesterol synthesis and its metabolites.
5.2. Effect of TQ on Reactive Oxygen Species (ROS)
Reactive oxygen species (ROS) are vigorously reactive oxygen molecules. They are produced in cells during normal mitochondrial oxidative metabolism and cellular respiration in response to xenobiotics. ROS can oxidatively damage the cellular components such as carbohydrates, lipids, proteins, and nucleic acids and change their function [67]. They are considered a key factor for many pathological conditions, including cancer. ROS contribute to several alterations of physiological function, structure, and mutagenesis in DNA, thus producing cancer [68]. ROS within a cell plays an essential role in controlling cell proliferation, apoptosis, and metabolic pathways [69]. ROS are produced by cancer cells and cellular components in the microenvironment via disturbed metabolism of the cancer cells [70]. They directly change the TME by activating cancer cells and inflammatory cells, which stimulates various inflammatory molecules to promote carcinogenesis [71]. Therefore, they are recognized as the hallmarks of cancer secreted in the tumor-promoting microenvironment [8].
ROS are master modulator of tumorigenesis [72]. Many cancer processes are redox-sensitive. These include proliferation, angiogenesis, cell motility, cell cycle progression, cell survival and apoptosis, cell morphology, energy metabolism, cell-to-cell adhesion, and tumor stemness [73]. Furthermore, it has been broadly described that oncogene activation promotes ROS accumulation in tumor cells [74]. It has been reported that ROS has dual actions, both tumor-suppressive or oncogenic functions, as various studies have shown contradictory results [75]. The functions are dependent on cancer stages, levels of ROS, and other trigger factors in cancer cells [76]. When ROS are high, they increase apoptosis, senescence, cellular damage, trigger microsatellite instability via DNA damage induction [77,78,79,80,81], and suppress tumorigenesis through the p38 pathway. Conversely, low to moderate levels of ROS promote proliferation via activation of various kinase cascades such as mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK)-Ras-Raf-MEK-ERK pathway phosphoinositide-3-kinase/protein kinase B (PI3K/Akt), cyclin D1 expression, and c-Jun N-terminal kinase (JNK) [82]. Moreover, ROS promotes the reversible deactivation of PTEN [83].
Several studies have reported that natural compounds such as catechins exert an antioxidant effect by directly scavenging free radicals through donating one electron of their phenolic group, and via chelating metal ions involved in radical production. Indirectly, catechins enhance the activity of antioxidant enzymes such as catalase, superoxide dismutase and glutathione peroxidase, and can inhibit pro-oxidant enzymes [84]. On the contrary, authors looking at Syringin induce oxidative stress to suppress the growth and proliferation of TNBC cells; however, the antioxidant N-acetylcysteine reversed the ROS levels and prevented apoptosis [85]. Similar studies have reported that ROS activates the antitumor activities of doxorubicin, resulting in the efficient inhibition of distant metastasis and tumor growth in TNBC cells [86].
Meanwhile, TQ has dual functions towards the regulation of ROS. Evidence shows that TQ inhibits proliferation and promotes apoptotic effects through upregulation of ROS in BC cells both in vivo and in vitro models [44,87]. The authors divulged TQ’s positive impact on ROS production and tumor suppression. Downregulation of XIAP(X-linked inhibitor of apoptosis protein), survivin, Bcl-xL, and Bcl-2 protein, as well as accelerating oxidative stress, can cause oxidative damage in mitochondria, triggering apoptosis [88].
On the other hand, TQ acts as antioxidant activity by inducing cytoprotective enzymes resulting in cells’ protection against ROS-induced cellular damage. Studies have reported that TQ upregulates cytoprotective enzymes, including superoxide dismutase, catalase, glutathione-S-transferase, glutathione reductase, and glutathione peroxidase, and functions by scavenging superoxide radicals and hydrogen peroxide and inhibiting lipid peroxidation [89,90,91]. TQ can also relay its antioxidant activity through increased activation of Nrf2 (nuclear factor-erythroid 2 related factor 2) in the nucleus, thus upregulating ARE gene activity [92].
These data suggest that TQ has anti-cancer activities via modulation of TME through suppression of ROS in TNBC, as shown in Figure 3.
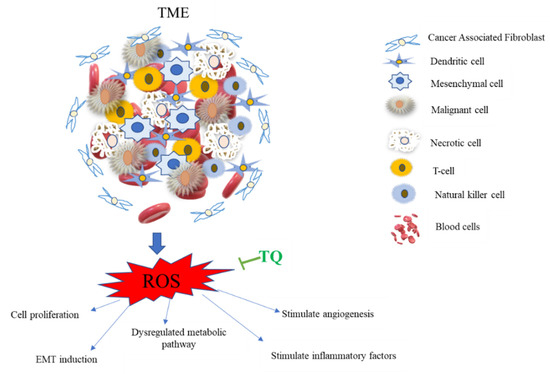
Figure 3.
TQ inhibition of reactive oxygen species (ROS) production in the tumor microenvironment (TME). The TME is a complicated web of inflammatory and non-inflammatory cells. ROS boosts cancer cell proliferation, EMT stimulation, metabolic system dysregulation, and angiogenesis stimulation through altering vascular endothelial growth factors like VEGF and promoting inflammatory chemicals. The metabolic processes of glycolysis and oxidative phosphorylation are strictly controlled by ROS, with an excess of ROS resulting in a dysregulated metabolic pathway [93]. TQ counteracts all of these effects by inhibiting the generation of reactive oxygen species (ROS) via activating cytoprotective enzymes and nuclear factor-erythroid 2 related factor 2 (Nrf2).
5.3. Effect of TQ on Eukaryotic Elongation Factor-2 Kinase (eEF-2K)
Eukaryotic elongation factor-2(eEF-2K) is the only known substrate for eEF-2kinase that promotes ribosomal translocation from A-site to P-site [94]. eEF-2k is a Ca2+/CaM-dependent member of the kinase family function, by phosphorylating/inactivating eEF-2K at threonine 56, leading to reduced peptide chain elongating [95], then increasing eEF-2K function, promoting tumor cell survival, growth, and metastasis [96]. Activation of eEF2K by oxidized low-density lipoprotein inhibits apoptosis in macrophages and promotes survival of cancer cells [97]. eEF-2K increases tumor necrosis factor-alpha (TNF-α), promotes TAM survival and function, induces nitric oxide (NO) in activated macrophages, and contributes to aggressive tumor behaviors [98]. eEF2K expression has been shown to protect cancer cells from hypoxia [99] by producing the expression of specific proteins, such as microtubule-associated proteins, resulting in the expression of tumor-promoting proteins and growth factors [100].
The TME is highly acidic and more hypoxic than normal tissues, leading to tumor aggressiveness [101]. eEF2K was proposed to be responsible for cell survival under acidic [102] and hypoxic conditions [99] in cancer cells and fibroblasts. Taken together, studies highlight the importance of eEF2K in both cancer cells and microenvironmental cells. Natural compounds have been used to target eEF2K via different mechanisms, such as interfering with the eEF2-binding site, CaM-binding site, or the ATP-binding site. For instance, rottlerin, a natural compound, acts as a non-specific inhibitor of eEF2K [103]. Studies showed that rottlerin suppressed the eEF2K gene and protein expression in cancer cells [104].
Studies have shown that eEF-2K significantly increased in TNBC cells associated with poor patient survival and prognosis, promoting cell proliferation, invasion, migration, chemotherapy resistance, and angiogenesis. In vivo studies of a highly aggressive TNBC MDA-MB-231 tumor in an orthotopic xenograft mice model, eEF-2K, were significantly elevated. This was confirmed by liposomal eEF-2K siRNA delivery, which resulted in down-regulation [105]. Similar studies also revealed that the loss/reduced expression of tumor suppressor gene-miR-603- leads to overexpression of eEF2K, and at the same time, when miR-360 is upregulated, results in downregulation of eEF2k by directly targeting the 3-UTR, then inhibits tumor cell migration, growth, and invasion in TNBC [106]. Overexpression of miR-603, which suppresses eEF-2k and inhibits cell migration, proliferation, and invasion in TQ-treated TNBC in mice models and the MDA-MB-231 cell line, has been found to reduce cell migration, proliferation, and invasion by targeting the miR-603/eEF-2k axis [107]. These findings show that utilizing TQ to prevent and treat TNBC that targets eEF2k could be a viable approach (Figure 4).
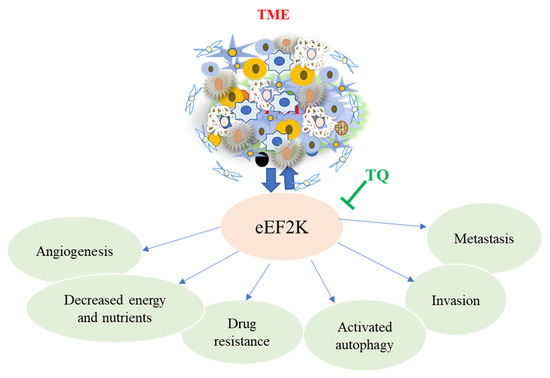
Figure 4.
The major role of eEF2K in promoting angiogenesis, metastasis, invasion, and drug resistance in cancer cells, including TNBC. These effects promote cancer progression, resulting in tumor growth, poor prognosis, and short survival in TNBC patients. TQ reverses this action by regulating and inhibiting the upregulation of eEF2K in the TME.
5.4. Effect of TQ on Inflammatory and Immune Cells
Inflammatory/immune cells in the TME play a role in cancer cell survival, development, proliferation, and differentiation by boosting growth and survival factors and encouraging angiogenesis, inducing mutation by damaging DNA and interfering with treatment [108]. In the TME, the prominent inflammatory and immune cells are tumor-associated macrophage (TAM), cancer-associated adipocytes (CAAs), cancer-associated fibroblasts (CAFs), tumor-infiltrating lymphocytes (TILs), tumor-associated neutrophils (TANs), myeloid-derived suppressor cells (MDSCs), T cells, B cells, natural killer (NK) cells, and dendritic cells (DCs). The cytokines and other factors and signaling molecules produced by these cells include interleukin-1 beta (IL-1β), interleukin-6 (IL-6), interleukin-10 (IL-10), interleukin-17 (IL-17), tumor necrosis factor-α (TNF-α), vascular endothelial growth factor (VEGF), transforming growth factor-β (TGF-β), and matrix metalloproteinases (MMPs). This review particularly focuses on the roles of TAMs, CAAs, CAFs, TILs, Il6, and TGF-β in the TME
5.4.1. Effect of TQ on Tumor-Associated Macrophage (TAM)
Macrophages are phagocytes that serve as a first-line defense against a disease that insults tissue. They are recruited in large numbers to tumors, resulting in immunosuppression. TAMs are derived from circulating monocytic precursors and are directed into tumors by chemokines. When TAMs are appropriately activated, they can kill tumor cells, but persistent expression causes tumorigenesis [109]. TAMs are related to the high expression of cytokines and chemokines that promote tumor growth progression, tumor cell proliferation, angiogenesis, favoring metastasis and invasion, and suppressing antitumor immunity [110].
TAMs are the most abundant immune cells and account for 30–50% of the total inflammatory cells in the TME. TAMs alter the TME by encouraging the proliferation of tumor cells via enrichment of the cytokines and growth factors and thus likely promote, rather than inhibit, cancer development and progression in TNBC [111]. In tumor cells, high TAM numbers are strongly associated with increased vascular density [112], inflammatory cytokines, and growth factors that help angiogenesis by upregulating endothelial cell survival, activation, and proliferation, and are also an important source of VEGF-A both in mice and human tumors [113]. VEGF-A derived from TAM also promotes vascular permeability, facilitating cancer cell intravasation and metastasis [114]. Evidence shows that VEGF-A deficiency in TAMs limits their ability to restore angiogenesis and relapse transplanted tumors after chemotherapy [115].
TAMs favor cancer cell tumor development, invasion, and metastasis into distant sites. TAMs promoted MDA-MB-231 TNBC cell proliferation through activating phosphoinositide 3-kinase (PI3K)-Akt signaling, as well as apoptosis inhibition through increased Bcl-2 and decreased Bax expression, according to in vitro studies. The effect was validated by suppressing PI3K-Akt signaling by adenoviral siRNA Akt1 transfection, which decreased PI3K-Akt signaling [116]. Furthermore, TAM recruited to the lung, which promotes tumor cell seeding and growth by uniquely expressing FMS-like tyrosine kinase 1 (Flt1, also known as VEGFR1), labels a subset of macrophages in human breast cancers that are significantly enriched in metastatic sites, according to studies using the murine TNBC cell line E0771 [117].
TAMs stimulate tumor angiogenesis, matrix remodeling, tumor cell migration and invasion, and immunosuppression in TNBC by secreting inhibitory cytokines, reducing the effector activities of Tumor-Infiltrating Lymphocytes (TILs), and increasing regulatory T cell proliferation, inflammatory molecules, and M2 markers [118]. Studies elucidated the mutual connection between TAMs and TNBC cells. In comparison to macrophages co-cultured with HR+ BC T47D cells, macrophages co-cultured with TNBC MDA-MB-231 cells had a different morphology, more aggressive behavior, higher levels of macrophage mannose receptor, and other M2 markers, and pro-inflammatory mediators such CCL2 [119].
Thus, TAMs and their downstream molecules may be attractive target sites for novel anti-cancer therapies. The best target would be to identify common proteins expressed or overexpressed only by TAM, and not by resident macrophages of distant, healthy cells, which are essential to face pathogens and could take part in anti-cancer actions. Considering this, chemokines and chemokine receptors, and pro-angiogenic factors are the potential target site for TNBC treatment. For instance, in BC, malignant cells recruit macrophages via the chemokine CCL5, and treatment with its receptor antagonist leads to a decreased number of infiltrating macrophages associated with a significantly reduced tumor size [120]. TAM pro-angiogenic functions have also been considered as another target site for anti-cancer development. Antiangiogenic factors have been demonstrated to inhibit macrophage recruitment and reduce tumor size [121]. Furthermore, in an orthotopic breast tumor model, anti-VEGF-A neutralizing antibody therapy reduced the creation of new blood and lymphatic vessels and the incidence of lymphatic and pulmonary metastasis [122]. VEGF promotes macrophage recruitment into tumors, and studies demonstrated that specific inhibition of VEGFR2 decreases tumor macrophage infiltration significantly [123].
TQ has been shown to have considerable anti-inflammatory effects on macrophages after oral administration by suppressing inducible nitric oxide synthase enzymes in response to inflammatory stimuli in mice [124]. According to research, TQ lowered tumor NF-β activity, M2 macrophages, and soluble VEGF levels, reducing proliferation and enhancing apoptosis in the ID8-NGL mouse model of ovarian cancer [125]. TQ’s anti-angiogenic activities were tested in a zebrafish angiogenesis model in vivo. The anti-angiogenic action of TQ was examined in zebrafish embryos by testing its inhibitory influence on blood vessel formation and identifying its molecular target. TQ decreased the formation of intersegmental vessels in zebrafish embryos in a dose-dependent manner, according to the findings. TQ was also discovered to suppress VEGF-A mRNA expression [126]. TQ has also been demonstrated to reduce cell proliferation, migration, invasion, and metastasis in TNBC by downregulating TAM-related chemokine receptor expression [127]. According to the findings, TQ may have an anti-cancer effect by modifying proteins that are either overexpressed or under-expressed by TAMs, indicating that it could be used to prevent and treat TNBC. The overall impact of TAM on TNBC carcinogenesis and the role of TQ is shown in Figure 5.
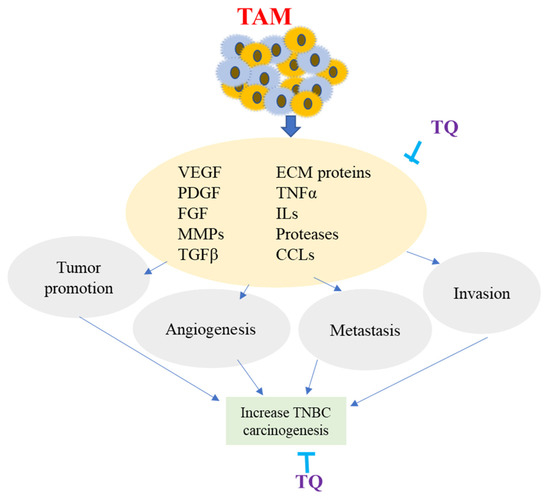
Figure 5.
Overview of Tumor-Associated Macrophage (TAM). TAM is produced from monocytes recruited at the tumor site by molecules produced by TME. Major factors involved in TAM are the chemokines (CCLs), vascular endothelial growth factor (VEGF), interleukins (ILs), transforming growth factor (TGFa), matrix metalloproteinases (MMPs), extracellular matrix proteins (ECM), and platelet-derived growth factors (PDGF). TAM produces several molecules that sustain malignant cell survival, modify neoplastic ECM proteins, enhance invasion and metastasis, promote the development of a newly formed vessel, and assist tumor cells in their progression. TQ potentially inhibits TAM-related expression of various factors and proteins, resulting in decreasing angiogenesis, invasion, and metastasis of TNBC.
