
Alcohol and Cancer: Epidemiology and Biological Mechanisms


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
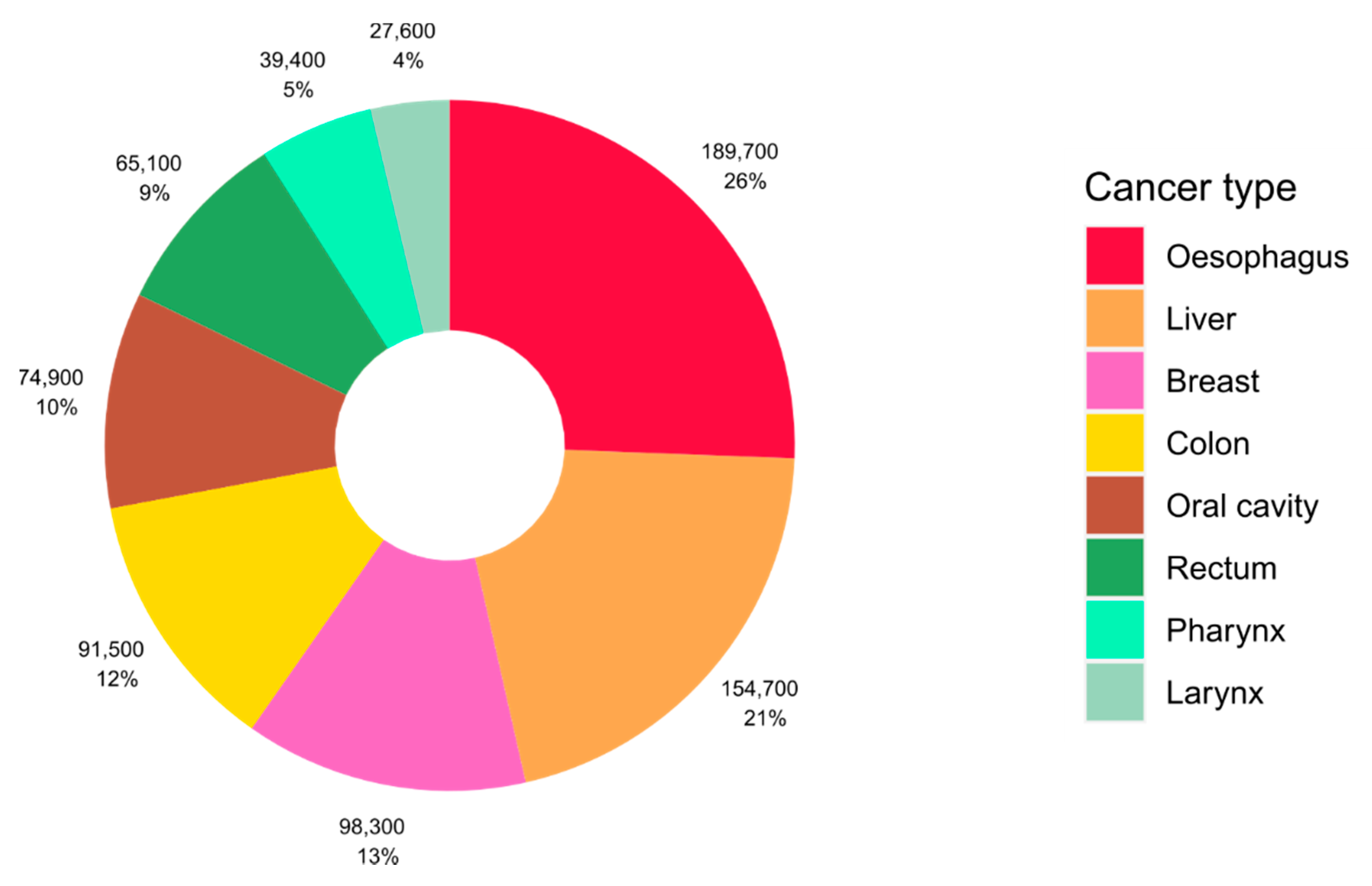
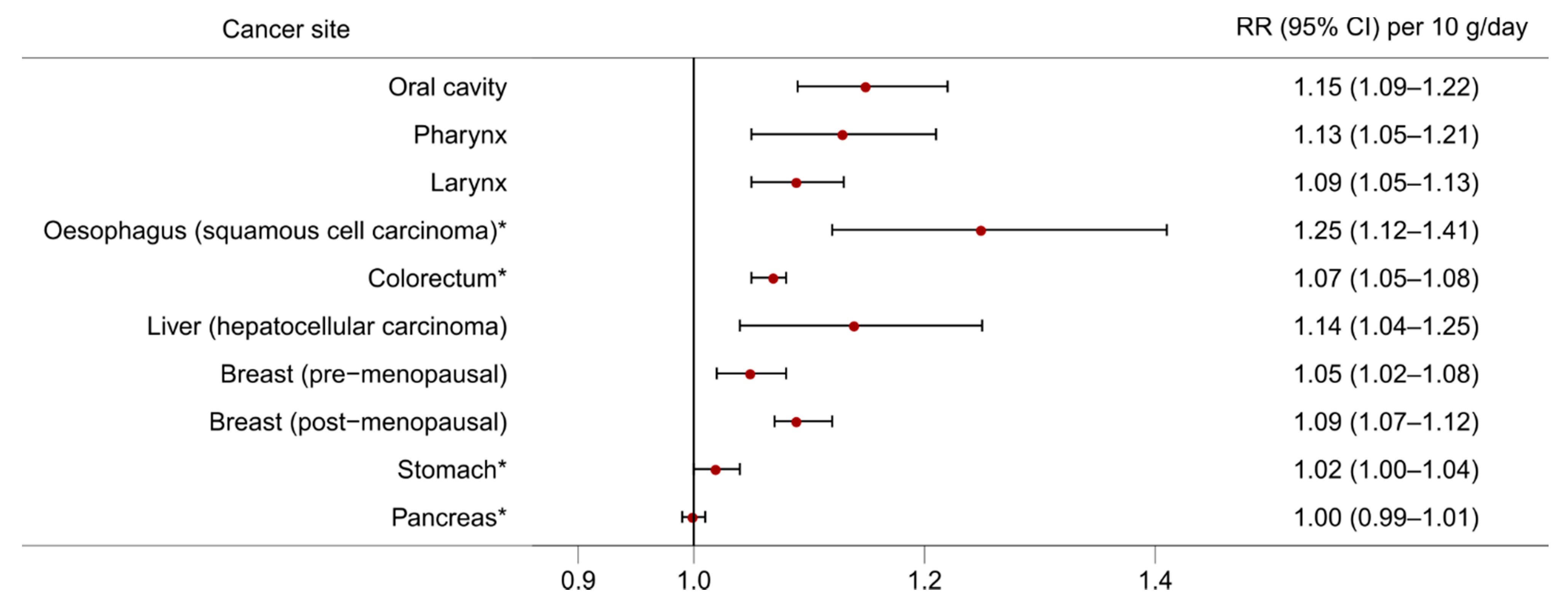
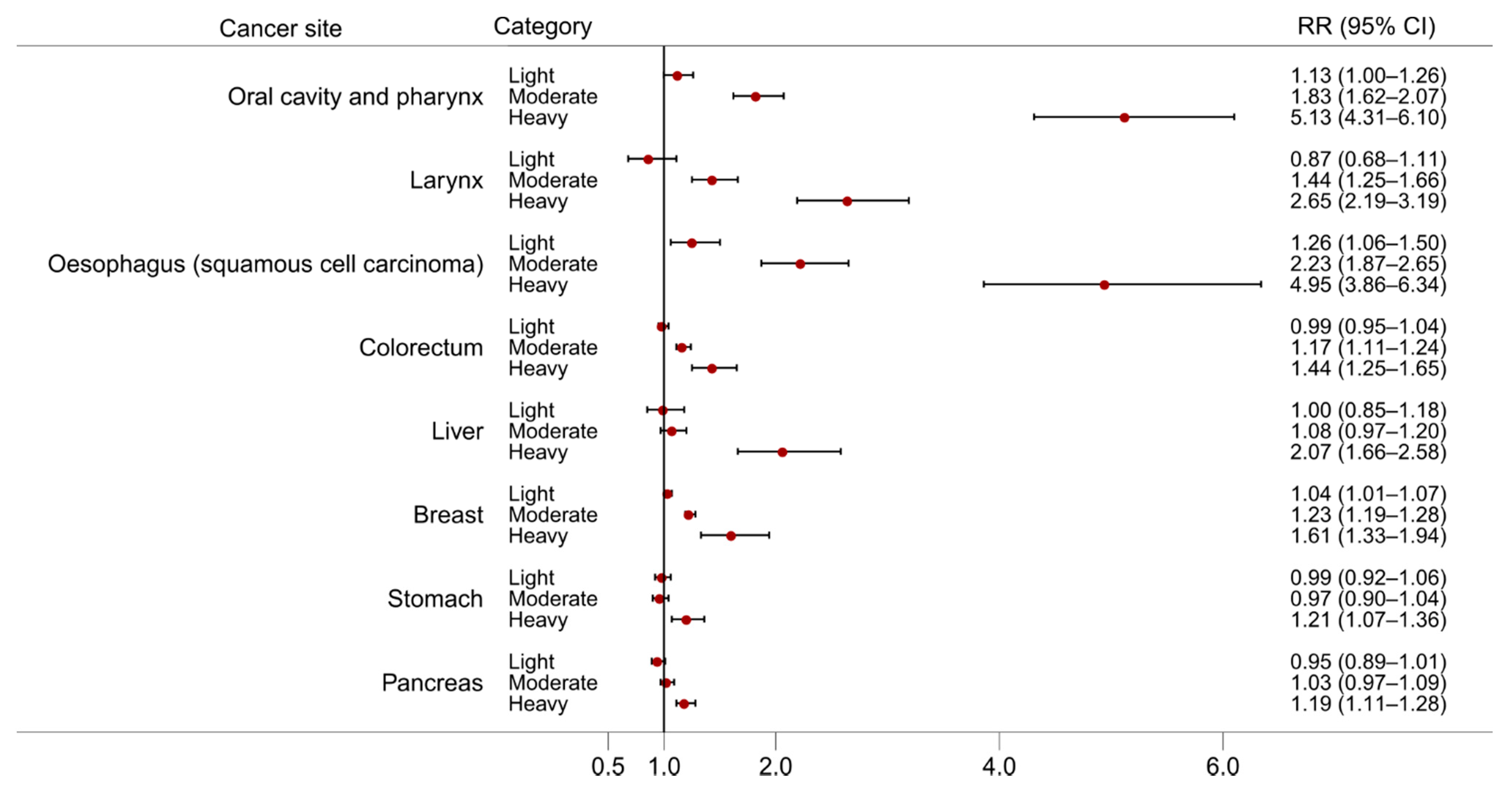
2. Alcohol and Cancer Risk
2.1. Oral Cavity Pharyngeal and Laryngeal Cancers
2.2. Oesophageal Cancer
2.3. Colorectal Cancer
2.4. Liver Cancer
2.5. Breast Cancer
2.6. Stomach Cancer
2.7. Pancreatic Cancer
2.8. Other Cancer Types
2.9. Confirming the Causal Relation Reported in Observational Studies
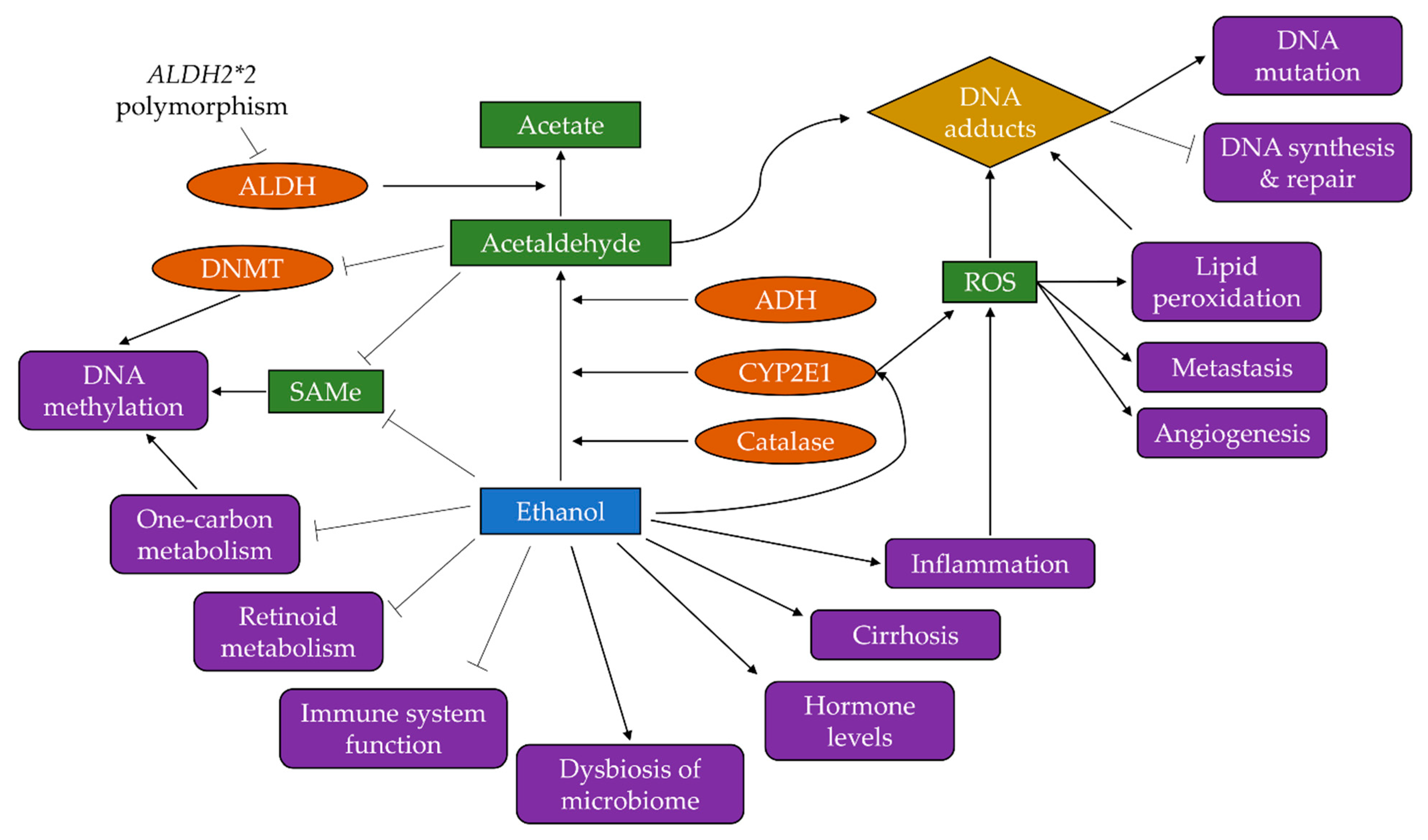
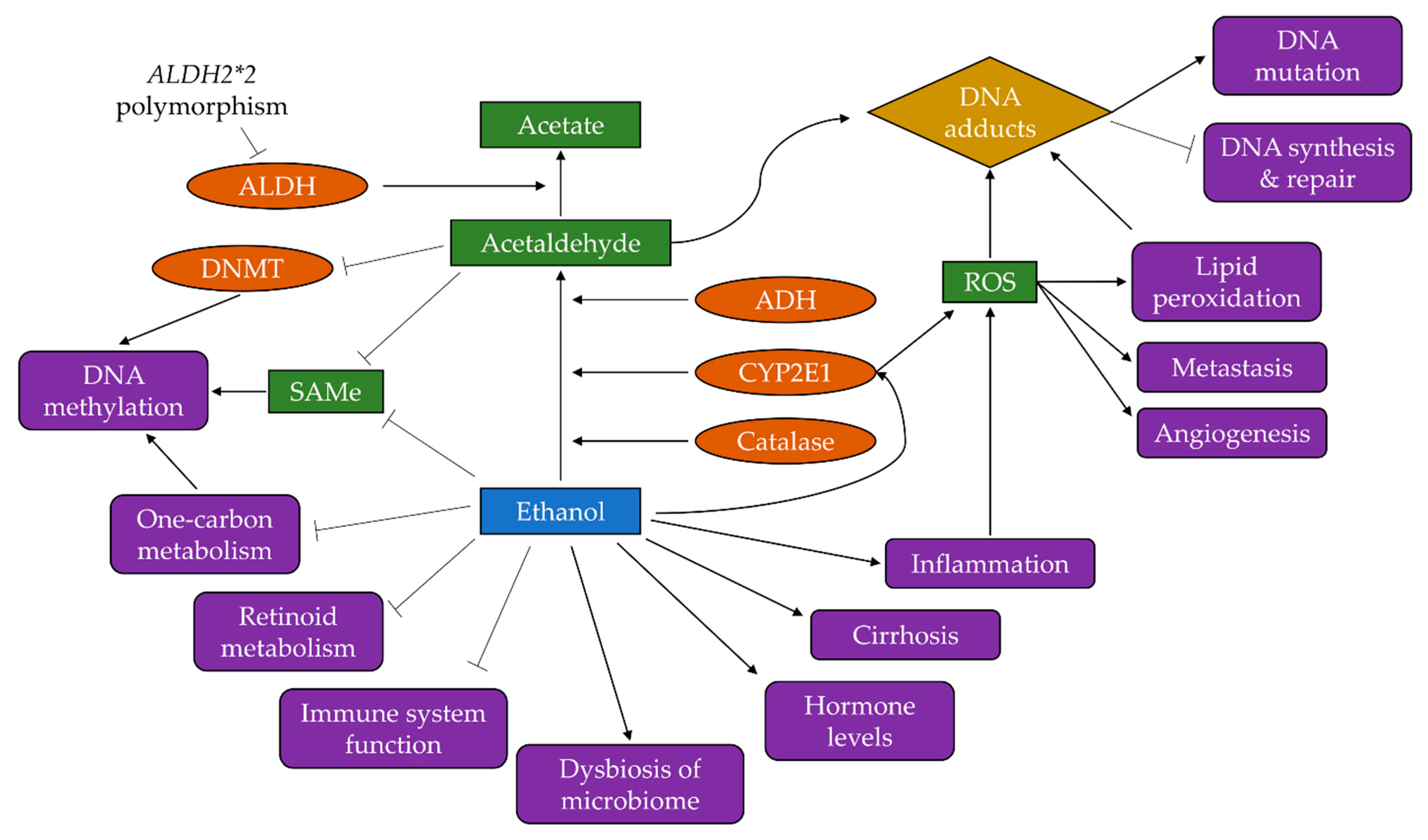
3. Mechanisms of Alcohol-Driven Carcinogenesis
