
Endocannabinoids and the Gut-Brain Control of Food Intake and Obesity


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction

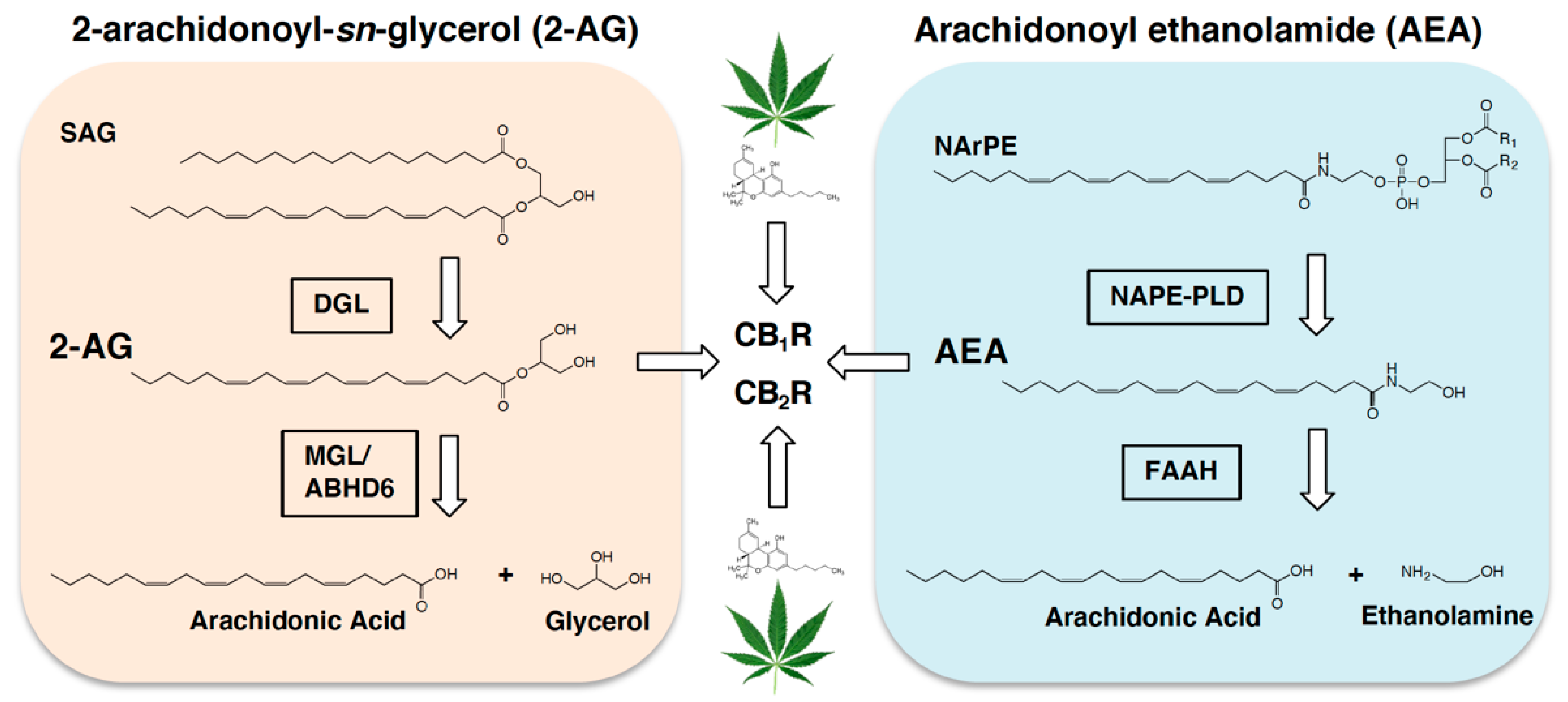
2. Gut-Brain Endocannabinoid Signaling Controls Intake of Palatable Foods
3. Endocannabinoids and Gut-Brain Neurotransmission: Indirect Mechanisms
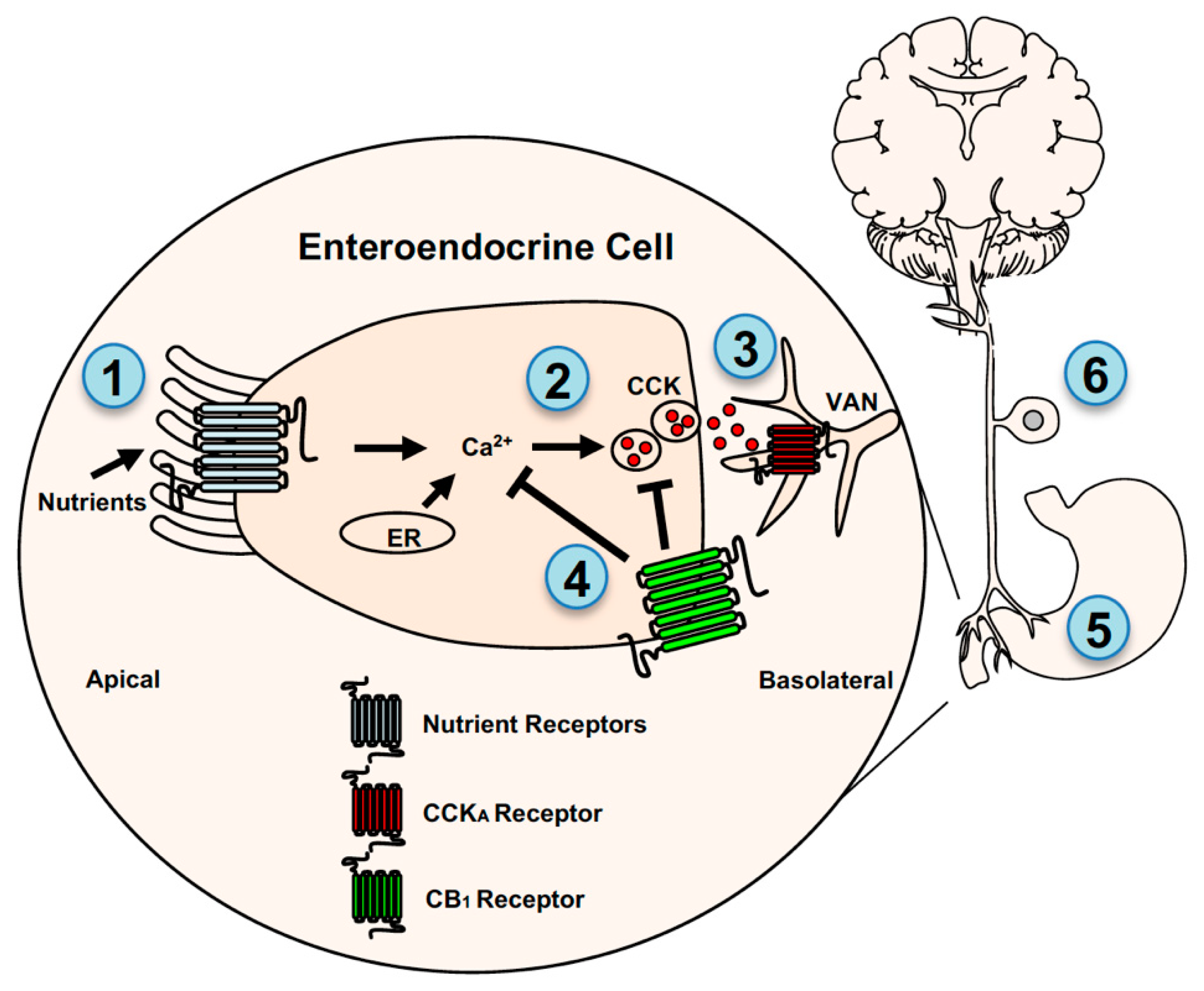
3.1. Interactions with Satiation Signaling Pathways
3.2. Interactions with Hunger Signaling Pathways
4. Endocannabinoids and Gut-Brain Neurotransmission: Direct Mechanisms
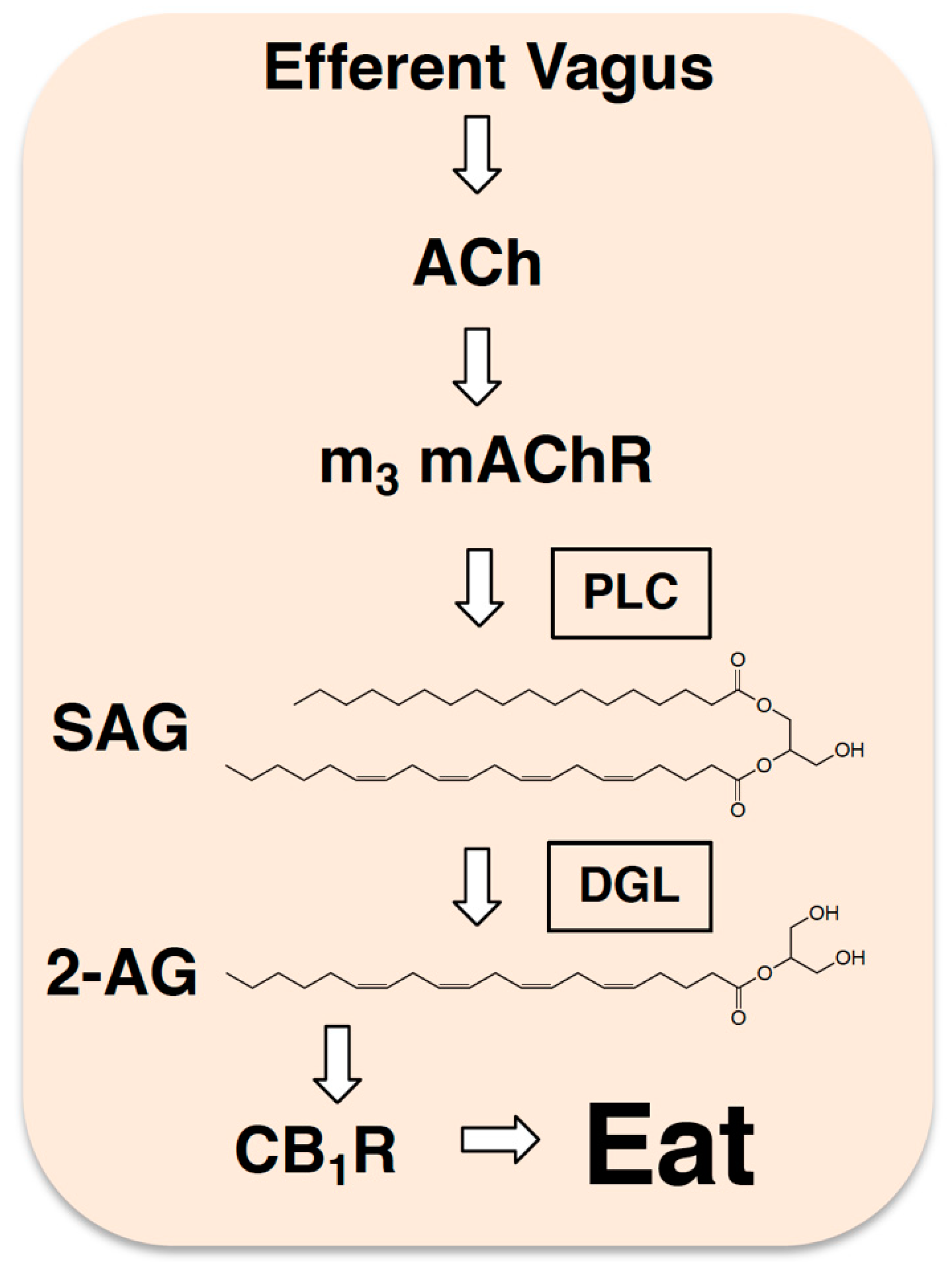
5. Endocannabinoids and Efferent Autonomic Neurotransmission
6. Targeting the Peripheral ECB System for Treatment of Human Obesity
7. Future Considerations
Author Contributions
Funding
Conflicts of Interest
References
- Schwartz, G.J. Roles for gut vagal sensory signals in determining energy availability and energy expenditure. Brain Res. 2018, 1693, 151–153. [Google Scholar] [CrossRef]
- Shechter, A.; Schwartz, G.J. Gut-brain nutrient sensing in food reward. Appetite 2018, 122, 32–35. [Google Scholar] [CrossRef] [PubMed]
- Clemmensen, C.; Muller, T.D.; Woods, S.C.; Berthoud, H.R.; Seeley, R.J.; Tschop, M.H. Gut-Brain Cross-Talk in Metabolic Control. Cell 2017, 168, 758–774. [Google Scholar] [CrossRef] [PubMed]
- Steinert, R.E.; Feinle-Bisset, C.; Asarian, L.; Horowitz, M.; Beglinger, C.; Geary, N. Ghrelin, CCK, GLP-1, and PYY(3-36): Secretory Controls and Physiological Roles in Eating and Glycemia in Health, Obesity, and After RYGB. Physiol. Rev. 2017, 97, 411–463. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.P.; Jerome, C.; Cushin, B.J.; Eterno, R.; Simansky, K.J. Abdominal vagotomy blocks the satiety effect of cholecystokinin in the rat. Science 1981, 213, 1036–1037. [Google Scholar] [CrossRef]
- Schwartz, G.J.; Moran, T.H. CCK elicits and modulates vagal afferent activity arising from gastric and duodenal sites. Ann. N. Y. Acad. Sci. 1994, 713, 121–128. [Google Scholar] [CrossRef]