5.4.2. Effect of TQ on Cancer-Associated Adipocytes (CAAs)
Adipose tissues are the basic supportive site of the energy storage depot. They have an endocrine function producing hormones, growth factors, cytokines, and other molecules named adipokines that regulate energy and metabolic homeostasis via various signaling processes [128,129] stimulate survival and growth of MDA-MB-231 TNBC cell [130]. One of the significant and primary cellular components of TME in BC is the adipose tissue that surrounds it and is involved in tumor initiation, growth, and metastasis via the promotion of angiogenesis [131]. It has been reported that tumor growth was increased when tumor cells were implanted in the brown or white tissue compared with the subcutaneous space of BC mice [132]. Consistent with these findings, the peritumoral adipose tissue is significantly vascularized and macrophage-rich. It produces higher proteases levels than normal adipose tissue [132,133].
Researchers have been examining how adipocytes interact with TNBC cells, since they are one of the most prominent resident cells in TNBC’s TME [134]. The study used TNBC model 4T1 cells from mouse mammary cancer and adipocytes. Fatty acid-binding protein 4, a hallmark exclusively expressed at high levels in mature adipocytes, was significantly downregulated, while cytokine CCL2, a potent chemokine for monocyte recruitment, was significantly upregulated in mature adipocytes cultured in a transwell system with or without 4T1 murine TNBC cells [135]. These findings suggest that TAAs have a role in TNBC progression and worsen the immunosuppressive TME by secreting an excessive amount of CCL2.
CAAs stimulates the secretion of various inflammatory factors such as chemokine (C-C motif) ligand 2(CCL2), CCL5, IL-1B, IL-6, TNF-α, VEGF, and leptin that contribute to promoting the metastasis and invasion of BC [136]. It has been reported that when TNBC cells (MDA-MB-231) were co-cultured with human adipocytes, studies showed that they boosted motility and invasiveness. The addition of glucose, which is utilized to stimulate the function of human adipocytes, furthered this effect [137]. It has been reported that adipokines produced by adipocytes, which secrete ILs, TNF-α, and hepatic growth factor, support the growth of BC by transferring fatty acids to enhance mitochondrial oxidation, extracellular matrix remodeling, and endotrophin production from collagen, supporting the growth of BC by transferring fatty acids to enhance mitochondrial oxidation, extracellular matrix remodeling, and endotrophin production from collagen [138]. Furthermore, adipocytes express programmed death-ligand 1 (PD-L1), which inhibits CD8+ T cell antitumor function; this was validated by an adipogenesis inhibitor, which reduces PD-L1 expression, enhancing the immune system’s ability to fight cancer [139].
TNBC treatment may target inflammatory factors, pro-angiogenic proteins, transporters, chemokines, cytokines, interleukins, and other downstream signaling molecules and factors released by CAAs. As a result, inhibiting these molecules offers significant promise for modifying the immunosuppressive TME and allowing for greater immunotherapy in the treatment of TNBC. TQ has been shown to have anti-inflammatory and immunomodulatory properties in both cellular and humoral immunity [140], as well as antiangiogenic effects [126].
According to a comprehensive review of studies, TQ has also been proven to protect against inflammatory factors by lowering pro-inflammatory cytokines such as IL-1, IL-6, and TNF-α, which are also produced by CAAs [141]. Because thymoquinone reduces the synthesis of these damaging pro-inflammatory cytokines, it also reduces the creation of free radicals in the TME. Furthermore, in vivo animal investigations demonstrated that TQ reduced adipocyte hypertrophy-induced inflammation [142], implying that TQ could be an alternative option to prevent TNBC associated with cancer-induced adipocytes in the microenvironment.
5.4.3. Effect of TQ on Cancer-Associated Fibroblasts (CAFs)
Fibroblasts are typically spindle-shaped cells with a flat oval nucleus located in tissues that synthesize collagen and other extracellular matrix proteins, providing a scaffold for cells, and playing significant roles in determining cell function and phenotype [143]. Fibroblasts provide oncogenic signals to malignant cells and to the transformed epithelia in a paracrine fashion. Carcinoma-associated fibroblast is an activated fibroblast, also known as myofibroblast, found in association with cancer cells which might facilitate angiogenesis and cancer progression [144]. CAFs in cancer cells promotes angiogenesis, decrease cancer cell dormancy and accelerate tumor growth in mice [145]. They are a significant source of tumor VEGFA [146] and support tumor angiogenesis in a VEGFA independent manner [147]. CAF-derived PDGFC (platelet-derived growth factor C) maintains angiogenesis by further secreting CAFs to stimulate pro-angiogenic growth factors, such as FGF2 (fibroblast growth factor) and osteopontin [148]. They indirectly modulate vascularization and blood flow in tumors, secrete enzymes such as lysyl oxidases and hydroxylases to catalyze the crosslinking of collagen to elastin and extracellular matrix molecules [149], and potentiate tumor angiogenesis by attracting vascular endothelial cells and recruiting monocytes from the bone marrow [150].
An in vitro investigation used patients’ specimens with primary invasive TNBC and BT-549, MDA-MB-231, and MDA-MB-468 cell lines cocultured with CAFs. According to their findings, CAFs enhance TNBC cell migration, invasion, and proliferation and participate in the EMT process via the Wnt/β-catenin pathway [151]. Other studies found that CAFs with high expression of leucine-rich repeat-containing protein 15 (LRRC15) increased cell migration and invasion by influencing the Wnt/β-catenin signaling pathway using MDA-MB-231 and MDA-MB-468 TNBC cell lines [152].
An in vivo animal model study reported that increased collagen expression is strongly associated with TNBC, and transforming growth factor-β (TGF-β) ligands produced by cancer cells and/or CAFs promote the accumulation of fibrotic desmoplastic tissues and the rate of cancer progression [153]. TGF- plays a critical role in TNBC epirubicin resistance by modulating stemness, EMT, and apoptosis in epirubicin-treated MDA-MB-231 TNBC cell lines, according to a study [154]. This was further supported by studies conducted both in vitro and in vivo that showed that TNBC tumors with enhanced collagen expression were reversed/prevented by pirfenidone, an antifibrotic agent that antagonizes TGF-β in patient-derived xenograft and 4T1 TNBC homograft tumors models [155]. TQ inhibited platelet-derived growth factor-BB (PDGF-BB), which resulted in lower proliferation and migration of vascular smooth muscle cells, according to a study conducted in vitro and in vivo [156]. TQ concentration-dependently inhibited several growth factors such as EGF and VEGF, the main CAF sources, in breast cancer cell lines in Balb/C mice [157]. In vitro studies on MDA-MB-231 cancer stem cells, TQ inhibited Wnt3a and PI3K and blunted the stimulatory effects of VEGF, EGF, and FGF. The study was further confirmed by demonstrating a lack of cellular response to pro-angiogenesis factors [158]. These data suggest that targeting CAFs are being considered to control TNBC using TQ.
5.4.4. Effect of TQ on Tumor-Infiltrating Lymphocytes (TILs)
TILs consist of T cells and are a significant player in breast TME [159], in which TNBC shows a higher rate of TIL infiltration within the TME [160]. Consequently, higher TIL tumors are more likely to be associated with estrogen negative [161]. Studies confirmed that TILs are most frequently found in highly proliferative tumors such as TNBC than in hormone-positive BC [162]. Clinical trials have demonstrated a strong association between TIL and TNBC. The excellent overall survival and disease-free survival rate were better for early-stage TNBC patients having higher expression of TILs after adjuvant therapy than low levels. This suggested that TILs improved outcomes, and TILs at diagnosis likely indicate an ongoing antitumor immune response [163]. In human and mouse datasets, activation of the Ras/MAPK pathway is significantly linked with a low level of TIL [164].
There is no information on how TQ affects TIL in TNBC. However, several investigations have shown that TQ improves the immune system by improving the immunological response to malignancies. T lymphocyte cell depletion was detected in rats after exposure to gamma radiation, which was recovered by the injection of TQ in mice by raising the percentage of CD4+ and CD8+ cells through modifying Bcl-2, Bax, PD-1, and caspase-3 expression [165]. When compared to the placebo or baseline group, N. Sativa oil alleviated autoimmune disease by boosting the expression of CD4+ cells in a clinical trial. TQ may be engaged in anti-inflammatory effects in TNBC patients by increasing the activity of FoxP3+ Treg cells [166]. TQ dramatically reduced diabetes complications in Streptozotocin-induced gestational diabetic mice by boosting T-cell proliferation and restoring thymus homing CD4+, CD8+ and circulating T-cells [167]. Foxp3+, CD3+, CD4+ and CD8+ T lymphocytes are the broadly studied major components of TILs [168]. Several pre-clinical and clinical trial studies supported that these high levels of TILs present at diagnosis were strongly associated with reduced distant recurrence rates and favorably altered the immune microenvironment [162,163,169]. This finding backs up TQ’s immunomodulatory properties in TNBC patients, suggesting that TQ will be important in increasing TILs through stimulating their core components such as Foxp3+, CD3+, CD4+, and CD8+.
5.4.5. Effect of TQ on Interleukin-6 (IL-6)
Inflammatory TME is regulated by several cytokines [170], including interleukin (IL)-1 and IL-6, which promote cancer cell proliferation and invasion, increasing intracellular signaling by NF-kB, and cytokine receptor activation accelerates tumor progression [171]. IL-6 is produced by several typical cell types, including monocytes, fibroblasts, and lymphocytes, and an autocrine production molecule that occurs in immune cells acts in a paracrine condition to promote neovascularization and inflammation-dependent carcinogenesis [172]. IL-6 participates in several biological activities, including immune regulation, hematopoiesis, and tumorigenesis [173]. An upregulated level of IL-6 in the TME is strongly associated with cancer cell proliferation, epithelial to mesenchymal transition, chemoresistance, invasion, metastasis survival of tumor cells, and angiogenesis through fueling STAT3 MAPK and Akt signaling [174]. Overexpression of IL-6 and its receptors in TME might be due to the increased cell proliferation by increasing Bcl-2 expression, which leads to changes in the proliferation/apoptosis balance towards neoplastic cell proliferation [175].
IL-6 is critical for the growth of TNBC, anchorage-independent colony formation, and resistance to apoptosis which contributes to poor prognosis in TNBC patients [176]. IL-6 is secreted by TNBC that increases expression of CCL5 in lymphatic endothelial cells by activating the IL-6 receptor and subsequently STAT3, which increases transcription of the CCL5 gene [177]. IL-6 is expressed in approximately 50% of BC, and its serum level was higher in TNBC than in hormone-positive BC patients [178]. To initiate signal transduction, IL-6 with IL-6 receptor recruits glycoprotein 130 kDa (GP130), a transmembrane protein, also called CD130, ubiquitously expressed in various tumor cells. This is a common receptor for well-known cytokines such as IL-6, 11, 27, LIF, CNTF, OSM, CT-1, and CLC) [179] forming a hexameric IL-6/IL-6R/GP130 complex, which further activates the downstream effector cascade, including phosphorylation of STAT3 [174]. These effects promote tumor survival, proliferation, differentiation, angiogenesis, metastasis, immunosuppression, apoptosis, and drug resistance and advance many human cancers [180].
Studies conducted using four different TNBC cell lines (SUM159, MDA-MB-231, MDA-MB-468, MDA-MB-436) have shown that inhibition of IL-6 expression by shRNA in TNBC leads to reduced cell survival and suppression of colony formation both in vivo and in vitro [176]. Similar studies reported that inhibitor of IL-6 receptor signaling strongly inhibits tumor growth and metastasis in TNBC cells (MDA-MB-231, SUM149, and SUM159 cells) [181]. TNBC cells mainly express the transmembrane GP130/IL-6 receptor, and developed specific molecule inhibitors targeting GP130 might be an alternative strategy for effective treatment or prevention of TNBC either in combination with chemotherapy or as a single agent; there are previous studies that have confirmed this scenario [182,183,184]. It has been reported that TQ downregulated IL6 induced phosphorylation of STAT3 and Akt in a concentration and time-dependent manner [185]; hence IL-6 activated Akt has been shown to play a major role in the mechanism of action of IL-6 [186].
5.4.6. Effect of TQ on Janus Kinases-Signal Transducer and Activator of Transcription Factor (JAK-STAT)
The JAK/STAT pathway functionally modulates the TME. JAK/STAT signaling pathway and contains four JAK domains (JAK1,2,3, and tyrosine kinase 2 (TYK2)) and seven STAT proteins (STAT1-4, 5A, 5B, and 6) [187]. Dysfunction of the JAK/STAT pathway leads to tumorigenesis, metastasis, invasion, survival of tumor cells, proliferation, anti-apoptosis, angiogenesis, and immune invasion [188]. The JAK/STAT pathway in BC is altered by down-regulation of phophotyrosine-specific phosphatase in TNBC [189]. It is also impaired by activation of PI3K/mTOR [190] and down-regulation negative regulators of STAT, including suppressor of cytokine signaling 3 [191]. Increasing the amount of JAK/STAT results in activating ligand IL-6 [192]. Precise regulation of STAT activation is critical for eliciting the appropriate responses to extracellular signals [193], but persistent activation of STAT3 via phosphorylation by cytokines is mediated through JAK [194] and is required for the survival of tumor cells by promoting the upregulation of genes that encode anti-apoptotic proteins, angiogenic factors, and cell cycle regulators [193].
Targeting JAK/STAT activation alone can lead to unanticipated effects on the TME. JAK/STAT inhibition and reverses the negative impacts within the TME on immune cell function. Authors reported that JAK2-specific inhibitors had been shown in reducing tumor growth in TNBC cells, but JAK1/STAT3 inhibitor had little effect, sometimes counteracting the JAK2 specific inhibition on TNBC in an in vivo model [195].
There are no good studies about the impact of TQ on this target site; it will serve as a potential alternative site for the treatment/prevention of TNBC. Still, studies in liver cancer confirmed that TQ strongly suppressed the phosphorylation of STAT3 associated with decreased JAK activity, thus enhancing the apoptotic death rate of cancer cells [196]. TQ has also been shown in studies to reduce cell proliferation by blocking the activation of the JAK2/STAT3 signaling pathway in gastric cancer cells both in vitro (HGC27, BGC823, and SGC7901 cell lines) and in a xenograft tumor mouse model. TQ reduced the activation and phosphorylation of the STAT3 pathway and the production of STAT3-dependent reporter genes such as survivin, cyclin D, and VEGF, preventing cell proliferation and inducing apoptosis [197]. TQ inhibited STAT3 phosphorylation at Tyr705 due to JAK2 and Src activity suppression in gastric cancer cell line (HGC27) investigations. TQ inhibited STAT3 and prevented the expression of multiple STAT3-regulated genes, indicating that it is an efficient factor for blocking the STAT3 pathway by preventing tyrosine kinase phosphorylation [197]. Furthermore, TQ inhibited STAT3 activation and the production of STAT3-regulated gene products such as cyclin D1, Bcl-2, Bcl-xL, survivin, Mcl-1, and vascular endothelial growth factor in human multiple myeloma cell lines (U266 and RPMI 8226) [185]. Based on these findings, TQ will serve as a potential therapy option for TNBC by addressing this signaling system.
5.5. Effect of TQ on Endothelial Cells
5.5.1. Effect of TQ on Vascular Endothelial Growth Factor-A (VEGF-A)
Vascular endothelial growth factor-A (VEGF-A), also referred to as vascular permeability factor, essential endothelial cells in angiogenesis and vasculo-genesis [198], is secreted by solid tumor for blood vessel formation to provide adequate blood to tumors for further stimulation of tumor proliferation and metastasis [199]. VEGF is highly involved in angiogenesis and increasing vascular permeability, and acts as a classical pro-tumorigenic NF-kB target gene. Overexpression of VEGF-A in BC is associated with reduced overall survival and disease-free survival mediated by endothelial growth factor receptors [200]. It is an essential molecular driver of highly aggressive cancers such as TNBC [201,202]. Upregulation of VEGF-A on endothelial cells increases cell survival, permeability, cell migration, and cell proliferation [203]. Parallel to the BC cell, it induces cell migration and cancer stem cell self-renewal; these two actions promote BC progression and metastasis [204]. VEGF is highly upregulated in the breast TME and activates oncogenic signaling pathways such as MAPK pathway and the phosphatidylinositol 3-kinase (PI3K)/Akt pathway, stimulating survival and proliferation, migration and angiogenesis [205].
Agents targeting VEGF help to prevent its interaction with vascular endothelial cells enabling increase apoptosis through decreasing cell proliferation, blood cell formation, and vascular permeability towards tumor cells [206]. TQ has confirmed anti-neoplastic activity via its ability to regulate several genetic pathways. In addition, TQ has established anti-cancerous effects by inhibiting the initiation, invasion, migration, and progression of cancer. TQ’s anti-cancer effects are chiefly mediated by regulating various cell signaling pathways such as VEGF, Bcl2/Bax ratio, p53, NF-kB, and other oncogenes [207]. Studies have shown that TQ down-regulated the expression of the VEGF-A gene and induced apoptosis in stomach cancer cells [208]. TQ considerably decreased VEGF-induced angiogenesis in vitro and in vivo, according to in vitro aortic ring assays and in vivo Matrigel plug assays. Furthermore, co-injection of TQ (6 mg/kg/day) for 15 days in a xenograft mice model with human prostate cancer cells (PC3 cells) resulted in a six-fold and 23-fold decrease in the size and weight of prostate tumors, respectively [209]. Another in-vitro study revealed that TQ down-regulates VEGF expression in hormone-positive BC cells [210]. Similarly, in vivo studies have shown that TQ, in combination with Resveratrol had a synergistic effect on induction of apoptosis anti-angiogenesis activity by decreasing expression of VEGF in mouse epithelial BC cells [211]. In line with this, TQ was also reported to inhibit the expression of NF-κβ-regulated genes VEGF-A in MDA-MB-231 TNBC cells [107]. These findings showed that TQ, which targets VEGF, could be a viable alternative in preventing and treating TNBC.