3.1. Production of Acetaldehyde
3.2. Induction of Oxidative Stress
3.3. Increased Inflammation
3.4. Disruption to One-Carbon Metabolism and Folate Absorption
3.5. Altered Retinoid Metabolism
3.6. Changes to Oestrogen Regulation
3.7. Reduced Function of the Immune System
3.8. Dysbiosis of the Microbiome
3.9. Liver Cirrhosis
3.10. Activation of Other Carcinogens
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Rumgay, H.; Shield, K.; Charvat, H.; Ferrari, P.; Sornpaisarn, B.; Obot, I.; Islami, F.; Lemmens, V.E.P.P.; Rehm, J.; Soerjomataram, I. Global burden of cancer in 2020 attributable to alcohol consumption: A population-based study. Lancet Oncol. 2021, 22, 1071–1080. [Google Scholar] [CrossRef]
- Buykx, P.; Li, J.; Gavens, L.; Hooper, L.; Lovatt, M.; Gomes de Matos, E.; Meier, P.; Holmes, J. Public awareness of the link between alcohol and cancer in England in 2015: A population-based survey. BMC Public Health 2016, 16, 1194. [Google Scholar] [CrossRef]
- Manthey, J.; Shield, K.D.; Rylett, M.; Hasan, O.S.M.; Probst, C.; Rehm, J. Global alcohol exposure between 1990 and 2017 and forecasts until 2030: A modelling study. Lancet 2019, 393, 2493–2502. [Google Scholar] [CrossRef]
- International Agency for Research on Cancer. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans. Volume 44. Alcohol Drinking; 1988; Volume 44, Available online: https://publications.iarc.fr/Book-And-Report-Series/Iarc-Monographs-On-The-Identification-Of-Carcinogenic-Hazards-To-Humans/Alcohol-Drinking-1988 (accessed on 20 June 2021).
- International Agency for Research on Cancer. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans. Volume 96. Alcohol Consumption and Ethyl Carbamate; 2010; Volume 96, Available online: https://publications.iarc.fr/Book-And-Report-Series/Iarc-Monographs-On-The-Identification-Of-Carcinogenic-Hazards-To-Humans/Alcohol-Consumption-And-Ethyl-Carbamate-2010 (accessed on 20 June 2021).
- International Agency for Research on Cancer. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans. Volume 100E. Personal Habits and Indoor Combustions; 2012; Available online: https://publications.iarc.fr/Book-And-Report-Series/Iarc-Monographs-On-The-Identification-Of-Carcinogenic-Hazards-To-Humans/Personal-Habits-And-Indoor-Combustions-2012 (accessed on 20 June 2021).
- World Cancer Research Fund/American Institute for Cancer Research. Diet, Nutrition, Physical Activity and Cancer: A Global Perspective; Continuous Update Project Expert Report; 2018; Available online: https://www.wcrf.org/wp-content/uploads/2021/02/Summary-of-Third-Expert-Report-2018.pdf (accessed on 20 June 2021).