- Raybould, H.E. Mechanisms of CCK signaling from gut to brain. Curr. Opin. Pharmcol. 2007, 7, 570–574. [Google Scholar] [CrossRef]
- Smith, G.P.; Jerome, C.; Norgren, R. Afferent axons in abdominal vagus mediate satiety effect of cholecystokinin in rats. Am. J. Physiol. 1985, 249, R638–R641. [Google Scholar] [CrossRef]
- Kaelberer, M.M.; Buchanan, K.L.; Klein, M.E.; Barth, B.B.; Montoya, M.M.; Shen, X.; Bohorquez, D.V. A gut-brain neural circuit for nutrient sensory transduction. Science 2018, 361. [Google Scholar] [CrossRef] [PubMed]
- Kaelberer, M.M.; Rupprecht, L.E.; Liu, W.W.; Weng, P.; Bohorquez, D.V. Neuropod Cells: The Emerging Biology of Gut-Brain Sensory Transduction. Ann. Rev. Neurosci. 2020, 43, 337–353. [Google Scholar] [CrossRef]
- Han, W.; Tellez, L.A.; Perkins, M.H.; Perez, I.O.; Qu, T.; Ferreira, J.; Ferreira, T.L.; Quinn, D.; Liu, Z.W.; Gao, X.B.; et al. A Neural Circuit for Gut-Induced Reward. Cell 2018, 175, 665–678.e23. [Google Scholar] [CrossRef]
- Han, W.; Tellez, L.A.; Niu, J.; Medina, S.; Ferreira, T.L.; Zhang, X.; Su, J.; Tong, J.; Schwartz, G.J.; van den Pol, A.; et al. Striatal Dopamine Links Gastrointestinal Rerouting to Altered Sweet Appetite. Cell Metab. 2016, 23, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Sclafani, A.; Ackroff, K. Role of gut nutrient sensing in stimulating appetite and conditioning food preferences. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2012, 302, R1119–R1133. [Google Scholar] [CrossRef] [PubMed]
- Sclafani, A. From appetite setpoint to appetition: 50years of ingestive behavior research. Physiol. Behav. 2018, 192, 210–217. [Google Scholar] [CrossRef]
- Wenzel, J.M.; Cheer, J.F. Endocannabinoid Regulation of Reward and Reinforcement through Interaction with Dopamine and Endogenous Opioid Signaling. Neuropsychopharmacology 2018, 43, 103–115. [Google Scholar] [CrossRef] [PubMed]
- DiPatrizio, N.V.; Piomelli, D. The thrifty lipids: Endocannabinoids and the neural control of energy conservation. Trends Neurosci. 2012, 35, 403–411. [Google Scholar] [CrossRef]