5.5.2. Effect of TQ on Transforming Growth Factor-β (TGFβ)
Transforming growth factor-β (TGFβ) is a multifunctional polypeptide that acts as a tumor suppressor in normal epithelial cells and the early stage of tumor cells. In contrast, in an advanced stage of tumor cells, it acts as tumorigenesis [212]; TGF-β signaling is mediated by two transmembrane receptors, TGF-β type I and II, and the ligand binding leads to SMAD2/3 phosphorylation and activation [213]. TGF-β binding to its receptor leads to active-like kinase recruitment and propagation of downstream intracellular signaling pathways to the nucleus through Smad proteins that act as a transcription activator [214]. TGF-β promotes tumor cell invasion and migration by inducing an epithelial–mesenchymal transition. It is a profound phenotypic conversion by which epithelial cells lose their cohesiveness and polarity that acquire motile and invasive properties. It plays a major role in the morphogenetic process during organogenesis [215].
In BC, TGF-kβ regulates several processes such as proliferation, differentiation, immunity, migration, and apoptosis. It has a dual function in the progression of tumors. It acts as tumorigenesis in the late stage that enhances invasion, migration, and survival of the tumor cells. A tumor suppressor in the early stage has an antiproliferative effect [216,217]. Authors have reported that TNBC is resistant to the growth-inhibitory activity of TGF-β [218]. In normal epithelial, endothelial, and neuronal cells, TGFβ has cytostatic effects and prevents hyper cell proliferation by controlling the G1/S phase transition events, mediated through cyclin-dependent kinase inhibitors [219,220]. The cell cycle arrest is also achieved by downregulation of the proliferative inducing transcription factor, c-Myc [221]; meanwhile, these antiproliferative effects can be opposing in an advanced stage. Evidence revealed that the TGF-β switches from tumor suppressor to pro-metastatic factor in BC progression [222].
The diversity effects of the TGF-β signaling pathway indicate a need for caution when targeting the design of effective TNBC therapeutics and/or prevention by identifying the factors that determine the balance between the opposing effects. It has been reported that TQ significantly reduced tumorigenic signaling such as TGF-β, with a consequence concentration-dependent inhibition of cancer cell growth, migration, and invasion [223]. According to the findings, TQ inhibited the metastatic phenotype and reversed EMT in prostate cancer cells in vitro by negatively modulating the TGF-/Smad2/3 signaling pathway [224]. These data imply that thymoquinone, which works by blocking TGF-β, could be a promising treatment agent for TNBC.
5.5.3. Effect of TQ on Insulin-Like Growth Factor I (IGF-I)
Insulin-like growth factor I (IGF-I) is essential for fast cell growth, cell differentiation, survival, migration, and invasion in BC [225]. IGF-1 binds to insulin-like growth factor receptor-type1(IGF-1R) and stimulates various signaling pathways, promoting the growth and survival of cancer cells [226]. More importantly, the IGF-1/IGF-1R system mediates stimulatory effects in cancer cells via different routes such as the phosphatidylinositol-3kinasw(PI3K)/Akt1, mTOR, and MAPK [227,228]. Expression of IGF-1 activates MAPK and Akt, leading to increased cell proliferation, tumor cell survival, invasion, migration, tumor cell growth, epithelial to mesenchymal transition, and anti-apoptotic effects [229,230].
IGF-I is a well-known activator of the Janus-activated kinase/signal transducer and activator of transcription (STAT) pathway [231] and has been implicated in NF-κB–mediated transcriptional regulation of inflammatory cytokines and vascular endothelial cell adhesion receptors. IGF-I promotes TNBC metastasis not only through a direct paracrine effect on tumor cell survival and proliferation but also through indirect effects, likely involving the TME and pro-inflammatory response [232].
It has been reported that upregulation of IGF-1R and increased IGF-1 levels are associated with a high BC risk and poor prognosis in BC patients [233]; furthermore, in TNBC patients, it has been associated with shorter survival [234]. Stimulation of IGF-I protects BC from the toxic effects of chemo- and radiotherapy [235] as well as contributing to disease progression through its anti-apoptotic and mitogenic effects on the mammary epithelial cells [236]. Authors have reported that patients diagnosed with BC showed a high level of IGF-I detection in 100% of TNBC cases [237]. High IGF-1 gene expression signature mediating cancer proliferation and short survival in TNBC cells, and TQ targeting this site suggests a rationale for prevention and/or treatment. In vitro studies targeting the IGF-IR gene in normal and malignant human breast cancer tissue specimens have demonstrated promising anti-neoplastic effect [238]. TQ significantly augments cisplatin-induced anti-tumor effects on gastric cancer both in vitro and in vivo by blocking the PI3K/AKT signaling pathway, according to research done with SGC-7901 human gastric cancer cells and BALB/c mice implanted with gastric cancer cells [239]. These signals are IGF-1’s downstream pathways, suggesting that TQ could be employed as an alternative to IGF-1 targeting in the treatment and prevention of TNBC.
5.5.4. Effect of TQ on Endoglin
Endoglin or CD105 is a homo-dimeric cell membrane glycoprotein receptor for TGF-β and bone morphogenetic protein-9(BMP-9), highly expressed in angiogenetic endothelial cells [240]. In addition to a co-receptor for TGF-β, endoglin is a proliferation-associated cell membrane antigen [241], indispensable for the development of angiogenesis [242], which promotes endothelial proliferation, and increases TGF-β/ALK1 (Activin receptor-like kinase 1) signaling transduction [243]. Since endoglin is expressed at a low level in quiescent endothelial cells, its expression is significantly upregulated in active vascular endothelial cells during tumor angiogenesis which directly involves cell proliferation, migration, capillary tube formation, and function as a pro-angiogenic role [244].
Overexpression of endoglin is strongly associated with metastasis, poor chemotherapy response, poor survival in BC patients, and the deletion of endoglin results in reversing carcinogenesis and chemoresistance [245,246]. Endothelial cells in the newly synthesized blood vessels in the TME significantly express the TGF-β co-receptor endoglin, which has been linked with poor prognosis and metastatic disease in many cancer cells, including TNBC [247].
Targeting endoglin and the VEGF pathway concurrently improves treatment in vitro and appears to reverse resistance in refractory cancer patients [248]. Studies targeting VEGF receptor kinase inhibitors as antiangiogenic therapies caused therapy resistance because of upregulation of another pro-angiogenic signaling pathway, such as the endoglin/transforming growth factor-β (TGF-β) pathway [249]. However, inhibiting VEGF and endoglin reduces tumor angiogenesis and decreases metastatic spread on BC cells [250]. It has been reported that agents that neutralize its antibodies, or a ligand trap targeting endoglin, inhibit BC metastatic spread and tumor angiogenesis in the mice model [250]. Preventing or blocking the tumor blood supply by disrupting the new blood cell formation has been considered as a general strategy to treat cancer [251]. TQ could be used to target TGF-B, VEGF, and other angiogenic factors in TNBC [158,209,252]. Targeting endoglin and its downstream pathways as an approach for treating TNBC with TQ would be another potential target site in this regard (Figure 6).
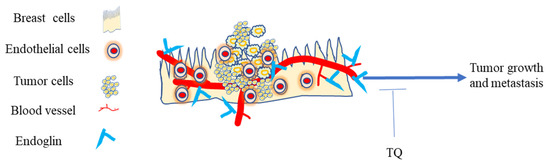
Figure 6.
Endoglin in the TME of TNBC. The expression of endoglin on endothelial cells is critical for promoting primary tumor growth and metastasis. Blocking endoglin in the TNBC TME with TQ exerts anti-cancer activity.
5.5.5. Effect of TQ on Indoleamine 2,3-dioxygenase (IDO)
Indoleamine 2,3-dioxygenase (IDO) is an immunocompromised enzyme that degrades tryptophan, essential for activation of T-cell, leading to inhibition of T-cell proliferation [253]. Following the catabolism of tryptophan, it generates different physiological active immunosuppressive metabolites such as kynurenine [254]. Kynurenine causes tumor-infiltrating lymphocytes to become anergic and die [255]. Blocking activation of T-cell reduces its prominent role in modulating host response to tumor cells and plays an immunosuppressive role following inflammatory stimuli and preventing immune activation [256]. Upregulation of IDO results in immunotolerance and immunosuppression and can obstruct the antitumor responses and enhance metastasis in BC [257]. IDO is expressed by macrophages [253], trophoblasts, and dendritic cells [258]. IDO expression in the TME has been correlated with the induction of multiple tolerogenic immune phenotypes, including the inhibition of effector T cell activation, enhanced infiltration of myeloid-derived suppressor cells, B cell dysfunction, and promotion of tumor angiogenesis [259].
Evidence suggests that various types of human tumor cell, including TNBC, express IDO, and inflammatory mediators, particularly interferon-γ (IFNγ), have the specific ability to enhance the expression of IDO [260]. Studies reported a high level of IDO expression in primary tumors and serum of BC patients [261]. Even so, further investigation is suggested, as it is not expressed in normal breast tissues [262]. TNBC cells express IDO in the presence of inflammation and T-cell infiltration [263]. Evidence from Pakistan showed that from a total of 100 BC patients, high IDO was observed in TNBC patients compared to non-TNBC patients, at 65.3% and 33.3%, respectively, and this overexpression of IDO was associated with decreased overall patient survival [264]. IDO expression has been shown to correlate with poor clinical prognosis in various cancers [265,266,267]. IDO in tumor cells can potentially suppress T-cells during tumor growth; it reduces not only the amount of T-cell infiltration but also compromises the cytotoxic function of effector CD8+ T-cells [268].
Since the immune system in BC patients is dysfunctional [269], targeting the IDO site is an alternative option for repopulating the host with immune cells, relieving IDO-mediated immune suppression in the TME and tumor cells themselves [270]. These inhibitory effects might converge to induce cytotoxic T-cell exhaustion and dampen antitumor immunity. It has been reported that IDO inhibitor combine with taxane augmented tumor-infiltrating lymphocytes to kill tumor cells and improve clinical outcomes in BC patients [271], since pro-inflammatory molecules such as IL-1, IL-6, and IFN induce expression of IDO through STAT independent pathway involving P38, MAPK, and NF-kβ [272] and these cytokines and signaling pathways are inhibited by TQ [185,273,274], so that IDO will be another potential target site for TQ.
6. Clinical Trials Have Shown the Importance of TQ in the Treatment of a Variety of Diseases
We highlighted the importance of TQ in the treatment of various diseases in various countries based on completed clinical trials, despite the fact that there are no published clinical trials of TQ on TNBC in the United States [275] (Table 1).

Table 1.
Summary of TQ application in clinical trials for various diseases.
7. Limitations of Thymoquinone (TQ) as a Natural Product
One of the most pressing concerns is TQ’s bioavailability. According to pharmacokinetic studies, TQ is rapidly eliminated and slowly absorbed, resulting in a decreased bioavailability of roughly 58% and a lag time of 23 min [282]. Several studies have been conducted to boost bioavailability by changing the chemical structure of the medicine and generating nanoparticles, and have proven to be successful. A thymoquinone-loaded nanostructured lipid carrier was devised in cancer cell lines to improve bioavailability (elimination half-life of 5 h) and induction of apoptosis and cell cycle arrest [283,284]. The investigators developed thymoquinone-encapsulated nanoparticles using biodegradable, hydrophilic polymers such as polyvinylpyrrolidone and polyethylene glycol to overcome thymoquinone’s poor solubility, thermal and light sensitivity, and minimal systemic bioavailability, which can significantly improve the cancer treatment’s efficiency. This nanoparticle can kill and inhibit the migration of breast cancer cells [285].
Another concern with clinical TQ is the issue of safety. Drugs are used to treat several degenerative diseases; however, many drugs are limited by side effects and toxicity. In many cases, bioactive compounds of eatable medicinal plants have a promising and prominent role in human health due to low toxicity [286], as with supplementation of TQ [287]. Studies indicate that TQ is not toxic in rats within the range of 10–100 mg/kg body, both in sub-chronic and subacute doses [288]. The LD50 of TQ was estimated to be 104.7 mg/kg after intraperitoneal injection and 870.9 mg/kg after oral intake in mice, and 57.5 mg/kg and 794.3 mg/kg in rats, respectively, following intraperitoneal and oral administration [289]. In another study, investigators revealed no side effect of thymoquinone on prolonged consumption of TQ (10–100 mg/kg up to 20 weeks) [290]. The acceptable dose of TQ for humans is 0.6 mg/kg/day [291]. On the other hand, TQ has anticancer action at relatively low concentrations, around 10 mg/kg [292] As a result, the issue of safety may not be a major concern.
8. Conclusions
TNBC is not simply a group of cancer cells, but rather a heterogeneous collection of various compounds that support the growth and progression of tumor cells. An emerging TME is quite a complicated and continuously evolving topic in the research area. We would like to focus on selected components in the microenvironment that directly or indirectly contributed to TNBC cell growth, proliferation, migration, invasion, and development of drug resistance. The composition of the TME is bulk and conventional therapy, even in combination, is not effective in the treatment of TNBC cases. The characteristics of the TME can be manipulated to design more effective therapies. TQ has been reported to down-regulate various tumorigenic induced associated signals and proteins. This review described essential features of the TME components and discussed strategies whereby TQ may be effective. Hence all these data suggest that TQ alone or in combination with an existing drug, focusing on the cancer-producing mechanisms of each TME component signaling process stage, can be a potential alternative for the success of therapeutic intervention against TNBCs. Since various components of the TME serve as a favorable shelter for TNBC cells, TQ will be helpful in prevention of TNBC through the modulation of the tumor microenvironment.
Author Contributions
Conceptualization, K.F.A.S. and G.M.A.; methodology, K.F.A.S. and G.M.A.; formal analysis, G.M.A.; writing—original draft preparation, G.M.A.; writing—review and editing, K.F.A.S., G.M.A., E.T., B.M., R.B.B., E.A.M. and R.E.; visualization, K.F.A.S., G.M.A., E.T., G.M.A., B.M., R.B.B., E.A.M. and R.E. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the National Institute of Minority Health and Health Disparities of the National Institutes of Health through Grant Number U54 MD 007582 and Grant Number P20 MD006738.
Acknowledgments
We thank The Title III program.
Conflicts of Interest
The authors declare that there is no conflict of interest.