- Bagnardi, V.; Rota, M.; Botteri, E.; Tramacere, I.; Islami, F.; Fedirko, V.; Scotti, L.; Jenab, M.; Turati, F.; Pasquali, E.; et al. Alcohol consumption and site-specific cancer risk: A comprehensive dose–response meta-analysis. Br. J. Cancer 2014, 112, 580. [Google Scholar] [CrossRef]
- Arnold, M.; Ferlay, J.; van Berge Henegouwen, M.I.; Soerjomataram, I. Global burden of oesophageal and gastric cancer by histology and subsite in 2018. Gut 2020, 69, 1564–1571. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.S.; Hsiao, J.-R.; Chen, C.-H. ALDH2 polymorphism and alcohol-related cancers in Asians: A public health perspective. J. Biomed. Sci. 2017, 24, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashibe, M.; Brennan, P.; Chuang, S.-C.; Boccia, S.; Castellsague, X.; Chen, C.; Curado, M.P.; Dal Maso, L.; Daudt, A.W.; Fabianova, E.; et al. Interaction between tobacco and alcohol use and the risk of head and neck cancer: Pooled analysis in the International Head and Neck Cancer Epidemiology Consortium. Cancer Epidemiol. Biomark. Prev. 2009, 18, 541–550. [Google Scholar] [CrossRef] [Green Version]
- Steevens, J.; Schouten, L.J.; Goldbohm, R.A.; van den Brandt, P.A. Alcohol consumption, cigarette smoking and risk of subtypes of oesophageal and gastric cancer: A prospective cohort study. Gut 2010, 59, 39–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben, Q.; Wang, L.; Liu, J.; Qian, A.; Wang, Q.; Yuan, Y. Alcohol drinking and the risk of colorectal adenoma: A dose–response meta-analysis. Eur. J. Cancer Prev. 2015, 24, 286–295. [Google Scholar] [CrossRef]
- Ferlay, J.; Ervik, M.; Lam, F.; Colombet, M.; Mery, L.; Piñeros, M.; Znaor, A.; Soerjomataram, I. Global Cancer Observatory: Cancer Today. Available online: https://gco.iarc.fr/today (accessed on 15 December 2020).
- Sun, Q.; Xie, W.; Wang, Y.; Chong, F.; Song, M.; Li, T.; Xu, L.; Song, C. Alcohol Consumption by Beverage Type and Risk of Breast Cancer: A Dose-Response Meta-Analysis of Prospective Cohort Studies. Alcohol Alcohol. 2020, 55, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Gormley, M.; Dudding, T.; Sanderson, E.; Martin, R.M.; Thomas, S.; Tyrrell, J.; Ness, A.R.; Brennan, P.; Munafò, M.; Pring, M.; et al. A multivariable Mendelian randomization analysis investigating smoking and alcohol consumption in oral and oropharyngeal cancer. Nat. Commun. 2020, 11, 6071. [Google Scholar] [CrossRef]
- Ingold, N.; Amin, H.A.; Drenos, F. Alcohol causes an increased risk of head and neck but not breast cancer in individuals from the UK Biobank study: A Mendelian randomisation analysis. medRxiv 2019, 19002832. [Google Scholar] [CrossRef]
- Larsson, S.C.; Carter, P.; Kar, S.; Vithayathil, M.; Mason, A.M.; Michaëlsson, K.; Burgess, S. Smoking, alcohol consumption, and cancer: A mendelian randomisation study in UK Biobank and international genetic consortia participants. PLoS Med. 2020, 17, e1003178. [Google Scholar] [CrossRef] [PubMed]
- Ong, J.-S.; Derks, E.M.; Eriksson, M.; An, J.; Hwang, L.-D.; Easton, D.F.; Pharoah, P.P.; Berchuck, A.; Kelemen, L.E.; Matsuo, K.; et al. Evaluating the role of alcohol consumption in breast and ovarian cancer susceptibility using population-based cohort studies and two-sample Mendelian randomization analyses. Int. J. Cancer 2021, 148, 1338–1350. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.; Jahanzaib Anwar, M.; Usman, A.; Keshavarzian, A.; Bishehsari, F. Colorectal Cancer and Alcohol Consumption-Populations to Molecules. Cancers 2018, 10, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seitz, H.K.; Stickel, F. Molecular mechanisms of alcohol-mediated carcinogenesis. Nat. Rev. Cancer 2007, 7, 599–612. [Google Scholar] [CrossRef]