- Simon, V.; Cota, D. MECHANISMS IN ENDOCRINOLOGY: Endocannabinoids and metabolism: Past, present and future. Eur. J. Endocrinol. 2017, 176, R309–R324. [Google Scholar] [CrossRef] [PubMed]
- Quarta, C.; Mazza, R.; Obici, S.; Pasquali, R.; Pagotto, U. Energy balance regulation by endocannabinoids at central and peripheral levels. Trends Mol. Med. 2011, 17, 518–526. [Google Scholar] [CrossRef]
- Di Marzo, V.; Goparaju, S.K.; Wang, L.; Liu, J.; Batkai, S.; Jarai, Z.; Fezza, F.; Miura, G.I.; Palmiter, R.D.; Sugiura, T.; et al. Leptin-regulated endocannabinoids are involved in maintaining food intake. Nature 2001, 410, 822–825. [Google Scholar] [CrossRef] [PubMed]
- Jarbe, T.U.; DiPatrizio, N.V. Delta9-THC induced hyperphagia and tolerance assessment: Interactions between the CB1 receptor agonist delta9-THC and the CB1 receptor antagonist SR-141716 (rimonabant) in rats. Behav. Pharmcol. 2005, 16, 373–380. [Google Scholar] [CrossRef]
- Ravinet Trillou, C.; Arnone, M.; Delgorge, C.; Gonalons, N.; Keane, P.; Maffrand, J.P.; Soubrie, P. Anti-obesity effect of SR141716, a CB1 receptor antagonist, in diet-induced obese mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2003, 284, R345–R353. [Google Scholar] [CrossRef] [PubMed]
- Gomez, R.; Navarro, M.; Ferrer, B.; Trigo, J.M.; Bilbao, A.; Del Arco, I.; Cippitelli, A.; Nava, F.; Piomelli, D.; Rodriguez de Fonseca, F. A peripheral mechanism for CB1 cannabinoid receptor-dependent modulation of feeding. J. Neurosci. 2002, 22, 9612–9617. [Google Scholar] [CrossRef] [PubMed]
- DiPatrizio, N.V.; Astarita, G.; Schwartz, G.; Li, X.; Piomelli, D. Endocannabinoid signal in the gut controls dietary fat intake. Proc. Natl. Acad. Sci. USA 2011, 108, 12904–12908. [Google Scholar] [CrossRef]
- DiPatrizio, N.V.; Igarashi, M.; Narayanaswami, V.; Murray, C.; Gancayco, J.; Russell, A.; Jung, K.M.; Piomelli, D. Fasting stimulates 2-AG biosynthesis in the small intestine: Role of cholinergic pathways. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2015, 309, R805–R813. [Google Scholar] [CrossRef] [PubMed]