References
- Hsu, Y.-L.; Uen, Y.-H.; Chen, Y.; Liang, H.-L.; Kuo, P.-L. Tricetin, a dietary flavonoid, inhibits proliferation of human breast adenocarcinoma mcf-7 cells by blocking cell cycle progression and inducing apoptosis. J. Agric. Food. Chem. 2009, 57, 8688–8695. [Google Scholar] [CrossRef]
- Lei, S.; Zheng, R.; Zhang, S.; Wang, S.; Chen, R.; Sun, K.; Zeng, H.; Zhou, J.; Wei, W. Global patterns of breast cancer incidence and mortality: A population-based cancer registry data analysis from 2000 to 2020. Cancer Commun. 2021, 41, 1183–1194. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Feng, Q.; Yang, H.; Wang, G.; Huang, L.; Ba, Q.; Zhang, C.; Wang, Y.; Chen, Y.; Cheng, Q.; et al. A Light-Triggered Mesenchymal Stem Cell Delivery System for Photoacoustic Imaging and Chemo-Photothermal Therapy of Triple Negative Breast Cancer. Adv. Sci. 2018, 5, 1800382. [Google Scholar] [CrossRef] [PubMed]
- Anders, C.; Carey, L.A. Understanding and Treating Triple-Negative Breast Cancer. Oncology 2008, 22, 1233–1243. [Google Scholar] [PubMed]
- Thakur, K.K.; Bordoloi, D.; Kunnumakkara, A.B. Alarming Burden of Triple-Negative Breast Cancer in India. Clin. Breast Cancer 2018, 18, e393–e399. [Google Scholar] [CrossRef] [PubMed]
- Hwang, K.-T.; Kim, J.; Jung, J.; Chang, J.H.; Chai, Y.J.; Oh, S.W.; Oh, S.; Kim, Y.A.; Park, S.B.; Hwang, K.R. Impact of Breast Cancer Subtypes on Prognosis of Women with Operable Invasive Breast Cancer: A Population-based Study Using SEER Database. Clin. Cancer Res. 2019, 25, 1970–1979. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Yu, T.; Di, G. Role of tumor microenvironment in triple-negative breast cancer and its prognostic significance. Chin. J. Cancer Res. 2017, 29, 237–252. [Google Scholar] [CrossRef]
- Lee, A.; Go, S.I.; Lee, W.S.; Lee, U.S.; Kim, M.J.; Kang, M.H.; Lee, G.W.; Kim, H.G.; Kang, J.H.; Jeon, K.N.; et al. Irinotecan and capecitabine combination chemotherapy in a patient with triple-negative breast cancer relapsed after adjuvant chemotherapy with anthracycline and taxane. Tumori 2015, 101, e9–e12. [Google Scholar] [CrossRef]
- Afghahi, A.; Timms, K.M.; Vinayak, S.; Jensen, K.C.; Kurian, A.W.; Carlson, R.W.; Chang, P.-J.; Schackmann, E.; Hartman, A.-R.; Ford, J.M.; et al. Tumor BRCA1 Reversion Mutation Arising During Neoadjuvant Platinum-Based Chemotherapy in Triple-Negative Breast Cancer Is Associated with Therapy Resistance. Clin. Cancer Res. 2017, 23, 3365–3370. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.D.; Siegel, R.L.; Lin, C.C.; Mariotto, A.B.; Kramer, J.L.; Rowland, J.H.; Stein, K.D.; Alteri, R.; Jemal, A. Cancer treatment and survivorship statistics, 2016. CA Cancer J. Clin. 2016, 66, 271–289. [Google Scholar] [CrossRef] [PubMed]
- Oakman, C.; Moretti, E.; Galardi, F.; Biagioni, C.; Santarpia, L.; Biganzoli, L.; Leo, A.D. Adjuvant systemic treatment for individual patients with triple negative breast cancer. Breast Cancer Res. 2011, 3, S135–S141. [Google Scholar] [CrossRef]
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef]
- Willumsen, N.; Thomsen, L.B.; Bager, C.L.; Jensen, C.; Karsdal, M.A. Quantification of altered tissue turnover in a liquid biopsy: A proposed precision medicine tool to assess chronic inflammation and desmoplasia associated with a pro-cancerous niche and response to immuno-therapeutic anti-tumor modalities. Cancer Immunol. Immunother. 2018, 67, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, M.; Werner, S. Cancer as an overhealing wound: An old hypothesis revisited. Nat. Rev. Mol. Cell Biol. 2008, 9, 628–638. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, H.F. Tumors: Wounds That Do Not Heal-Redux. Cancer Immunol. Res. 2015, 3, 1–11. [Google Scholar] [CrossRef]
- Wong, A.Y.; Whited, J.L. Parallels between wound healing, epimorphic regeneration and solid tumors. Development 2020, 147, dev181636. [Google Scholar] [CrossRef]
- Joyce, J.A.; Pollard, J.W. Microenvironmental regulation of metastasis. Nat. Rev. Cancer 2009, 9, 239–252. [Google Scholar] [CrossRef]
- Liu, S.; Cong, Y.; Wang, D.; Sun, Y.; Deng, L.; Liu, Y.; Martin-Trevino, R.; Shang, L.; McDermott, S.P.; Landis, M.D.; et al. Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts. Stem. Cell Rep. 2014, 2, 78–91. [Google Scholar] [CrossRef]
- Matsumoto, H.; Koo, S.L.; Dent, R.; Tan, P.H.; Iqbal, J. Role of inflammatory infiltrates in triple negative breast cancer. J. Clin. Pathol. 2015, 68, 506–510. [Google Scholar] [CrossRef]
- Esquivel-Velázquez, M.; Ostoa-Saloma, P.; Palacios-Arreola, M.I.; Nava-Castro, K.E.; Castro, J.I.; Morales-Montor, J. The role of cytokines in breast cancer development and progression. J. Interferon Cytokine Res. 2015, 35, 1–16. [Google Scholar] [CrossRef]
- Salehi, B.; Stojanović-Radić, Z.; Matejić, J.; Sharifi-Rad, M.; Anil Kumar, N.V.; Martins, N.; Sharifi-Rad, J. The therapeutic potential of curcumin: A review of clinical trials. Eur. J. Med. Chem. 2019, 163, 527–545. [Google Scholar] [CrossRef] [PubMed]
- Zaynab, M.; Fatima, M.; Abbas, S.; Sharif, Y.; Umair, M.; Zafar, M.H.; Bahadar, K. Role of secondary metabolites in plant defense against pathogens. Microb. Pathog. 2018, 124, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Zubair, H.; Azim, S.; Ahmad, A.; Khan, M.A.; Patel, G.K.; Singh, S.; Singh, A.P. Cancer Chemoprevention by Phytochemicals: Nature’s Healing Touch. Molecules 2017, 22, 395. [Google Scholar] [CrossRef] [PubMed]
- Gali-Muhtasib, H.; Roessner, A.; Schneider-Stock, R. Thymoquinone: A promising anti-cancer drug from natural sources. Int. J. Biochem. Cell Biol. 2006, 38, 1249–1253. [Google Scholar] [CrossRef] [PubMed]
- Vuorela, P.; Leinonen, M.; Saikku, P.; Tammela, P.; Rauha, J.-P.; Wennberge, T.; Vuorela, H. Natural Products in the Process of Finding New Drug Candidates. Curr. Med. Chem. 2004, 11, 1375–1389. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs over the Last 25 Years. J. Nat. Prod. 2007, 70, 461–477. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zheng, Z.; Wu, M.; Zhang, L.; Wang, J.; Fu, W.; Xu, N.; Zhao, Z.; Lao, Y.; Xu, H. The natural compound neobractatin inhibits tumor metastasis by upregulating the RNA-binding-protein MBNL2. Cell Death Dis. 2019, 10, 554. [Google Scholar] [CrossRef]
- Casey, S.C.; Amedei, A.; Aquilano, K.; Azmi, A.S.; Benencia, F.; Bhakta, D.; Bilsland, A.E.; Boosani, C.S.; Chen, S.; Ciriolo, M.R.; et al. Cancer prevention and therapy through the modulation of the tumor microenvironment. Semin. Cancer Biol. 2015, 35, S199–S223. [Google Scholar] [CrossRef] [PubMed]
- Deepak, K.G.K.; Vempati, R.; Nagaraju, G.P.; Dasari, V.R.; Nagini, S.; Rao, D.N.; Malla, R.R. Tumor microenvironment: Challenges and opportunities in targeting metastasis of triple negative breast cancer. Pharmacol. Res. 2020, 153, 104683. [Google Scholar] [CrossRef]
- Sharma, S.H.; Thulasingam, S.; Nagarajan, S. Chemopreventive agents targeting tumor microenvironment. Life Sci. 2016, 145, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Trang, N.t.D.; Wanner, M.J.; Phuong, L.V.N.; Koomen, G.J.; Dung, N.X. Thymoquinone from Eupatorium ayapana. Planta Med. 1999, 59, 99. [Google Scholar] [CrossRef] [PubMed]
- Al-Ghamdi, M. A review of pharmaco-therapeutics effects of Nigella Sativa. Pak. J. Med. Res. 2002, 41, 77–83. [Google Scholar]
- Randhawa, M.A.; Alghamdi, M.S. Anticancer Activity of Nigella sativa (Black Seed)—A Review. Am. J. Chin. Med. 2011, 39, 1075–1091. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, Y.K.; Abdelrazek, H.M. Cancer: Thymoquinone antioxidant/pro-oxidant effect as potential anticancer remedy. Biomed. Pharmacother. 2019, 115, 108783. [Google Scholar] [CrossRef]
- El-Mahdy, M.A.; Zhu, Q.; Wang, Q.E.; Wani, G.; Wani, A.A. Thymoquinone induces apoptosis through activation of caspase-8 and mitochondrial events in p53-null myeloblastic leukemia HL-60 cells. Int. J. Cancer 2005, 117, 409–417. [Google Scholar] [CrossRef]
- Rooney, S.; Ryan, M.F. Effects of alpha-hederin and thymoquinone, constituents of Nigella sativa, on human cancer cell lines. Anticancer Res. 2005, 25, 2199–2204. [Google Scholar]
- Adinew, G.M.; Taka, E.; Mendonca, P.; Messeha, S.S.; Soliman, K.F.A. The Anticancer Effects of Flavonoids through miRNAs Modulations in Triple-Negative Breast Cancer. Nutrients 2021, 13, 1212. [Google Scholar] [CrossRef]
- Alhebshi, A.H.; Gotoh, M.; Suzuki, I. Thymoquinone protects cultured rat primary neurons against amyloid#²-induced neurotoxicity. Biochem. Biophys. Res. Commun. 2013, 433, 362–367. [Google Scholar]
- Talib, W.H. Regressions of Breast Carcinoma Syngraft Following Treatment with Piperine in Combination with Thymoquinone. Scientia Pharmaceutica 2017, 85, 27. [Google Scholar] [CrossRef]
- Sutton, K.M.; Greenshields, A.L.; Hoskin, D.W. Thymoquinone, a bioactive component of black caraway seeds, causes G1 phase cell cycle arrest and apoptosis in triple-negative breast cancer cells with mutant p53. Nutr. Cancer 2014, 66, 408–418. [Google Scholar] [CrossRef] [PubMed]
- Rajput, S.; Kumar, B.N.; Sarkar, S.; Das, S.; Azab, B.; Santhekadur, P.K.; Das, S.K.; Emdad, L.; Sarkar, D.; Fisher, P.B.; et al. Targeted apoptotic effects of thymoquinone and tamoxifen on XIAP mediated Akt regulation in breast cancer. PLoS ONE 2013, 8, e61342. [Google Scholar] [CrossRef]
- Woo, C.C.; Hsu, A.; Kumar, A.P.; Sethi, G.; Tan, K.H. Thymoquinone inhibits tumor growth and induces apoptosis in a breast cancer xenograft mouse model: The role of p38 MAPK and ROS. PLoS ONE 2013, 8, e75356. [Google Scholar] [CrossRef]
- Effenberger-Neidnicht, K.; Schobert, R. Combinatorial effects of thymoquinone on the anti-cancer activity of doxorubicin. Cancer Chemother. Pharmacol. 2011, 67, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Arafael, S.A.; Zhu, Q.; Shah, Z.I.; Wani, G.; Barakat, B.M.; Racoma, I.; El-Mahdy, M.A.; Wani, A.A. Thymoquinone up-regulates PTEN expression and induces apoptosis in doxorubicin-resistant human breast cancer cells. Mutat. Res. 2011, 706, 28–35. [Google Scholar] [CrossRef]
- Odeh, F.; Ismail, S.; Abu-Dahab, R.; Mahmoud, I.; Al Bawab, A. Thymoquinone in liposomes: A study of loading efficiency and biological activity towards breast cancer. Drug Deliv. 2012, 19, 371–377. [Google Scholar] [CrossRef]
- Ikonen, E. Cellular cholesterol trafficking and compartmentalization. Nat. Rev. Mol. Cell Biol. 2008, 9, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Murai, T. Cholesterol lowering: Role in cancer prevention and treatment. Biol. Chem. 2015, 396, 1–11. [Google Scholar] [CrossRef]
- Gorin, A.; Gabitova, L.; Astsaturov, I. Regulation of cholesterol biosynthesis and cancer signaling. Curr. Opin. Pharmacol. 2012, 12, 710–716. [Google Scholar] [CrossRef]
- Kuzu, O.F.; Gowda, R.; Noory, M.A.; Robertson, G.P. Modulating cancer cell survival by targeting intracellular cholesterol transport. Br. J. Cancer 2017, 117, 513–524. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Liu, X.; Liu, S.; Cao, Q. Cholesterol promotes the migration and invasion of renal carcinoma cells by regulating the KLF5/miR-27a/FBXW7 pathway. Biochem. Biophys. Res. Commun. 2018, 502, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Kuzu, O.F.; Noory, M.A.; Robertson, G.P. The Role of Cholesterol in Cancer. Cancer Res. 2016, 76, 2063. [Google Scholar] [CrossRef]
- Raccosta, L.; Fontana, R.; Corna, G.; Maggioni, D.; Moresco, M.; Russo, V. Cholesterol metabolites and tumor microenvironment: The road towards clinical translation. Cancer Immunol. Immunother. 2016, 65, 111–117. [Google Scholar] [CrossRef]
- Fararjeh, A.F.S.; Al Khader, A.; Kaddumi, E.; Obeidat, M.; Al-Fawares, O.l. Differential Expression and Prognostic Significance of STARD3 Gene in Breast Carcinoma. Int. J. Mol. Cell Med. 2021, 10, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Montero, J.; Morales, A.; Llacuna, L.; Lluis, J.M.; Terrones, O.; Basañez, G.; Antonsson, B.; Prieto, J.; García-Ruiz, C.; Colell, A.; et al. Mitochondrial Cholesterol Contributes to Chemotherapy Resistance in Hepatocellular Carcinoma. Cancer Res. 2008, 68, 5246–5256. [Google Scholar] [CrossRef]
- Vassilev, B.; Sihto, H.; Li, S.; Hölttä-Vuori, M.; Ilola, J.; Lundin, J.; Isola, J.; Kellokumpu-Lehtinen, P.-L.; Joensuu, e.; Ikonen, E. Elevated levels of StAR-related lipid transfer protein 3 alter cholesterol balance and adhesiveness of breast cancer cells: Potential mechanisms contributing to progression of HER2-positive breast cancers. Am. J. Pathol. 2015, 185, 987–1000. [Google Scholar] [CrossRef]
- Dong, F.; Mo, Z.; Eid, W.; Courtney, K.C.; Zha, X. Akt inhibition promotes ABCA1-mediated cholesterol efflux to ApoA-I through suppressing mTORC1. PLoS ONE 2014, 9, e113789. [Google Scholar] [CrossRef]
- Porstmann, T.; Santos, C.R.; Griffiths, B.; Cully, M.; Wu, M.; Leevers, S.; Griffiths, J.R.; Chung, Y.-L.; Schulze, A. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab. 2008, 8, 224–236. [Google Scholar] [CrossRef]
- Freed-Pastor, W.A.; Mizuno, H.; Zhao, X.; Langerød, A.; Moon, S.-H.; Rodriguez-Barrueco, R.; Barsotti, A.; Chicas, A.; Li, W.; Polotskaia, A.; et al. Mutant p53 Disrupts Mammary Tissue Architecture via the Mevalonate Pathway. Cell 2012, 148, 244–258. [Google Scholar] [CrossRef]
- Giró-Perafita, A.; Rabionet, M.; Planas, M.; Feliu, L.; Ciurana, J.; Ruiz-Martínez, S.; Puig, T. EGCG-Derivative G28 Shows High Efficacy Inhibiting the Mammosphere-Forming Capacity of Sensitive and Resistant TNBC Models. Molecules 2019, 24, 1027. [Google Scholar] [CrossRef]
- Giró-Perafita, A.; Palomeras, S.; Lum, D.H.; Blancafort, A.; Viñas, G.; Oliveras, G.; Pérez-Bueno, F.; Sarrats, A.; Welm, A.L.; Puig, T. Preclinical Evaluation of Fatty Acid Synthase and EGFR Inhibition in Triple-Negative Breast Cancer. Clin. Cancer Res. 2016, 22, 4687–4697. [Google Scholar] [CrossRef] [PubMed]
- Antalis, C.J.; Arnold, T.; Rasool, T.; Lee, B.; Buhman, K.K.; Siddiqui, R.A. High ACAT1 expression in estrogen receptor negative basal-like breast cancer cells is associated with LDL-induced proliferation. Breast Cancer Res. Treat. 2010, 122, 661–670. [Google Scholar] [CrossRef]