- Garaycoechea, J.I.; Crossan, G.P.; Langevin, F.; Mulderrig, L.; Louzada, S.; Yang, F.; Guilbaud, G.; Park, N.; Roerink, S.; Nik-Zainal, S.; et al. Alcohol and endogenous aldehydes damage chromosomes and mutate stem cells. Nature 2018, 553, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; McIntee, E.J.; Cheng, G.; Shi, Y.; Villalta, P.W.; Hecht, S.S. Identification of DNA Adducts of Acetaldehyde. Chem. Res. Toxicol. 2000, 13, 1149–1157. [Google Scholar] [CrossRef]
- Brooks, P.J.; Theruvathu, J.A. DNA adducts from acetaldehyde: Implications for alcohol-related carcinogenesis. Alcohol 2005, 35, 187–193. [Google Scholar] [CrossRef]
- Varela-Rey, M.; Woodhoo, A.; Martinez-Chantar, M.-L.; Mato, J.M.; Lu, S.C. Alcohol, DNA methylation, and cancer. Alcohol Res. 2013, 35, 25–35. [Google Scholar]
- Barve, S.; Chen, S.-Y.; Kirpich, I.; Watson, W.H.; McClain, C. Development, Prevention, and Treatment of Alcohol-Induced Organ Injury: The Role of Nutrition. Alcohol Res. 2017, 38, 289–302. [Google Scholar]
- Millonig, G.; Wang, Y.; Homann, N.; Bernhardt, F.; Qin, H.; Mueller, S.; Bartsch, H.; Seitz, H.K. Ethanol-mediated carcinogenesis in the human esophagus implicates CYP2E1 induction and the generation of carcinogenic DNA-lesions. Int. J. Cancer 2011, 128, 533–540. [Google Scholar] [CrossRef]
- Albano, E. Alcohol, oxidative stress and free radical damage. Proc. Nutr. Soc. 2006, 65, 278–290. [Google Scholar] [CrossRef] [Green Version]
- Haorah, J.; Ramirez, S.H.; Floreani, N.; Gorantla, S.; Morsey, B.; Persidsky, Y. Mechanism of alcohol-induced oxidative stress and neuronal injury. Free. Radic. Biol. Med. 2008, 45, 1542–1550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linhart, K.; Bartsch, H.; Seitz, H.K. The role of reactive oxygen species (ROS) and cytochrome P-450 2E1 in the generation of carcinogenic etheno-DNA adducts. Redox Biol. 2014, 3, 56–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ratna, A.; Mandrekar, P. Alcohol and Cancer: Mechanisms and Therapies. Biomolecules 2017, 7, 61. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Yang, J.-L.; Yu, K.-K.; Xu, M.; Xu, Y.-Z.; Chen, L.; Lu, Y.-M.; Fang, H.-S.; Wang, X.-Y.; Hu, Z.-Q.; et al. Activation of the NF-κB pathway as a mechanism of alcohol enhanced progression and metastasis of human hepatocellular carcinoma. Mol. Cancer 2015, 14, 10. [Google Scholar] [CrossRef] [Green Version]
- Molina, P.E.; Happel, K.I.; Zhang, P.; Kolls, J.K.; Nelson, S. Focus on: Alcohol and the immune system. Alcohol Res. Health 2010, 33, 97–108. [Google Scholar]
- Zimmermann, H.W.; Seidler, S.; Gassler, N.; Nattermann, J.; Luedde, T.; Trautwein, C.; Tacke, F. Interleukin-8 is activated in patients with chronic liver diseases and associated with hepatic macrophage accumulation in human liver fibrosis. PLoS ONE 2011, 6, e21381. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.-T.; Juan, C.-Y.; Chang, K.-J.; Chen, W.-J.; Kuo, M.-L. IL-6 inhibits apoptosis and retains oxidative DNA lesions in human gastric cancer AGS cells through up-regulation of anti-apoptotic gene mcl-1. Carcinogenesis 2001, 22, 1947–1953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Persson, E.C.; Schwartz, L.M.; Park, Y.; Trabert, B.; Hollenbeck, A.R.; Graubard, B.I.; Freedman, N.D.; McGlynn, K.A. Alcohol consumption, folate intake, hepatocellular carcinoma, and liver disease mortality. Cancer Epidemio. Biomark. Prev. 2013, 22, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Sellers, T.A.; Kushi, L.H.; Cerhan, J.R.; Vierkant, R.A.; Gapstur, S.M.; Vachon, C.M.; Olson, J.E.; Therneau, T.M.; Folsom, A.R. Dietary Folate Intake, Alcohol, and Risk of Breast Cancer in a Prospective Study of Postmenopausal Women. Epidemiology 2001, 12, 420–428. [Google Scholar] [CrossRef] [PubMed]