- Argueta, D.A.; DiPatrizio, N.V. Peripheral endocannabinoid signaling controls hyperphagia in western diet-induced obesity. Physiol. Behav. 2017, 171, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Cota, D.; Marsicano, G.; Tschop, M.; Grubler, Y.; Flachskamm, C.; Schubert, M.; Auer, D.; Yassouridis, A.; Thone-Reineke, C.; Ortmann, S.; et al. The endogenous cannabinoid system affects energy balance via central orexigenic drive and peripheral lipogenesis. J. Clin. Investig. 2003, 112, 423–431. [Google Scholar] [CrossRef]
- LoVerme, J.; Duranti, A.; Tontini, A.; Spadoni, G.; Mor, M.; Rivara, S.; Stella, N.; Xu, C.; Tarzia, G.; Piomelli, D. Synthesis and characterization of a peripherally restricted CB1 cannabinoid antagonist, URB447, that reduces feeding and body-weight gain in mice. Bioorg. Med. Chem. Lett. 2009, 19, 639–643. [Google Scholar] [CrossRef]
- Randall, P.A.; Vemuri, V.K.; Segovia, K.N.; Torres, E.F.; Hosmer, S.; Nunes, E.J.; Santerre, J.L.; Makriyannis, A.; Salamone, J.D. The novel cannabinoid CB1 antagonist AM6545 suppresses food intake and food-reinforced behavior. Pharmcol. Biochem. Behav. 2010, 97, 179–184. [Google Scholar] [CrossRef]
- Cluny, N.L.; Vemuri, V.K.; Chambers, A.P.; Limebeer, C.L.; Bedard, H.; Wood, J.T.; Lutz, B.; Zimmer, A.; Parker, L.A.; Makriyannis, A.; et al. A novel peripherally restricted cannabinoid receptor antagonist, AM6545, reduces food intake and body weight, but does not cause malaise, in rodents. Br. J. Pharmcol. 2011, 161, 629–642. [Google Scholar] [CrossRef]
- Tam, J.; Vemuri, V.K.; Liu, J.; Batkai, S.; Mukhopadhyay, B.; Godlewski, G.; Osei-Hyiaman, D.; Ohnuma, S.; Ambudkar, S.V.; Pickel, J.; et al. Peripheral CB1 cannabinoid receptor blockade improves cardiometabolic risk in mouse models of obesity. J. Clin. Investig. 2010, 120, 2953–2966. [Google Scholar] [CrossRef]