- Li, J.; Gu, D.; Lee, S.S.Y.; Song, B.; Bandyopadhyay, S.; Chen, S.; Konieczny, S.F.; Ratliff, T.L.; Liu, X.; Xie, J.; et al. Abrogating cholesterol esterification suppresses growth and metastasis of pancreatic cancer. Oncogene 2016, 35, 6378–6388. [Google Scholar] [CrossRef] [PubMed]
- Rajput, S.; Kumar, B.N.; Dey, K.K.; Pal, I.; Parekh, A.; Mandal, M. Molecular targeting of Akt by thymoquinone promotes G(1) arrest through translation inhibition of cyclin D1 and induces apoptosis in breast cancer cells. Life Sci. 2013, 93, 783–790. [Google Scholar] [CrossRef] [PubMed]
- Dastjerdi, M.N.; Mehdiabady, E.M.; Iranpour, F.G.; Bahramian, H. Effect of Thymoquinone on P53 Gene Expression and Consequence Apoptosis in Breast Cancer Cell Line. Int. J. Prev. Med. 2016, 7, 66. [Google Scholar] [CrossRef]
- Ray, P.D.; Huang, B.-W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef]
- Waris, G.; Ahsan, H. Reactive oxygen species: Role in the development of cancer and various chronic conditions. J. Carcinog. 2006, 5, 14. [Google Scholar] [CrossRef]
- Diebold, L.; Chandel, N.S. Mitochondrial ROS regulation of proliferating cells. Free Radic. Biol. Med. 2016, 100, 86–93. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-inducible factors in physiology and medicine. Cell 2012, 148, 399–408. [Google Scholar] [CrossRef]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef] [PubMed]
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef]
- Lee, A.C.; Fenster, B.E.; Ito, H.; Takeda, K.; Bae, N.S.; Hirai, T.; Yu, Z.X.; Ferrans, V.J.; Howard, B.H.; Finkel, T. Ras proteins induce senescence by altering the intracellular levels of reactive oxygen species. J. Biol. Chem. 1999, 274, 7936–7940. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.-S.; Hong, M.-Z.; Ren, J.-L. Reactive oxygen species: A double-edged sword in oncogenesis. World J. Gastroenterol. 2009, 15, 1702–1707. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, V.; Tuli, H.S.; Varol, A.; Thakral, F.; Yerer, M.B.; Sak, K.; Varol, M.; Jain, A.; Khan, M.A.; Sethi, G. Role of Reactive Oxygen Species in Cancer Progression: Molecular Mechanisms and Recent Advancements. Biomolecules 2019, 9, 735. [Google Scholar] [CrossRef]
- Kodama, R.; Kato, M.; Furuta, S.; Ueno, S.; Zhang, Y.; Matsuno, K.; Yabe-Nishimura, C.; Tanaka, E.; Kamata, T. ROS-generating oxidases Nox1 and Nox4 contribute to oncogenic Ras-induced premature senescence. Genes Cells 2013, 18, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Scherz-Shouval, R.; Elazar, Z. Regulation of autophagy by ROS: Physiology and pathology. Trends Biochem. Sci. 2011, 36, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Chandra, J.; Samali, A.; Orrenius, S. Triggering and modulation of apoptosis by oxidative stress. Free Radic. Biol. Med. 2000, 29, 323–333. [Google Scholar] [CrossRef]
- López-Lázaro, M. Dual role of hydrogen peroxide in cancer: Possible relevance to cancer chemoprevention and therapy. Cancer Lett. 2007, 252, 1–8. [Google Scholar] [CrossRef]
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in cancer therapy: The bright side of the moon. Exp. Mol. Med. 2020, 52, 192–203. [Google Scholar] [CrossRef]
- Son, Y.; Cheong, Y.-K.; Kim, N.-H.; Chung, H.-T.; Kang, D.G.; Pae, H.-O. Mitogen-Activated Protein Kinases and Reactive Oxygen Species: How Can ROS Activate MAPK Pathways? J. Signal. Transduct. 2011, 2011, 792639. [Google Scholar] [CrossRef]
- Leslie, N.R.; Bennett, D.; Lindsay, Y.E.; Stewart, H.; Gray, A.; Downes, C.P. Redox regulation of PI 3-kinase signalling via inactivation of PTEN. EMBO J. 2003, 22, 5501–5510. [Google Scholar] [CrossRef]
- Bernatoniene, J.; Kopustinskiene, D.M. The Role of Catechins in Cellular Responses to Oxidative Stress. Molecules 2018, 23, 965. [Google Scholar] [CrossRef]
- Lee, C.H.; Huang, C.W.; Chang, P.C.; Shiau, J.P.; Lin, I.P.; Lin, M.Y.; Chen, C.Y. Reactive oxygen species mediate the chemopreventive effects of syringin in breast cancer cells. Phytomedicine 2019, 61, 152844. [Google Scholar] [CrossRef]
- Zhou, F.; Feng, B.; Wang, T.; Wang, D.; Cui, Z.; Wang, S.; Ding, C.; Zhang, Z.; Liu, J.; Yu, H.; et al. Theranostic Prodrug Vesicles for Reactive Oxygen Species-Triggered Ultrafast Drug Release and Local-Regional Therapy of Metastatic Triple-Negative Breast Cancer. Adv. Funct. Mater. 2017, 27, 1703674. [Google Scholar] [CrossRef]
- Khader, M.; Eckl, P.M. Thymoquinone: An emerging natural drug with a wide range of medical applications. Iran. J. Basic Med. Sci. 2014, 17, 950–957. [Google Scholar]
- Khan, M.A.; Tania, M.; Fu, S.; Fu, J. Thymoquinone, as an anticancer molecule: From basic research to clinical investigation. Oncotarget 2017, 8, 1907–51919. [Google Scholar]
- Kassab, R.B.; El-Hennamy, R.E. The role of thymoquinone as a potent antioxidant in ameliorating the neurotoxic effect of sodium arsenate in female rat. Egypt. J. Basic Appl. Sci. 2017, 4, 160–167. [Google Scholar] [CrossRef]
- Sayed-Ahmed, M.M.; Aleisa, A.M.; Al-Rejaie, S.S.; Al-Yahya, A.A.; Al-Shabanah, O.A.; Hafez, M.M.; Nagi, M.N. Thymoquinone attenuates diethylnitrosamine induction of hepatic carcinogenesis through antioxidant signaling. Oxid. Med. Cell Longev. 2010, 3, 254–261. [Google Scholar] [CrossRef]
- Badary, O.A.; Taha, R.A.; Gamal el-Din, A.M.; Abdel-Wahab, M.H. Thymoquinone is a potent superoxide anion scavenger. Drug Chem. Toxicol. 2003, 26, 87–98. [Google Scholar] [CrossRef]
- Kundu, J.; Kim, D.-H.; Kundu, J.K.; Chun, K.-S. Thymoquinone induces heme oxygenase-1 expression in HaCaT cells via Nrf2/ARE activation: Akt and AMPKα as upstream targets. Food Chem. Toxicol. 2014, 65, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Ushio-Fukai, M.; Ash, D.; Nagarkoti, S.; Belin de Chantemèle, E.J.; Fulton, D.J.R.; Fukai, T. Interplay Between Reactive Oxygen/Reactive Nitrogen Species and Metabolism in Vascular Biology and Disease. Antioxid. Redox Signal. 2021, 34, 1319–1354. [Google Scholar] [CrossRef] [PubMed]
- Ryazanov, A.G. Ca2+/calmodulin-dependent phosphorylation of elongation factor 2. FEBS Lett. 1987, 214, 331–334. [Google Scholar] [CrossRef]
- Leprivier, G.; Remke, M.; Rotblat, B.; Dubuc, A.; Mateo, A.-R.F.; Kool, M.; Agnihotri, S.; El-Naggar, A.; Yu, B.; Somasekharan, S.P.; et al. The eEF2 Kinase Confers Resistance to Nutrient Deprivation by Blocking Translation Elongation. Cell 2013, 153, 1064–1079. [Google Scholar] [CrossRef]
- Kenney, J.W.; Moore, C.E.; Wang, X.; Proud, C.G. Eukaryotic elongation factor 2 kinase, an unusual enzyme with multiple roles. Adv. Biol. Regul. 2014, 55, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.H.; Riazy, M.; Smith, E.M.; Proud, C.G.; Steinbrecher, U.P.; Duronio, V. Oxidized LDL-mediated macrophage survival involves elongation factor-2 kinase. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 92–98. [Google Scholar] [CrossRef]
- Xu, Z.; Zhao, L.; Yang, X.; Ma, S.; Ge, Y.; Liu, Y.; Liu, S.; Shi, J.; Zheng, D. Mmu-miR-125b overexpression suppresses NO production in activated macrophages by targeting eEF2K and CCNA2. BMC Cancer 2016, 16, 252. [Google Scholar] [CrossRef][Green Version]
- Moore, C.E.; Mikolajek, H.; Regufe da Mota, S.; Wang, X.; Kenney, J.W.; Werner, J.M.; Proud, C.G. Elongation Factor 2 Kinase Is Regulated by Proline Hydroxylation and Protects Cells during Hypoxia. Mol. Cell Biol. 2015, 35, 1788–1804. [Google Scholar] [CrossRef]
- Kenney, J.W.; Genheden, M.; Moon, K.M.; Wang, X.; Foster, L.J.; Proud, C.G. Eukaryotic elongation factor 2 kinase regulates the synthesis of microtubule-related proteins in neurons. J. Neurochem. 2016, 136, 276–284. [Google Scholar] [CrossRef]
- Neri, D.; Supuran, C.T. Interfering with pH regulation in tumours as a therapeutic strategy. Nat. Rev. Drug Discov. 2011, 10, 767–777. [Google Scholar] [CrossRef]
- Xie, J.; Mikolajek, H.; Pigott, C.R.; Hooper, K.J.; Mellows, T.; Moore, C.E.; Mohammed, H.; Werner, J.M.; Thomas, G.J.; Proud, C.G. Molecular Mechanism for the Control of Eukaryotic Elongation Factor 2 Kinase by pH: Role in Cancer Cell Survival. Mol. Cell. Biol. 2015, 35, 1805–1824. [Google Scholar] [CrossRef]
- Gschwendt, M.; Muller, H.J.; Kielbassa, K.; Zang, R.; Kittstein, W.; Rincke, G.; Marks, F. Rottlerin, a Novel Protein Kinase Inhibitor. Biochem. Biophys. Res. Commun. 1994, 199, 93–98. [Google Scholar] [CrossRef]
- Gschwendt, M.; Kittstein, W.; Marks, F. Elongation factor-2 kinase: Effective inhibition by the novel protein kinase inhibitor rottlerin and relative insensitivity towards staurosporine. FEBS Lett. 1994, 338, 85–88. [Google Scholar] [CrossRef]
- Tekedereli, I.; Alpay, S.N.; Tavares, C.D.J.; Cobanoglu, Z.E.; Kaoud, T.S.; Sahin, I.; Sood, A.K.; Lopez-Berestein, G.; Dalby, K.N.; Ozpolat, B. Targeted silencing of elongation factor 2 kinase suppresses growth and sensitizes tumors to doxorubicin in an orthotopic model of breast cancer. PLoS ONE 2012, 7, e41171. [Google Scholar] [CrossRef] [PubMed]
- Bayraktar, R.; Pichler, M.; Kanlikilicer, P.; Ivan, C.; Bayraktar, E.; Kahraman, N.; Aslan, B.; Oguztuzun, S.; Ulasli, M.; Arslan, A.; et al. MicroRNA 603 acts as a tumor suppressor and inhibits triple-negative breast cancer tumorigenesis by targeting elongation factor 2 kinase. Oncotarget 2017, 8, 11641–11658. [Google Scholar] [CrossRef]
- Kabil, N.; Bayraktar, R.; Kahraman, N.; Mokhlis, H.A.; Calin, G.A.; Lopez-Berestein, G.; Ozpolat, B. Thymoquinone inhibits cell proliferation, migration, and invasion by regulating the elongation factor 2 kinase (eEF-2K) signaling axis in triple-negative breast cancer. Breast Cancer Res. Treat. 2018, 171, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F.; Mantovani, A. Inflammation and cancer: Back to Virchow? Lancet 2001, 357, 539–545. [Google Scholar] [CrossRef]
- Mantovani, A.; Bussolino, F.; Dejana, E. Cytokine regulation of endothelial cell function. FASEB J. 1992, 6, 2591–2599. [Google Scholar] [CrossRef]
- Pathria, P.; Louis, T.L.; Varner, J.A. Targeting Tumor-Associated Macrophages in Cancer. Trends Immunol. 2019, 40, 177. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.-Y.; Luo, R.-Z.; Peng, R.-J.; Wang, S.-S.; Xue, C. High infiltration of tumor-associated macrophages in triple-negative breast cancer is associated with a higher risk of distant metastasis. Onco Targets Ther. 2014, 7, 1475–1480. [Google Scholar] [CrossRef] [PubMed]
- Leek, R.D.; Lewis, C.E.; Whitehouse, R.; Greenall, M.; Clarke, J.; Harris, A.L. Association of Macrophage Infiltration with Angiogenesis and Prognosis in Invasive Breast Carcinoma. Cancer Res. 1996, 56, 4625–4629. [Google Scholar]
- Priceman, S.J.; Sung, J.L.; Shaposhnik, Z.; Burton, J.B.; Torres-Collado, A.X.; Moughon, D.L.; Johnson, M.; Lusis, A.J.; Cohen, D.A.; Iruela-Arispe, M.L.; et al. Targeting distinct tumor-infiltrating myeloid cells by inhibiting CSF-1 receptor: Combating tumor evasion of antiangiogenic therapy. Blood 2010, 115, 1461–1471. [Google Scholar] [CrossRef] [PubMed]
- Harney, A.S.; Arwert, E.N.; Entenberg, D.; Wang, Y. Real-Time Imaging Reveals Local, Transient Vascular Permeability, and Tumor Cell Intravasation Stimulated by TIE2hi Macrophage-Derived VEGFA. Cancer Discov. 2015, 5, 932–943. [Google Scholar] [CrossRef] [PubMed]
- Hughes, R.; Qian, B.-Z.; Rowan, C.; Muthana, M.; Keklikoglou, I.; Olson, O.C.; Tazzyman, S.; Danson, S.; Addison, C.; Clemons, M.; et al. Perivascular M2 Macrophages Stimulate Tumor Relapse after Chemotherapy. Cancer Res. 2015, 75, 3479–3491. [Google Scholar] [CrossRef]
- Li, H.; Yang, B.; Huang, J.; Lin, Y.; Xiang, T.; Wan, J.; Li, H.; Chouaib, S.; Ren, G. Cyclooxygenase-2 in tumor-associated macrophages promotes breast cancer cell survival by triggering a positive-feedback loop between macrophages and cancer cells. Oncotarget 2015, 6, 29637–29650. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.-Z.; Zhang, H.; Li, J.; He, T.; Yeo, E.-J.; Soong, D.Y.H.; Carragher, N.O.; Munro, A.; Chang, A.; Bresnick, A.R.; et al. FLT1 signaling in metastasis-associated macrophages activates an inflammatory signature that promotes breast cancer metastasis. J. Exp. Med. 2015, 212, 1433–1448. [Google Scholar] [CrossRef]
- Qian, B.Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef]
- Hollmén, M.; Roudnicky, F.; Karaman, S.; Detmar, M. Characterization of macrophage--cancer cell crosstalk in estrogen receptor positive and triple-negative breast cancer. Sci. Rep. 2015, 5, 9188. [Google Scholar] [CrossRef]
- Robinson, S.C.; Scott, K.A.; Wilson, J.L.; Thompson, R.G.; Proudfoot, A.E.I.; Balkwill, F.R. A Chemokine Receptor Antagonist Inhibits Experimental Breast Tumor Growth. Cancer Res. 2003, 63, 8360. [Google Scholar]
- Vukanovic, J.; Isaacs, J.T. Linomide Inhibits Angiogenesis, Growth, Metastasis, and Macrophage Infiltration within Rat Prostatic Cancers. Cancer Res. 1995, 55, 1499. [Google Scholar]
- Whitehurst, B.; Flister, M.J.; Bagaitkar, J.; Volk, L.; Bivens, C.M.; Pickett, B.; Castro-Rivera, E.; Brekken, R.A.; Gerard, R.D.; Ran, S. Anti-VEGF-A therapy reduces lymphatic vessel density and expression of VEGFR-3 in an orthotopic breast tumor model. Int. J. Cancer 2007, 121, 2181–2191. [Google Scholar] [CrossRef]
- Dineen, S.P.; Lynn, K.D.; Holloway, S.E.; Miller, A.F.; Sullivan, J.P.; Shames, D.S.; Beck, A.W.; Barnett, C.C.; Fleming, J.B.; Brekken, R.A. Vascular Endothelial Growth Factor Receptor 2 Mediates Macrophage Infiltration into Orthotopic Pancreatic Tumors in Mice. Cancer Res. 2008, 68, 4340. [Google Scholar] [CrossRef] [PubMed]
- El-Mahmoudy, A.; Matsuyama, H.; Borgan, M.A.; Shimizu, Y.; El-Sayed, M.G.; Minamoto, N.; Takewaki, T. Thymoquinone suppresses expression of inducible nitric oxide synthase in rat macrophages. Int. Immunopharmacol. 2002, 2, 1603–1611. [Google Scholar] [CrossRef]
- Wilson, A.J.; Saskowski, J.; Barham, W.; Khabele, D.; Yull, F. Microenvironmental effects limit efficacy of thymoquinone treatment in a mouse model of ovarian cancer. Mol. Cancer 2015, 14, 192. [Google Scholar] [CrossRef]
- Paramasivam, A.; Kalaimangai, M.; Sambantham, S.; Anandan, B.; Jayaraman, G. Anti-angiogenic activity of thymoquinone by the down-regulation of VEGF using zebrafish (Danio rerio) model. Biomed. Prev. Nutr. 2012, 2, 169–173. [Google Scholar] [CrossRef]