- Dumitrescu, R.G.; Shields, P.G. The etiology of alcohol-induced breast cancer. Alcohol 2005, 35, 213–225. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Nguyen, N.; Colditz, G.A. Links between alcohol consumption and breast cancer: A look at the evidence. Womens Health 2015, 11, 65–77. [Google Scholar] [CrossRef] [Green Version]
- Singletary, K.W.; Gapstur, S.M. Alcohol and Breast CancerReview of Epidemiologic and Experimental Evidence and Potential Mechanisms. JAMA 2001, 286, 2143–2151. [Google Scholar] [CrossRef]
- Endogenous Hormones Breast Cancer Collaborative Group; Key, T.J.; Appleby, P.N.; Reeves, G.K.; Roddam, A.W.; Helzlsouer, K.J.; Alberg, A.J.; Rollison, D.E.; Dorgan, J.F.; Brinton, L.A.; et al. Circulating sex hormones and breast cancer risk factors in postmenopausal women: Reanalysis of 13 studies. Br. J. Cancer 2011, 105, 709–722. [Google Scholar] [CrossRef] [PubMed]
- Assi, N.; Rinaldi, S.; Viallon, V.; Dashti, S.G.; Dossus, L.; Fournier, A.; Cervenka, I.; Kvaskoff, M.; Turzanski-Fortner, R.; Bergmann, M.; et al. Mediation analysis of the alcohol-postmenopausal breast cancer relationship by sex hormones in the EPIC cohort. Int. J. Cancer 2020, 146, 759–768. [Google Scholar] [CrossRef]
- Pessione, F.; Degos, F.; Marcellin, P.; Duchatelle, V.; Njapoum, C.; Martinot-Peignoux, M.; Degott, C.; Valla, D.; Erlinger, S.; Rueff, B. Effect of alcohol consumption on serum hepatitis C virus RNA and histological lesions in chronic hepatitis C. Hepatology 1998, 27, 1717–1722. [Google Scholar] [CrossRef]
- Cao, Y.; Willett, W.C.; Rimm, E.B.; Stampfer, M.J.; Giovannucci, E.L. Light to moderate intake of alcohol, drinking patterns, and risk of cancer: Results from two prospective US cohort studies. BMJ 2015, 351, h4238. [Google Scholar] [CrossRef] [Green Version]
- Shin, N.-R.; Whon, T.W.; Bae, J.-W. Proteobacteria: Microbial signature of dysbiosis in gut microbiota. Trends Biotechnol. 2015, 33, 496–503. [Google Scholar] [CrossRef] [PubMed]
- Fedirko, V.; Tran, H.Q.; Gewirtz, A.T.; Stepien, M.; Trichopoulou, A.; Aleksandrova, K.; Olsen, A.; Tjønneland, A.; Overvad, K.; Carbonnel, F.; et al. Exposure to bacterial products lipopolysaccharide and flagellin and hepatocellular carcinoma: A nested case-control study. BMC Med. 2017, 15, 72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.M.; Kim, S.Y.; Seki, E. Inflammation and Liver Cancer: Molecular Mechanisms and Therapeutic Targets. Semin. Liver Dis. 2019, 39, 26–42. [Google Scholar] [CrossRef] [PubMed]
- Howie, N.; Trigkas, T.; Cruchley, A.; Wertz, P.; Squier, C.; Williams, D. Short-term exposure to alcohol increases the permeability of human oral mucosa. Oral Dis. 2001, 7, 349–354. [Google Scholar] [CrossRef] [PubMed]
- van Roekel, E.H.; Trijsburg, L.; Assi, N.; Carayol, M.; Achaintre, D.; Murphy, N.; Rinaldi, S.; Schmidt, J.A.; Stepien, M.; Kaaks, R.; et al. Circulating Metabolites Associated with Alcohol Intake in the European Prospective Investigation into Cancer and Nutrition Cohort. Nutrients 2018, 10, 654. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rumgay, H.; Murphy, N.; Ferrari, P.; Soerjomataram, I. Alcohol and Cancer: Epidemiology and Biological Mechanisms. Nutrients 2021, 13, 3173. https://doi.org/10.3390/nu13093173
Rumgay H, Murphy N, Ferrari P, Soerjomataram I. Alcohol and Cancer: Epidemiology and Biological Mechanisms. Nutrients. 2021; 13(9):3173. https://doi.org/10.3390/nu13093173
Chicago/Turabian StyleRumgay, Harriet, Neil Murphy, Pietro Ferrari, and Isabelle Soerjomataram. 2021. "Alcohol and Cancer: Epidemiology and Biological Mechanisms" Nutrients 13, no. 9: 3173. https://doi.org/10.3390/nu13093173
APA StyleRumgay, H., Murphy, N., Ferrari, P., & Soerjomataram, I. (2021). Alcohol and Cancer: Epidemiology and Biological Mechanisms. Nutrients, 13(9), 3173. https://doi.org/10.3390/nu13093173