- Tam, J.; Cinar, R.; Liu, J.; Godlewski, G.; Wesley, D.; Jourdan, T.; Szanda, G.; Mukhopadhyay, B.; Chedester, L.; Liow, J.S.; et al. Peripheral cannabinoid-1 receptor inverse agonism reduces obesity by reversing leptin resistance. Cell Metab. 2012, 16, 167–179. [Google Scholar] [CrossRef]
- Maccarrone, M.; Bab, I.; Biro, T.; Cabral, G.A.; Dey, S.K.; Di Marzo, V.; Konje, J.C.; Kunos, G.; Mechoulam, R.; Pacher, P.; et al. Endocannabinoid signaling at the periphery: 50 years after THC. Trends Pharmcol. Sci. 2015, 36, 277–296. [Google Scholar] [CrossRef]
- Izzo, A.A.; Sharkey, K.A. Cannabinoids and the gut: New developments and emerging concepts. Pharmacol. Ther. 2010, 126, 21–38. [Google Scholar] [CrossRef] [PubMed]
- Cluny, N.L.; Reimer, R.A.; Sharkey, K.A. Cannabinoid signalling regulates inflammation and energy balance: The importance of the brain-gut axis. Brain Behav. Immun. 2012, 26, 691–698. [Google Scholar] [CrossRef]
- Cani, P.D.; Plovier, H.; Hul, M.V.; Geurts, L.; Delzenne, N.M.; Druart, C.; Everard, A. Endocannabinoids—At the crossroads between the gut microbiota and host metabolism. Nat. Rev. Endocrinol. 2016, 12, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Artmann, A.; Petersen, G.; Hellgren, L.I.; Boberg, J.; Skonberg, C.; Nellemann, C.; Hansen, S.H.; Hansen, H.S. Influence of dietary fatty acids on endocannabinoid and N-acylethanolamine levels in rat brain, liver and small intestine. Biochim. Biophys. Acta 2008, 1781, 200–212. [Google Scholar] [CrossRef] [PubMed]
- Tam, J.; Szanda, G.; Drori, A.; Liu, Z.; Cinar, R.; Kashiwaya, Y.; Reitman, M.L.; Kunos, G. Peripheral cannabinoid-1 receptor blockade restores hypothalamic leptin signaling. Mol. Metab. 2017, 6, 1113–1125. [Google Scholar] [CrossRef]
- Christensen, R.; Kristensen, P.K.; Bartels, E.M.; Bliddal, H.; Astrup, A. Efficacy and safety of the weight-loss drug rimonabant: A meta-analysis of randomised trials. Lancet 2007, 370, 1706–1713. [Google Scholar] [CrossRef]
- DiPatrizio, N.V.; Joslin, A.; Jung, K.M.; Piomelli, D. Endocannabinoid signaling in the gut mediates preference for dietary unsaturated fats. FASEB J. 2013, 27, 2513–2520. [Google Scholar] [CrossRef] [PubMed]