- Shanmugam, M.K.; Ahn, K.S.; Hsu, A.; Woo, C.C.; Yuan, Y.; Tan, K.H.B.; Chinnathambi, A.; Alahmadi, T.A.; Alharbi, S.A.; Koh, A.P.F.; et al. Thymoquinone Inhibits Bone Metastasis of Breast Cancer Cells Through Abrogation of the CXCR4 Signaling Axis. Front. Pharmacol. 2018, 9, 1294. [Google Scholar] [CrossRef] [PubMed]
- Nieman, K.M.; Romero, I.L.; Houten, B.V.; Lengyel, E. Adipose tissue and adipocytes support tumorigenesis and metastasis. Biochim. Biophys. Acta 2013, 1831, 1533–1541. [Google Scholar] [CrossRef]
- Poulos, S.P.; Hausman, D.B.; Hausman, G.J. The development and endocrine functions of adipose tissue. Mol. Cell Endocrinol. 2010, 323, 20–34. [Google Scholar] [CrossRef]
- Vona-Davis, L.; Rose, D.P. Adipokines as endocrine, paracrine, and autocrine factors in breast cancer risk and progression. Endocr. Relat. Cancer 2007, 14, 189–206. [Google Scholar] [CrossRef]
- Bifulco, M.; Pisanti, S. “Adiponcosis”: A new term to name the obesity and cancer link. J. Clin. Endocrinol. Metab. 2013, 98, 4664–4665. [Google Scholar] [CrossRef]
- Dirat, B.; Bochet, L.; Dabek, M.; Daviaud, D.; Dauvillier, S.; Majed, B.; Wang, Y.Y.; Meulle, A.; Salles, B.; Gonidec, S.L.; et al. Cancer-Associated Adipocytes Exhibit an Activated Phenotype and Contribute to Breast Cancer Invasion. Cancer Res. 2011, 71, 2455–2465. [Google Scholar] [CrossRef]
- Wagner, M.; Bjerkvig, R.; Wiig, H.; Melero-Martin, J.M.; Lin, R.-Z.; Klagsbrun, M.; Dudley, A.C. Inflamed tumor-associated adipose tissue is a depot for macrophages that stimulate tumor growth and angiogenesis. Angiogenesis 2012, 15, 481–495. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Li, B.; Li, Z.; Li, J.; Sun, S.; Sun, S. Cancer-associated adipocytes: Key players in breast cancer progression. J. Hematol. Oncol. 2019, 12, 95. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Tiruthani, K.; Wang, M.; Zhou, X.; Qiu, N.; Xiong, Y.; Pecot, C.V.; Liu, R.; Huang, L. Tumor-targeted gene therapy with lipid nanoparticles inhibits tumor-associated adipocytes and remodels the immunosuppressive tumor microenvironment in triple-negative breast cancer. Nanoscale Horiz. 2021, 6, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Palma, M.D.; Biziato, D.; Petrova, T.V. Microenvironmental regulation of tumour angiogenesis. Nat. Rev. Cancer 2017, 17, 457–474. [Google Scholar] [CrossRef]
- D’Esposito, V.; Liguoro, D.; Ambrosio, M.R.; Collina, F.; Cantile, M.; Spinelli, R.; Raciti, G.A.; Miele, C.; Valentino, R.; Campiglia, P.; et al. Adipose microenvironment promotes triple negative breast cancer cell invasiveness and dissemination by producing CCL5. Oncotarget 2016, 7, 24495–24509. [Google Scholar] [CrossRef]
- Choia, J.; Chab, Y.J.; Koo, J.S. Adipocyte biology in breast cancer: From silent bystander to active facilitator. Prog. Lipid Res. 2018, 69, 11–20. [Google Scholar] [CrossRef]
- Pandolfi, J.B.; Ferraro, A.A.; Sananez, I.; Gancedo, M.C.; Baz, P.; Billordo, L.A.; Fainboim, L.; Arruvito, L. ATP-induced inflammation drives tissue-resident Th17 cells in metabolically unhealthy obesity. J. Immunol. 2016, 196, 3287–3296. [Google Scholar] [CrossRef]
- Shaterzadeh-Yazdi, H.; Noorbakhsh, M.-F.; Hayati, F.; Samarghandian, S.; Farkhondeh, T. Immunomodulatory and Anti-inflammatory Effects of Thymoquinone. Cardiovasc. Hematol. Disord Drug Targets 2018, 18, 52–60. [Google Scholar] [CrossRef]
- Pannu, A.; Goyal, R.K.; Ojha, S.; Nandave, M. Chapter 33—Therapeutic Potential of Thymoquinone in Treatment of Rheumatoid Arthritis and Related Autoimmune Diseases. In Bioactive Food as Dietary Interventions for Arthritis and Related Inflammatory Diseases, 2nd ed.; Watson, R.R., Preedy, V.R., Eds.; Academic Press: Cambridge, MA, USA, 2019; pp. 575–587. [Google Scholar] [CrossRef]
- Shen, H.H.; Peterson, S.J.; Bellner, L.; Choudhary, A.; Levy, L.; Gancz, L.; Sasson, A.; Trainer, J.; Rezzani, R.; Resnick, A.; et al. Cold-Pressed Nigella Sativa Oil Standardized to 3% Thymoquinone Potentiates Omega-3 Protection against Obesity-Induced Oxidative Stress, Inflammation, and Markers of Insulin Resistance Accompanied with Conversion of White to Beige Fat in Mice. Antioxidants 2020, 9, 489. [Google Scholar] [CrossRef] [PubMed]
- Virchow, R. As Based upon Physiological and Pathological Histology. Nutr. Rev. 1989, 47, 23–25. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Zeisberg, M. Fibroblasts in cancer. Nat. Rev. Cancer 2006, 6, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Tuxhorn, J.A.; McAlhany, S.J.; Dang, T.D.; Ayala, G.E.; Rowley, D.R. Stromal Cells Promote Angiogenesis and Growth of Human Prostate Tumors in a Differential Reactive Stroma (DRS) Xenograft Model. Cancer Res. 2002, 62, 3298–3307. [Google Scholar] [PubMed]
- Gerber, H.P.; Kowalski, J.; Sherman, D.; Eberhard, D.A.; Ferrara, N. Complete inhibition of rhabdomyosarcoma xenograft growth and neovascularization requires blockade of both tumor and host vascular endothelial growth factor. Cancer Res. 2000, 60, 6253–6258. [Google Scholar] [PubMed]
- Crawford, Y.; Kasman, I.; Yu, L.; Zhong, C.; Wu, X.; Modrusan, Z.; Kaminker, J.; Ferrara, N. PDGF-C mediates the angiogenic and tumorigenic properties of fibroblasts associated with tumors refractory to anti-VEGF treatment. Cancer Cell 2009, 15, 21–34. [Google Scholar] [CrossRef]
- Pietras, K.; Pahler, J.; Bergers, G.; Hanahan, D. Functions of paracrine PDGF signaling in the proangiogenic tumor stroma revealed by pharmacological targeting. PLoS Med. 2008, 5, e19. [Google Scholar] [CrossRef]
- Egeblad, M.; Rasch, M.G.; Weaver, V.M. Dynamic interplay between the collagen scaffold and tumor evolution. Curr. Opin. Cell Biol. 2010, 22, 697–706. [Google Scholar] [CrossRef]
- Orimo, A.; Gupta, P.B.; Sgroi, D.C.; Arenzana-Seisdedos, F.; Delaunay, T.; Naeem, R.; Carey, V.J.; Richardson, A.L.; Weinberg, R.A. Stromal Fibroblasts Present in Invasive Human Breast Carcinomas Promote Tumor Growth and Angiogenesis through Elevated SDF-1/CXCL12 Secretion. Cell 2005, 121, 335–348. [Google Scholar] [CrossRef]
- Wang, M.; Zhang, J.; Huang, Y.; Ji, S.; Shao, G.; Feng, S.; Chen, D.; Zhao, K.; Wang, Z.; Wu, A. Cancer-Associated Fibroblasts Autophagy Enhances Progression of Triple-Negative Breast Cancer Cells. Med. Sci. Monit. 2017, 23, 3904–3912. [Google Scholar] [CrossRef]
- Yang, Y.; Wu, H.; Fan, S.; Bi, Y.; Hao, M.; Shang, J. Cancer-associated fibroblast-derived LRRC15 promotes the migration and invasion of triple-negative breast cancer cells via Wnt/β-catenin signalling pathway regulation. Mol. Med. Rep. 2022, 25, 2. [Google Scholar] [CrossRef]
- Bhowmick, N.A.; Chytil, A.; Plieth, D.; Gorska, A.E.; Dumont, N.; Shappell, S.; Washington, M.K.; Neilson, E.G.; Moses, H.L. TGF-beta signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. Science 2004, 3030, 848–851. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Zhang, L.; He, X.; Zhang, P.; Sun, C.; Xu, X.; Lu, Y.; Li, F. TGF-β plays a vital role in triple-negative breast cancer (TNBC) drug-resistance through regulating stemness, EMT and apoptosis. Biochem. Biophys. Res. Commun. 2018, 502, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Takai, K.; Le, A.; Weaver, V.M.; Werb, Z. Targeting the cancer-associated fibroblasts as a treatment in triple-negative breast cancer. Oncotarget 2016, 7, 82889–82901. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Xiang, Y.; Zhao, X.; Cai, C.; Chen, H.; Jiang, W.; Wang, Y.; Zeng, C. Thymoquinone suppresses platelet-derived growth factor-BB–induced vascular smooth muscle cell proliferation, migration and neointimal formation. J. Cell. Mol. Med. 2019, 23, 8482–8492. [Google Scholar] [CrossRef]
- Imrana, M.; Raufb, A.; Khanc, I.A.; Shahbazd, M.; Qaisranie, T.B.; Fatmawatif, S.; Abu-Izneidg, T.; Imranh, A.; Rahmanb, K.U.; Gondal, T.A. Thymoquinone: A novel strategy to combat cancer: A review. Biomed. Pharmacother. 2018, 106, 390–402. [Google Scholar] [CrossRef]
- Haiaty, S.; Rashidi, M.-R.; Akbarzadeh, M.; Bazmani, A.; Mostafazadeh, M.; Nikanfar, S.; Zibaei, Z.; Rahbarghazi, R.; Nouri, M. Thymoquinone inhibited vasculogenic capacity and promoted mesenchymal-epithelial transition of human breast cancer stem cells. BMC Complement. Med. Ther. 2021, 21, 83. [Google Scholar] [CrossRef]
- Cruz-Merino, L.d.l.; Barco-Sánchez, A.; Carrasco, F.H.; Fernández, E.N.; Benítez, A.V.; Molina, J.B.; Peinado, A.M.; López, A.G.; Borrego, M.R.; Villena, M.C.M.d.; et al. New insights into the role of the immune microenvironment in breast carcinoma. Clin. Dev. Immunol. 2013, 2013, 785317. [Google Scholar] [CrossRef]
- Stanton, S.E.; Disis, M.L. Clinical significance of tumor-infiltrating lymphocytes in breast cancer. J. Immunother. Cancer 2016, 4, 59. [Google Scholar] [CrossRef]
- Dieci, M.V.; Mathieu, M.C.; Guarneri, V.; Conte, P.; Delaloge, S.; Andre, F.; Goubar, A. Prognostic and predictive value of tumor-infiltrating lymphocytes in two phase III randomized adjuvant breast cancer trials. Ann. Oncol. 2015, 26, 1698–1704. [Google Scholar] [CrossRef]
- Loi, S.; Sirtaine, N.; Piette, F.; Salgado, R.; Viale, G.; Eenoo, F.V.; Rouas, G.; Francis, P.; Crown, J.P.A.; Hitre, E.; et al. Prognostic and predictive value of tumor-infiltrating lymphocytes in a phase III randomized adjuvant breast cancer trial in node-positive breast cancer comparing the addition of docetaxel to doxorubicin with doxorubicin-based chemotherapy: BIG 02-98. J. Clin. Oncol. 2013, 31, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Adams, S.; Gray, R.J.; Demaria, S.; Goldstein, L.; Perez, E.A.; Shulman, L.N.; Martino, S.; Wang, M.; Jones, V.E.; Saphner, T.J.; et al. Prognostic Value of Tumor-Infiltrating Lymphocytes in Triple-Negative Breast Cancers From Two Phase III Randomized Adjuvant Breast Cancer Trials: ECOG 2197 and ECOG 1199. J. Clin. Oncol. 2014, 32, 2959–2966. [Google Scholar] [CrossRef] [PubMed]
- Loi, S.; Dushyanthen, S.; Beavis, P.A.; Salgado, R.; Denkert, C.; Savas, P.; Combs, S.; Rimm, D.L.; Giltnane, J.M.; Estrada, M.V.; et al. RAS/MAPK activation is associated with reduced tumor-infiltrating lymphocytes in triple-negative breast cancer: Therapeutic cooperation between MEK and PD-1/PD-L1 immune checkpoint inhibitors. Clin. Cancer Res. 2016, 22, 1499–1509. [Google Scholar] [CrossRef] [PubMed]
- Guida, M.S.; El-Aal, A.A.; Kafafy, Y.; Salama, S.F.; Badr, B.M.; Badr, G. Thymoquinone Rescues T Lymphocytes from Gamma Irradiation-Induced Apoptosis and Exhaustion by Modulating Pro-Inflammatory Cytokine Levels and PD-1, Bax, and Bcl-2 Signaling. Cell. Physiol. Biochem. 2016, 38, 786–800. [Google Scholar] [CrossRef]
- Kheirouri, S.; Hadi, V.; Alizadeh, M. Immunomodulatory Effect of Nigella sativa Oil on T Lymphocytes in Patients with Rheumatoid Arthritis. Immunol. Investig. 2016, 45, 271–283. [Google Scholar] [CrossRef]
- Badr, G.; Alwasel, S.; Ebaid, H.; Mohany, M.; Alhazza, I. Perinatal supplementation with thymoquinone improves diabetic complications and T cell immune responses in rat offspring. Cell. Immunol. 2011, 267, 133–140. [Google Scholar] [CrossRef]
- Zheng, X.; Jin, W.; Wang, S.; Ding, H. Progression on the Roles and Mechanisms of Tumor-Infiltrating T Lymphocytes in Patients With Hepatocellular Carcinoma. Front. Immunol. 2021, 12, 729705. [Google Scholar] [CrossRef]
- Loi, S.; Michiels, S.; Salgado, R.; Sirtaine, N.; Jose, V.; Fumagalli, D.; KellokumpuLehtinen, P.-L.; Bono, V.K.; Desmedt, C.; Piccart, M.J.; et al. Tumor infiltrating lymphocytes are prognostic in triple negative breast cancer and predictive for trastuzumab benefit in early breast cancer: Results from the FinHER trial. Ann. Oncol. 2014, 25, 1544–1550. [Google Scholar] [CrossRef]
- Nicolini, A.; Carpi, A.; Rossi, G. Cytokine & Growth Factor Reviews. Cytokine Growth Factor Rev. 2006, 17, 325–337. [Google Scholar]
- Karin, M.; Greten, F.R. NF-kappaB: Linking inflammation and immunity to cancer development and progression. Nat. Rev. Immunol. 2005, 5, 749–759. [Google Scholar] [CrossRef]
- Kuilman, T.; Michaloglou, C.; Vredeveld, L.C.W.; Douma, S.; Doorn, R.v.; Desmet, C.J.; Aarden, L.A.; Mooi, W.J.; Peeper, D.S. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 2008, 133, 1019–1031. [Google Scholar] [CrossRef]
- Guo, Y.; Xu, F.; Lu, T.; Duan, Z.; Zhang, Z. Interleukin-6 signaling pathway in targeted therapy for cancer. Cancer Treat. Rev. 2012, 38, 904–910. [Google Scholar] [CrossRef]
- Bharti, R.; Dey, G.; Mandal, M. Cancer development, chemoresistance, epithelial to mesenchymal transition and stem cells: A snapshot of IL-6 mediated involvement. Cancer Lett. 2016, 375, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Tunon, I.; Ricote, M.; Ruiz, A.; Fraile, B.; Paniagua, R.; Royuela, M. IL-6, its receptors and its relationship with bcl-2 and bax proteins in infiltrating and in situ human breast carcinoma. Histopathology 2005, 47, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Hartman, Z.C.; Poage, G.M.; Hollander, P.d.; Tsimelzon, A.; Hill, J.; Panupinthu, N.; Zhang, Y.; Mazumdar, A.; Hilsenbeck, S.G.; Mills, G.B.; et al. Growth of triple-negative breast cancer cells relies upon coordinate autocrine expression of the pro-inflammatory cytokines IL-6 and IL-8. Cancer Res. 2013, 73, 3470–3480. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Fertig, E.J.; Jin, K.; Sukumar, S.; Pandey, N.B.; Popela, A.S. Breast cancer cells condition lymphatic endothelial cells within pre-metastatic niches to promote metastasis. Nat. Commun. 2014, 5, 4715. [Google Scholar] [CrossRef] [PubMed]
- Won, H.S.; Kim, Y.A.; Lee, J.S.; Jeon, E.K.; An, H.J.; Sun, D.S.; Ko, Y.H.; Kim, J.S. Soluble Interleukin-6 Receptor is a Prognostic Marker for Relapse-Free Survival in Estrogen Receptor-Positive Breast Cancer. J. Cancer Investig. 2013, 31, 516–521. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.A.; Scheller, J.; Rose-John, S. Therapeutic strategies for the clinical blockade of IL-6/gp130 signaling. J. Clin. Investig. 2011, 121, 3375–3383. [Google Scholar] [CrossRef] [PubMed]
- Saini, U.; Naidu, S.; ElNaggar, A.C.; Bid, H.K.; Wallbillich, J.J.; Bixel, K.; Bolyard, C.; Suarez, A.A.; Kaur, B.; Kuppusamy, P.; et al. Elevated STAT3 expression in ovarian cancer ascites promotes invasion and metastasis: A potential therapeutic target. Oncogene 2017, 36, 168–181. [Google Scholar] [CrossRef]
- Jin, K.; Pandey, N.B.; Popel, A.S. Simultaneous blockade of IL-6 and CCL5 signaling for synergistic inhibition of triple-negative breast cancer growth and metastasis. Breast Cancer Res. Treat. 2018, 20, 54. [Google Scholar] [CrossRef]
- Oshima, T.; Cao, X.; Grande, F.; Yamada, R.; Garofalo, A.; Louie, S.; Neamati, N. Combination effects of SC144 and cytotoxic anticancer agents. Anticancer Drugs 2009, 20, 312–320. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Grande, F.; Garofalo, A.; Neamati, N. Discovery of a Novel Orally Active Small-Molecule gp130 Inhibitor for the Treatment of Ovarian Cancer. Mol. Cancer Ther. 2013, 12, 937–949. [Google Scholar] [CrossRef]
- Heo, T.-H.; Wahler, J.; Suh, N. Potential therapeutic implications of IL-6/IL-6R/gp130-targeting agents in breast cancer. Oncotarget 2016, 7, 15460–15473. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Rajendran, P.; Sethi, G. Thymoquinone inhibits proliferation, induces apoptosis and chemosensitizes human multiple myeloma cells through suppression of signal transducer and activator of transcription 3 activation pathway. Br. J. Pharmacol. 2010, 161, 541–554. [Google Scholar] [CrossRef] [PubMed]
- Tu, Y.; Gardner, A.; Lichtenstein, A. The phosphatidylinositol 3-kinase/AKT kinase pathway in multiple myeloma plasma cells: Roles in cytokine-dependent survival and proliferative responses. Cancer Res. 2000, 60, 6763–6770. [Google Scholar] [PubMed]
- Shuai, K.; Liu, B. Regulation of JAK-STAT signalling in the immune system. Nat. Rev. Immunol. 2003, 3, 900–911. [Google Scholar] [CrossRef]
- Sonnenblick, A.; Uziely, B.; Nechushtan, H.; Kadouri, L.; Galun, E.; Axelrod, J.H.; Katz, D.; Daum, H.; Hamburger, T.; Maly, B.; et al. Tumor STAT3 tyrosine phosphorylation status, as a predictor of benefit from adjuvant chemotherapy for breast cancer. Breast Cancer Res. Treat. 2013, 138, 407–413. [Google Scholar] [CrossRef]
- Shields, B.J.; Wiede, F.; Gurzov, E.N.; Wee, K.; Hauser, C.; Zhu, H.-J.; Molloy, T.J.; O’Toole, S.A.; Daly, R.J.; Sutherland, R.L.; et al. TCPTP Regulates SFK and STAT3 Signaling and Is Lost in Triple-Negative Breast Cancers. Mol. Cell. Biol. 2013, 3, 557–570. [Google Scholar] [CrossRef]
- Yokogami, K.; Wakisaka, S.; Avruch, J.; Reeves, S.A. Serine phosphorylation and maximal activation of STAT3 during CNTF signaling is mediated by the rapamycin target mTOR. Curr. Biol. 2000, 10, 47–50. [Google Scholar] [CrossRef]
- Sutherland, K.D.; Lindeman, G.J.; Choong, D.Y.H.; Wittlin, S.; Brentzell, L.; Phillips, W.; Campbell, I.G.; Visvader, J.E. Differential hypermethylation of SOCS genes in ovarian and breast carcinomas. Oncogene 2004, 23, 7726–7733. [Google Scholar] [CrossRef]
- Bachelot, T.; Ray-Coquard, I.; Menetrier-Caux, C.; Rastkha, M.; Duc, A.; Blay, J.-Y. Prognostic value of serum levels of interleukin 6 and of serum and plasma levels of vascular endothelial growth factor in hormone-refractory metastatic breast cancer patients. Br. J. Cancer 2003, 88, 1721–1726. [Google Scholar] [CrossRef]
- Bowman, T.; Garcia, R.; Turkson, J.; Jove, R. STATs in oncogenesis. Oncogene 2000, 19, 2474–2488. [Google Scholar] [CrossRef]
- Yu, H.; Jove, R. The STATs of cancer--new molecular targets come of age. Nat. Rev. Cancer 2004, 4, 97–105. [Google Scholar] [CrossRef]
- Balko, J.M.; Schwarz, L.J.; Luo, N.; Estrada, M.V.; Giltnane, J.M.; Dávila-González, D.; Wang, K.; Sánchez, V.; Dean, P.T.; Combs, S.E.; et al. Triple-negative breast cancers with amplification of JAK2 at the 9p24 locus demonstrate JAK2-specific dependence. Sci. Transl. Med. 2016, 8, 334ra353. [Google Scholar] [CrossRef]
- Cikman, O.; Taysi, S.; Gulsen, M.; Demir, E.; Akan, M.; Diril, H.; Kiraz, H.; Karaayvaz, M.; Tarakcioglu, M. The Radioprotective Effects of Caffeic Acid Phenethyl Ester and Thymoquinone on Oxidative and Nitrosative Stress in Liver Tissue of Rats Exposed to Total Head Irradiation. West. Indian Med. J. 2016, 65, 1–7. [Google Scholar]
- Zhu, W.-Q.; Wang, J.; Guo, X.-F.; Liu, Z.; Dong, W.-G. Thymoquinone inhibits proliferation in gastric cancer via the STAT3 pathway in vivo and in vitro. World J. Gastroenterol. 2016, 22, 4149–4159. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.-L.; Eichmann, A. The power of VEGF (vascular endothelial growth factor) family molecules. Cell Mol. Life Sci. 2013, 70, 1673–1674. [Google Scholar] [CrossRef]
- Chung, A.S.; Lee, J.; Ferrara, N. Targeting the tumour vasculature: Insights from physiological angiogenesis. Nat. Rev. Cancer 2010, 10, 505–514. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Sullivan, C.A.W.; Zerkowski, M.P.; Molinaro, A.M.; Rimm, D.L.; Camp, R.L.; Chung, G.G. High levels of vascular endothelial growth factor and its receptors (VEGFR-1, VEGFR-2, neuropilin-1) are associated with worse outcome in breast cancer. Hum. Pathol. 2008, 39, 1835–1843. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.C.; Voisin, V.; Wang, S.; Wang, D.-Y.; Jones, R.A.; Datti, A.; Uehling, D.; Al-awar, R.; Egan, S.E.; Bader, G.D.; et al. Combined deletion of Pten and p53 in mammary epithelium accelerates triple-negative breast cancer with dependency on eEF2K. EMBO Mol. Med. 2014, 6, 1542–1560. [Google Scholar] [CrossRef]
- Hamurcu, Z.; Ashour, A.; Kahraman, N.; Ozpolat, B. FOXM1 regulates expression of eukaryotic elongation factor 2 kinase and promotes proliferation, invasion and tumorgenesis of human triple negative breast cancer cells. Oncotarget 2016, 7, 16619–16635. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Pan, C.; Sun, J.; Gilbert, C.; Drews-Elger, K.; Azzam, D.J.; Picon-Ruiz, M.; Kim, M.; Ullmer, W.; El-Ashry, D.; et al. VEGF drives cancer-initiating stem cells through VEGFR-2/Stat3 signaling to upregulate Myc and Sox2. Oncogene 2015, 34, 3107–3119. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.-X.; Chen, S.; Huang, L.; Zhou, Y.; Shao, Z.-M. Monitoring Serum VEGF in Neoadjuvant Chemotherapy for Patients with Triple-Negative Breast Cancer: A New Strategy for Early Prediction of Treatment Response and Patient Survival. Oncologist 2019, 24, 753–761. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Gerber, H.-P.; LeCouter, J. The biology of VEGF and its receptors. Nat. Med. 2003, 9, 669–676. [Google Scholar] [CrossRef]
- Marty, M.; Pivot, X. The potential of anti-vascular endothelial growth factor therapy in metastatic breast cancer: Clinical experience with anti-angiogenic agents, focusing on bevacizumab. Eur. J. Cancer 2008, 44, 912–920. [Google Scholar] [CrossRef] [PubMed]
- Almatroodi, S.A.; Almatroudi, A.; Alsahli, M.A.; Khan, A.A.; Rahmani, A.H. Thymoquinone, an Active Compound of Nigella sativa: Role in Prevention and Treatment of Cancer. Curr. Pharm. Biotechnol. 2020, 21, 1028–1041. [Google Scholar] [CrossRef]
- Rashid, M.; Sanjarin, F.; Sabouni, F. Thymoquinone Effects on Cell Viability, Apoptosis and VEGF-A Gene Expression Level in AGS(CRL-1739) Cell Line. Anti-Cancer Agents Med. Chem. 2019, 19, 820–826. [Google Scholar] [CrossRef]
- Peng, L.; Liu, A.; Shen, Y.; Xu, H.-Z.; Yang, S.-Z.; Ying, X.-Z.; Liao, W.; Liu, H.-X.; Lin, Z.-Q.; Chen, Q.-Y.; et al. Antitumor and anti-angiogenesis effects of thymoquinone on osteosarcoma through the NF-κB pathway. Oncol. Rep. 2013, 29, 571–578. [Google Scholar] [CrossRef]
- Yegin, Z.; Duran, T.; Yildirim, I.H. Thymoquinone Down-regulates VEGFA and Up-regulates FLT1 Transcriptional Levels in Human Breast Cancer Cells. Int. J. Hum. Genet. 2020, 20, 19–24. [Google Scholar] [CrossRef]
- Alobaedi, O.H.; Alib, W.H.T.; Basheti, I.A. Antitumor effect of thymoquinone combined with resveratrol on mice transplanted with breast cancer. Asian Pac. J. Trop. Med. 2017, 10, 400–408. [Google Scholar] [CrossRef]
- Bierie, B.; Moses, H.L. Gain or loss of TGFbeta signaling in mammary carcinoma cells can promote metastasis. Cell Cycle 2009, 8, 3319–3327. [Google Scholar] [CrossRef][Green Version]
- Papageorgis, P.; Stylianopoulos, T. Role of epithelial-mesenchymal transition markers in triple-negative breast cancer (Review). Int. J. Oncol. 2015, 46, 933–943. [Google Scholar] [CrossRef]
- Dallas, N.A.; Samuel, S.; Xia, L.; Fan, F.; Gray, M.J.; Lim, S.J.; Ellis, L.M. Endoglin (CD105): A Marker of Tumor Vasculature and Potential Target for Therapy. Clin. Cancer Res. 2008, 14, 1931–1937. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.J.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Joshi, A.; Cao, D. TGF-beta signaling, tumor microenvironment and tumor progression: The butterfly effect. Front. Biosci. 2010, 15, 180–194. [Google Scholar] [CrossRef]
- Band, A.M.; Laiho, M. Crosstalk of TGF-β and Estrogen Receptor Signaling in Breast Cancer. J. Mammary Gland. Biol. Neoplasia 2011, 16, 109–115. [Google Scholar] [CrossRef]
- Lin, S.; Yang, J.; Elkahloun, A.G.; Bandyopadhyay, A.; Wang, L.; Cornell, J.E.; Yeh, I.-T.; Agyin, J.; Tomlinson, G.; Suna, L.-Z. Attenuation of TGF-β signaling suppresses premature senescence in a p21-dependent manner and promotes oncogenic Ras-mediated metastatic transformation in human mammary epithelial cells. Biol. Cell 2012, 23, 1569–1581. [Google Scholar] [CrossRef] [PubMed]
- Datto, M.B.; Li, Y.; Panus, J.F.; Howe, D.J.; Xiong, Y.; Wang, X.F. Transforming growth factor beta induces the cyclin-dependent kinase inhibitor p21 through a p53-independent mechanism. Proc. Natl. Acad. Sci. USA 1995, 92, 5545–5549. [Google Scholar] [CrossRef]
- Hannon, G.J.; Beach, D. pl5INK4B is a potentia| effector of TGF-β-induced cell cycle arrest. Nature 1994, 371, 257–261. [Google Scholar] [CrossRef]
- Pietenpol, J.A.; Stein, R.W.; Moran, E.; Yaciuk, P.; Schlegel, R.; Lyons, R.M.; Pittelkow, M.R.; Münger, K.; Howley, P.M.; Moses, H.L. TGF-β1 inhibition of c-myc transcription and growth in keratinocytes is abrogated by viral transforming proteins with pRB binding domains. Cell 1990, 61, 777–785. [Google Scholar] [CrossRef]
- Tang, B.; Vu, M.; Booke, T.; Santner, S.J.; Miller, F.R.; Anver, M.R.; Wakefield, L.M. TGF-beta switches from tumor suppressor to prometastatic factor in a model of breast cancer progression. J. Clin. Investig. 2003, 112, 1116–1124. [Google Scholar] [CrossRef]
- Yi, T.; Cho, S.-G.; Yi, Z.; Pang, X.; Rodriguez, M.; Wang, Y.; Sethi, G.; Aggarwal, B.B.; Liu, M. Thymoquinone inhibits tumor angiogenesis and tumor growth through suppressing AKT and extracellular signal-regulated kinase signaling pathways. Mol. Cancer Ther. 2008, 7, 1789–1796. [Google Scholar] [CrossRef]
- Kou, B.; Liu, W.; Zhao, W.; Duan, P.; Yang, Y.; Yi, Q.; Guo, F.; Li, J.; Zhou, J.; Kou, Q. Thymoquinone inhibits epithelial-mesenchymal transition in prostate cancer cells by negatively regulating the TGF-β/Smad2/3 signaling pathway. Oncol. Rep. 2017, 38, 3592–3598. [Google Scholar] [CrossRef]
- Pollak, M.N.; Schernhammer, E.S.; Hankinson, S.E. Insulin-like growth factors and neoplasia. Nat. Rev. Cancer 2004, 4, 505–518. [Google Scholar] [CrossRef] [PubMed]
- Pollak, M. Insulin and insulin-like growth factor signalling in neoplasia. Nat. Rev. Cancer 2008, 8, 915–928. [Google Scholar] [CrossRef]
- Yang, Y.; Yee, D. Targeting Insulin and Insulin-Like Growth Factor Signaling in Breast Cancer. J. Mammary Gland. Biol. Neoplasia 2012, 17, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Crudden, C.; Song, D.; Cismas, S.; Trocmé, E.; Pasca, S.; Calin, G.A.; Girnita, A.; Girnita, L. Below the Surface: IGF-1R Therapeutic Targeting and Its Endocytic Journey. Cells 2019, 8, 1223. [Google Scholar] [CrossRef] [PubMed]
- MyersJr, M.G.; Sun, X.J.; White, M.F. The IRS-1 signaling system. Trends Biochem. Sci. 1994, 19, 289–293. [Google Scholar] [CrossRef]
- Rosenzweig, S.A.; Atreya2, H.S. Defining the pathway to insulin-like growth factor system targeting in cancer. Biochem. Pharmacol. 2010, 10, 1115–1124. [Google Scholar] [CrossRef]
- LeRoith, D. Insulin-like growth factor I receptor signaling--overlapping or redundant pathways? Endocrinology 2000, 141, 1287–1288. [Google Scholar] [CrossRef]
- Wu, Y.; Brodt, P.; Sun, H.; Mejia, W.; Novosyadlyy, R.; Nunez, N.; Chen, X.; Mendoza, A.; Hong, S.H.; Khanna, C.; et al. Insulin-like growth factor-I regulates the liver microenvironment in obese mice and promotes liver metastasis. Cancer Res. 2010, 70, 57–67. [Google Scholar] [CrossRef]
- Law, J.H.; Habibi, G.; Kaiji Hu, H.M.; Wang, M.Y.C.; Stratford, A.L.; Park, E.; Gee, J.M.W.; Finlay, P.; Jones, H.E.; Nicholson, R.I.; et al. Phosphorylated Insulin-Like Growth Factor-I/Insulin Receptor Is Present in All Breast Cancer Subtypes and Is Related to Poor Survival. Cancer Res. 2008, 68, 10238–10246. [Google Scholar] [CrossRef] [PubMed]
- Hartog, H.; Horlings, H.M.; Vegt, B.v.d.; Kreike, B.; Ajouaou, A.; Vijver, M.J.v.d.; Boezen, H.M.; Bock, G.H.d.; Graaf, W.T.A.v.d.; Wesseling, J. Divergent effects of insulin-like growth factor-1 receptor expression on prognosis of estrogen receptor positive versus triple negative invasive ductal breast carcinoma. Breast Cancer Res. Treat. 2011, 129, 725–736. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Reiss, K.; Wang, J.-Y.; Romano, G.; Furnari, F.B.; Cavenee, W.K.; Morrione, A.; Tu, X.; Baserga, R. IGF-I receptor signaling in a prostatic cancer cell line with a PTEN mutation. Oncogene 2000, 19, 2687–2694. [Google Scholar] [CrossRef]
- Hankinson, S.E. Circulating levels of sex steroids and prolactin in premenopausal women and risk of breast cancer. Adv. Exp. Med. Biol. 2008, 617, 161–169. [Google Scholar]
- Bahhnassy, A.; Mohanad, M.; Shaarawy, S.; Ismail, M.F.; El-Bastawisy, A.; Ashmawy, A.M.; Zekri, A.-R. Transforming growth factor-β, insulin-like growth factor I/insulin-like growth factor I receptor and vascular endothelial growth factor-A: Prognostic and predictive markers in triple-negative and non-triple-negative breast cancer. Mol. Med. Rep. 2015, 12, 851–864. [Google Scholar] [CrossRef] [PubMed]
- Resnik, J.L.; Reichart, D.B.; Huey, K.; Webster, N.J.; Seely, B.L. Elevated insulin-like growth factor I receptor autophosphorylation and kinase activity in human breast cancer. Cancer Res. 1998, 58, 1159–1164. [Google Scholar]