- DiPatrizio, N.V.; Piomelli, D. Intestinal lipid-derived signals that sense dietary fat. J. Clin. Investig. 2015, 125, 891–898. [Google Scholar] [CrossRef]
- Argueta, D.A.; Perez, P.A.; Makriyannis, A.; DiPatrizio, N.V. Cannabinoid CB1 Receptors Inhibit Gut-Brain Satiation Signaling in Diet-Induced Obesity. Front. Physiol. 2019, 10, 704. [Google Scholar] [CrossRef] [PubMed]
- Piomelli, D. The molecular logic of endocannabinoid signalling. Nat. Rev. 2003, 4, 873–884. [Google Scholar] [CrossRef] [PubMed]
- Pertwee, R.G. Endocannabinoids and Their Pharmacological Actions. Handb. Exp. Pharmcol. 2015, 231, 1–37. [Google Scholar] [CrossRef]
- Higuchi, S.; Irie, K.; Yamaguchi, R.; Katsuki, M.; Araki, M.; Ohji, M.; Hayakawa, K.; Mishima, S.; Akitake, Y.; Matsuyama, K.; et al. Hypothalamic 2-arachidonoylglycerol regulates multistage process of high-fat diet preferences. PLoS ONE 2012, 7, e38609. [Google Scholar] [CrossRef]
- Higuchi, S.; Ohji, M.; Araki, M.; Furuta, R.; Katsuki, M.; Yamaguchi, R.; Akitake, Y.; Matsuyama, K.; Irie, K.; Mishima, K.; et al. Increment of hypothalamic 2-arachidonoylglycerol induces the preference for a high-fat diet via activation of cannabinoid 1 receptors. Behav. Brain Res. 2011, 216, 477–480. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, R.R.; Sharma, P.L. Stimulation of accumbens shell cannabinoid CB(1) receptors by noladin ether, a putative endocannabinoid, modulates food intake and dietary selection in rats. Pharmcol. Res. 2012, 66, 276–282. [Google Scholar] [CrossRef] [PubMed]
- DiPatrizio, N.V.; Simansky, K.J. Activating parabrachial cannabinoid CB1 receptors selectively stimulates feeding of palatable foods in rats. J. Neurosci. 2008, 28, 9702–9709. [Google Scholar] [CrossRef]
- DiPatrizio, N.V.; Simansky, K.J. Inhibiting parabrachial fatty acid amide hydrolase activity selectively increases the intake of palatable food via cannabinoid CB1 receptors. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 295, R1409–R1414. [Google Scholar] [CrossRef]