- Ma, J.; Hu, X.; Li, J.; Wu, D.; Lan, Q.; Wang, Q.; Tian, S.; Dong, W. Enhancing conventional chemotherapy drug cisplatin-induced anti-tumor effects on human gastric cancer cells both in vitro and in vivo by Thymoquinone targeting PTEN gene. Oncotarget 2017, 8, 85926–85939. [Google Scholar] [CrossRef] [PubMed]
- Gougos, A.; Letarte, M. Primary Structure of Endoglin, an RGD-containing Glycoprotein of Human Endothelial Cells. J. Biol. Chem. 1990, 265, 8361–8364. [Google Scholar] [CrossRef]
- Miller, D.W.; Graulich, W.; Karges, B.; Stahl, S.; Ernst, M.; Ramaswamy, A.; Sedlacek, H.H.; Müller, R.; Adamkiewicz, J. Elevated expression of endoglin, a component of the TGF-β-receptor complex, correlates with proliferation of tumor endothelial cells. IJC 1999, 81, 568–572. [Google Scholar] [CrossRef]
- Li, D.Y.; Sorensen, L.K.; Brooke, B.S.; Urness, L.D.; Davis, E.C.; Taylor, D.G.; Boak, B.B.; Wendel, D.P. Defective Angiogenesis in Mice Lacking Endoglin. Science 1999, 284, 1534–1537. [Google Scholar] [CrossRef] [PubMed]
- Lebrin, F.; Goumans, M.-J.; Jonker, L.; Carvalho, R.L.C.; Valdimarsdottir, G.; Thorikay, M.; Mummery, C.; Arthur, H.M.; Dijke, P.t. Endoglin promotes endothelial cell proliferation and TGF-beta/ALK1 signal transduction. EMBO J. 2004, 23, 4018–4028. [Google Scholar] [CrossRef]
- Bernabeu, C.; Conley, B.A.; Vary, C.P.H. Novel Biochemical Pathways of Endoglin in Vascular Cell Physiology. J. Cell Biochem. 2007, 102, 1375–1388. [Google Scholar] [CrossRef] [PubMed]
- Beresford, M.J.; Harris, A.L.; Ah-See, M.; Daley, F.; Padhani, A.R.; Makris, A. The relationship of the neo-angiogenic marker, endoglin, with response to neoadjuvant chemotherapy in breast cancer. Br. J. Cancer 2006, 95, 1683–1688. [Google Scholar] [CrossRef] [PubMed]
- Dales, J.-P.; Garcia, S.; Bonnier, P.; Duffaud, F.; Andrac-Meyer, L.; Ramuz, O.; Lavaut, M.-N.; Allasia, C.; Charpin, C. CD105 Expression Is a Marker of High Metastatic Risk and Poor Outcome in Breast Carcinomas: Correlations Between Immunohistochemical Analysis and Long-Term Follow-up in a Series of 929 Patients. Am. J. Clin. Pathol. 2003, 119, 374–380. [Google Scholar] [CrossRef][Green Version]
- Schoonderwoerd, M.J.A.; Koops, M.F.M.; Angela, R.A.; Koolmoes, B.; Toitou, M.; Paauwe, M.; Barnhoorn, M.C.; Liu, Y.; Sier, C.F.M.; Hardwick, J.C.H.; et al. Targeting Endoglin-Expressing Regulatory T Cells in the Tumor Microenvironment Enhances the Effect of PD1 Checkpoint Inhibitor Immunotherapy. Clin. Cancer Res. 2020, 26, 3831–3842. [Google Scholar] [CrossRef] [PubMed]
- Rosen, L.S.; Gordon, M.S.; Robert, F.; Matei, D.E. Endoglin for Targeted Cancer Treatment. Curr. Oncol. Rep. 2014, 16, 365. [Google Scholar] [CrossRef] [PubMed]
- Bergers, G.; Hanahan, D. Modes of resistance to anti-angiogenic therapy. Nat. Rev. Cancer 2008, 8, 592–603. [Google Scholar] [CrossRef] [PubMed]
- Paauwe, M.; Heijkants, R.; Oudt, C.; Pelt, G.v.; Cui, C.; Theuer, C.; Hardwick, J.; Sier, C.; Hawinkels, L. Endoglin targeting inhibits tumor angiogenesis and metastatic spread in breast cancer. Oncogene 2016, 35, 4069–4079. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J. New perspectives in clinical oncology from angiogenesis research. Eur. J. Cancer 1996, 32A, 2534–2539. [Google Scholar] [CrossRef]
- Lim, S.O.; Li, C.W.; Xia, W.; Lee, H.H.; Chang, S.S.; Shen, J.; Hsu, J.L.; Raftery, D.; Djukovic, D.; Gu, H.; et al. EGFR Signaling Enhances Aerobic Glycolysis in Triple-Negative Breast Cancer Cells to Promote Tumor Growth and Immune Escape. Cancer Res. 2016, 76, 1284–1296. [Google Scholar] [CrossRef]
- Munn, D.H.; Shafizadeh, E.; Attwood, J.T.; Bondarev, I.; Pashine, A.; Mellor, A.L. Inhibition of T Cell Proliferation by Macrophage Tryptophan Catabolism. J. Exp. Med. 1999, 189, 1363–1372. [Google Scholar] [CrossRef] [PubMed]
- Prendergast, G.C. Immune escape as a fundamental trait of cancer: Focus on IDO. Oncogene 2008, 27, 3889–3900. [Google Scholar] [CrossRef]
- Fallarino, F.; Grohmann, U.; Vacca, C.; Bianchi, R.; Orabona, C.; Spreca, A.; Fioretti, M.C.; Puccetti, P. T cell apoptosis by tryptophan catabolism. Cell Death Differ. 2002, 9, 1069–1077. [Google Scholar] [CrossRef]
- Dill, E.A.; Dillon, P.M.; Bullock, T.N.; Mills, A.M. IDO expression in breast cancer: An assessment of 281 primary and metastatic cases with comparison to PD-L1. Mod. Pathol. 2018, 31, 1513–1522. [Google Scholar] [CrossRef] [PubMed]
- Grohmann, U.; Fallarino, F.; Puccetti, P. Tolerance, DCs and tryptophan: Much ado about IDO. Trends Immunol. 2003, 24, 242–248. [Google Scholar] [CrossRef]
- Munn, D.H.; Sharma, M.D.; Hou, D.; Baban, B.; Lee, J.R.; Antonia, S.J.; Messina, J.L.; Chandler, P.; Koni, P.A.; Mellor, A.L. Expression of indoleamine 2,3-dioxygenase by plasmacytoid dendritic cells in tumor-draining lymph nodes. J. Clin. Investig. 2004, 114, 280–290. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Du, W.; Yan, F.; Wang, Y.; Li, H.; Cao, S.; Yu, W.; Shen, C.; Liu, J.; Ren, X. Myeloid-derived suppressor cells suppress antitumor immune responses through IDO expression and correlate with lymph node metastasis in patients with breast cancer. J. Immunol. 2013, 190, 3783–3797. [Google Scholar] [CrossRef] [PubMed]
- Gu, T.; Rowswell-Turner, R.B.; Kilinc, M.O.; Egilmez, N.K. Central role of IFNgamma-indoleamine 2,3-dioxygenase axis in regulation of interleukin-12-mediated antitumor immunity. Cancer Res. 2010, 70, 129–138. [Google Scholar] [CrossRef]
- Sakurai, K.; Amano, S.; Enomoto, K.; Kashio, M.; Saito, Y.; Sakamoto, A.; Matsuo, S.; Suzuki, M.; Kitajima, A.; Hirano, T.; et al. Study of indoleamine 2,3-dioxygenase expression in patients with breast cancer. Gan Kagaku Ryoho. Cancer Chemother. 2005, 32, 1546–1549. [Google Scholar]
- Théate, I.; Baren, N.v.; Pilotte, L.; Moulin, P.; Larrieu, P.; Renauld, J.-C.; Hervé, C.; Gutierrez-Roelens, I.; Marbaix, E.; Sempoux, C.; et al. Extensive profiling of the expression of the indoleamine 2,3-dioxygenase 1 protein in normal and tumoral human tissues. Cancer Immunol. Res. 2015, 3, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Godin-Ethier, J.; Pelletier, S.; Hanafi, L.A.; Gannon, P.O.; Forget, M.A.; Routy, J.P.; Boulassel, M.R.; Krzemien, U.; Tanguay, S.; Lattouf, J.B.; et al. Human activated T lymphocytes modulate IDO expression in tumors through Th1/Th2 balance. J. Immunol. 2009, 183, 7752–7760. [Google Scholar] [CrossRef]
- Asghar, K.; Loya, A.; Rana, I.A.; Tahseen, M.; Ishaq, M.; Farooq, A.; Bakar, M.A.; Masood, I. Indoleamine 2,3-dioxygenase expression and overall survival in patients diagnosed with breast cancer in Pakistan. Cancer Manag. Res. 2019, 11, 475–481. [Google Scholar] [CrossRef]
- Brandacher, G.; Perathoner, A.; Ladurner, R.; Schneeberger, S.; Obrist, P.; Winkler, C.; Werner, E.R.; Werner-Felmayer, G.; Weiss, H.G.; Göbel, G.; et al. Prognostic value of indoleamine 2,3-dioxygenase expression in colorectal cancer: Effect on tumor-infiltrating T cells. Clin. Cancer Res. 2006, 12, 1144–1151. [Google Scholar] [CrossRef] [PubMed]
- Ino, K.; Yoshida, N.; Kajiyama, H.; Shibata, K.; Yamamoto, E.; Kidokoro, K.; Takahashi, N.; Terauchi, M.; Nawa, A.; Nomura, S.; et al. Indoleamine 2,3-dioxygenase is a novel prognostic indicator for endometrial cancer. Br. J. Cancer 2006, 95, 1555–1561. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, A.; Nikaido, T.; Ochiai, K.; Takakura, S.; Saito, M.; Aoki, Y.; Ishii, N.; Yanaihara, N.; Yamada, K.; Takikawa, O.; et al. Indoleamine 2,3-dioxygenase serves as a marker of poor prognosis in gene expression profiles of serous ovarian cancer cells. Clin. Cancer Res. 2005, 11, 6030–6039. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Liu, L.; Liu, K.; Bizargity, P.; Hancock, W.W.; Visner, G.A. Reduced cytotoxic function of effector CD8+ T cells is responsible for indoleamine 2,3-dioxygenase-dependent immune suppression. J. Immunol. 2009, 183, 1022–1031. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, T.L. Immune suppression in cancer: Effects on immune cells, mechanisms and future therapeutic intervention. Semin. Cancer Biol. 2006, 16, 3–15. [Google Scholar] [CrossRef]
- Ling, W.; Zhang, J.; Yuan, Z.; Ren, G.; Zhang, L.; Chen, X.; Rabson, A.B.; Roberts, A.I.; Wang, Y.; Shi, Y. Mesenchymal stem cells use IDO to regulate immunity in tumor microenvironment. Cancer Res. 2014, 74, 1576–1587. [Google Scholar] [CrossRef]
- Muller, A.J.; DuHadaway, J.B.; Donover, P.S.; Sutanto-Ward, E.; Prendergast, G.C. Inhibition of indoleamine 2,3-dioxygenase, an immunoregulatory target of the cancer suppression gene Bin1, potentiates cancer chemotherapy. Nat. Med. 2005, 11, 312–319. [Google Scholar] [CrossRef]
- Fujigaki, H.; Saito, K.; Fujigaki, S.; Takemura, M.; Sudo, K.; Ishiguro, H.; Seishima, M. The signal transducer and activator of transcription 1alpha and interferon regulatory factor 1 are not essential for the induction of indoleamine 2,3-dioxygenase by lipopolysaccharide: Involvement of p38 mitogen-activated protein kinase and nuclear factor-kappaB pathways, and synergistic effect of several proinflammatory cytokines. J. Biochem. 2006, 139, 655–662. [Google Scholar]
- Alam, M.; Zameer, S.; Najmi, A.K.; Ahmad, F.J.; Imam, S.S.; Akhtar, M. Thymoquinone Loaded Solid Lipid Nanoparticles Demonstrated Antidepressant-Like Activity in Rats via Indoleamine 2, 3- Dioxygenase Pathway. Drug Res. 2020, 70, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Sethi, G.; Ahn, K.S.; Aggarwal, B.B. Targeting nuclear factor-kappa B activation pathway by thymoquinone: Role in suppression of antiapoptotic gene products and enhancement of apoptosis. Mol. Cancer Res. 2008, 6, 1059–1070. [Google Scholar] [CrossRef] [PubMed]
- NIH.US. National Library of Medicinev ClnicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/results?cond=thymoquinone&term=&cntry=&state=&city=&dist= (accessed on 20 December 2021).
- Akhondian, J.; Parsa, A.; Rakhshande, H. The effect of Nigella sativa L. (black cumin seed) on intractable pediatric seizures. Med. Sci. Monit. 2007, 13, Cr555–Cr559. [Google Scholar] [PubMed]
- Al Amri, A.M.; Bamosa, A.O. Phase i safety and clinical activity study of thymoquinone in patients with advanced refractory malignant disease. Shiraz E Med. J. 2009, 10, 107–111. [Google Scholar]
- Ammar, I.M.M.; Salem, M.A.A. Amelioration of polycystic ovary syndrome-related disorders by supplementation of thymoquinone and metformin. Middle East. Fertil. Soc. J. 2021, 26, 29. [Google Scholar] [CrossRef]
- Heshmati, J.; Namazi, N.; Memarzadeh, M.-R.; Taghizadeh, M.; Kolahdooz, F. Nigella sativa oil affects glucose metabolism and lipid concentrations in patients with type 2 diabetes: A randomized, double-blind, placebo-controlled trial. Food Res. Int. 2015, 70, 87–93. [Google Scholar] [CrossRef]
- Nikkhah-Bodaghi, M.; Darabi, Z.; Agah, S.; Hekmatdoost, A. The effects of Nigella sativa on quality of life, disease activity index, and some of inflammatory and oxidative stress factors in patients with ulcerative colitis. Phytother. Res. 2019, 33, 1027–1032. [Google Scholar] [CrossRef]
- Boskabady, M.H.; Javan, H.; Sajady, M.; Rakhshandeh, H. The possible prophylactic effect of Nigella sativa seed extract in asthmatic patients. Fundam. Clin. Pharmacol. 2007, 21, 559–566. [Google Scholar] [CrossRef]
- Alkharfy, K.M.; Ahmad, A.; Khan, R.M.; Al-Shagha, W.M. Pharmacokinetic plasma behaviors of intravenous and oral bioavailability of thymoquinone in a rabbit model. Eur. J. Drug Metab. Pharmacokinet. 2015, 40, 319–323. [Google Scholar] [CrossRef]
- Ng, W.K.; Saiful Yazan, L.; Yap, L.H.; Wan Nor Hafiza, W.A.; How, C.W.; Abdullah, R. Thymoquinone-loaded nanostructured lipid carrier exhibited cytotoxicity towards breast cancer cell lines (MDA-MB-231 and MCF-7) and cervical cancer cell lines (HeLa and SiHa). BioMed Res. Int. 2015, 2015, 263131. [Google Scholar] [CrossRef] [PubMed]
- Abdelwahab, S.I.; Sheikh, B.Y.; Taha, M.M.; How, C.W.; Abdullah, R.; Yagoub, U.; El-Sunousi, R.; Eid, E.E. Thymoquinone-loaded nanostructured lipid carriers: Preparation, gastroprotection, in vitro toxicity, and pharmacokinetic properties after extravascular administration. Int. J. Nanomed. 2013, 8, 2163–2172. [Google Scholar] [CrossRef]
- Dehghani, H.; Hashemi, M.; Entezari, M.; Mohsenifar, A. The comparison of anticancer activity of thymoquinone and nanothymoquinone on human breast adenocarcinoma. Iran. J. Pharm. Res. 2015, 14, 539–546. [Google Scholar]
- Mu, G.G.; Zhang, L.L.; Li, H.Y.; Liao, Y.; Yu, H.G. Thymoquinone Pretreatment Overcomes the Insensitivity and Potentiates the Antitumor Effect of Gemcitabine Through Abrogation of Notch1, PI3K/Akt/mTOR Regulated Signaling Pathways in Pancreatic Cancer. Dig. Dis. Sci. 2015, 60, 1067–1080. [Google Scholar] [CrossRef]
- Vance, S.H.; Benghuzzi, H.; Wilson-Simpson, F.; Tucci, M. Thymoquinone supplementation and its effect on kidney tubule epithelial cells in vitro. Biomed. Sci. Instrum. 2008, 44, 477–482. [Google Scholar]
- Abukhader, M.M. The effect of route of administration in thymoquinone toxicity in male and female rats. Indian J. Pharm. Sci. 2012, 74, 195–200. [Google Scholar] [CrossRef]
- Al-Ali, A.; Alkhawajah, A.A.; Randhawa, M.A.; Shaikh, N.A. Oral and intraperitoneal LD50 of thymoquinone, an active principle of Nigella sativa, in mice and rats. J. Ayub. Med. Coll. Abbottabad. 2008, 20, 25–27. [Google Scholar]
- Gali-Muhtasib, H.; Kuester, D.; Mawrin, C.; Bajbouj, K.; Diestel, A.; Ocker, M.; Habold, C.; Foltzer-Jourdainne, C.; Schoenfeld, P.; Peters, B.; et al. Thymoquinone triggers inactivation of the stress response pathway sensor CHEK1 and contributes to apoptosis in colorectal cancer cells. Cancer Res. 2008, 68, 5609. [Google Scholar] [CrossRef] [PubMed]
- Khader, M.; Bresgen, N.; Eckl, P. In Vitro toxicological properties of thymoquinone. Food Chem. Toxicol. Int. J. Publ. Br. Ind. Biol. Res. Assoc. 2008, 47, 129–133. [Google Scholar] [CrossRef]
- Khan, M.A.; Tania, M.; Wei, C.; Mei, Z.; Fu, S.; Cheng, J.; Xu, J.; Fu, J. Thymoquinone inhibits cancer metastasis by downregulating TWIST1 expression to reduce epithelial to mesenchymal transition. Oncotarget 2015, 6, 19580–19591. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).