- Mahler, S.V.; Smith, K.S.; Berridge, K.C. Endocannabinoid hedonic hotspot for sensory pleasure: Anandamide in nucleus accumbens shell enhances ‘liking’ of a sweet reward. Neuropsychopharmacology 2007, 32, 2267–2278. [Google Scholar] [CrossRef]
- Wei, D.; Lee, D.; Li, D.; Daglian, J.; Jung, K.M.; Piomelli, D. A role for the endocannabinoid 2-arachidonoyl-sn-glycerol for social and high-fat food reward in male mice. Psychopharmacology 2016, 233, 1911–1919. [Google Scholar] [CrossRef]
- Mendez-Diaz, M.; Rueda-Orozco, P.E.; Ruiz-Contreras, A.E.; Prospero-Garcia, O. The endocannabinoid system modulates the valence of the emotion associated to food ingestion. Addict. Biol. 2012, 17, 725–735. [Google Scholar] [CrossRef]
- De Luca, M.A.; Solinas, M.; Bimpisidis, Z.; Goldberg, S.R.; Di Chiara, G. Cannabinoid facilitation of behavioral and biochemical hedonic taste responses. Neuropharmacology 2012, 63, 161–168. [Google Scholar] [CrossRef]
- Jarrett, M.M.; Scantlebury, J.; Parker, L.A. Effect of delta9-tetrahydrocannabinol on quinine palatability and AM251 on sucrose and quinine palatability using the taste reactivity test. Physiol. Behav. 2007, 90, 425–430. [Google Scholar] [CrossRef]
- Melis, T.; Succu, S.; Sanna, F.; Boi, A.; Argiolas, A.; Melis, M.R. The cannabinoid antagonist SR 141716A (Rimonabant) reduces the increase of extra-cellular dopamine release in the rat nucleus accumbens induced by a novel high palatable food. Neurosci. Lett. 2007, 419, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Droste, S.M.; Saland, S.K.; Schlitter, E.K.; Rodefer, J.S. AM 251 differentially effects food-maintained responding depending on food palatability. Pharmcol. Biochem. Behav. 2010, 95, 443–448. [Google Scholar] [CrossRef] [PubMed]
- South, T.; Deng, C.; Huang, X.F. AM 251 and beta-Funaltrexamine reduce fat intake in a fat-preferring strain of mouse. Behav. Brain Res. 2007, 181, 153–157. [Google Scholar] [CrossRef] [PubMed]
- Thornton-Jones, Z.D.; Vickers, S.P.; Clifton, P.G. The cannabinoid CB1 receptor antagonist SR141716A reduces appetitive and consummatory responses for food. Psychopharmacology 2005, 179, 452–460. [Google Scholar] [CrossRef]
- Feja, M.; Leigh, M.P.K.; Baindur, A.N.; McGraw, J.J.; Wakabayashi, K.T.; Cravatt, B.F.; Bass, C.E. The novel MAGL inhibitor MJN110 enhances responding to reward-predictive incentive cues by activation of CB1 receptors. Neuropharmacology 2020, 162, 107814. [Google Scholar] [CrossRef]
- Salamone, J.D.; McLaughlin, P.J.; Sink, K.; Makriyannis, A.; Parker, L.A. Cannabinoid CB1 receptor inverse agonists and neutral antagonists: Effects on food intake, food-reinforced behavior and food aversions. Physiol. Behav. 2007, 91, 383–388. [Google Scholar] [CrossRef]
- Williams, C.M.; Kirkham, T.C. Anandamide induces overeating: Mediation by central cannabinoid (CB1) receptors. Psychopharmacology 1999, 143, 315–317. [Google Scholar] [CrossRef]
- DiPatrizio, N.V. Endocannabinoids in the Gut. Cannabis Cannabinoid Res. 2016, 1, 67–77. [Google Scholar] [CrossRef]
- Greenberg, D.; Smith, G.P. The controls of fat intake. Psychosom. Med. 1996, 58, 559–569. [Google Scholar] [CrossRef]
- Avalos, B.; Argueta, D.A.; Perez, P.A.; Wiley, M.; Wood, C.; DiPatrizio, N.V. Cannabinoid CB1 Receptors in the Intestinal Epithelium Are Required for Acute Western-Diet Preferences in Mice. Nutrients 2020, 12, 2874. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention (CDC); National Center for Health Statistics (NCHS). National Health and Nutrition Examination Survey Data; U.S. Department of Health and Human: Hyattsville, MD, USA. Available online: https://www.cdc.gov/nchs/nhanes/index.htm (accessed on 6 April 2021).
- Levine, A.S.; Kotz, C.M.; Gosnell, B.A. Sugars and fats: The neurobiology of preference. J. Nutr. 2003, 133, 831S–834S. [Google Scholar] [CrossRef] [PubMed]
- Monteleone, P.; Piscitelli, F.; Scognamiglio, P.; Monteleone, A.M.; Canestrelli, B.; Di Marzo, V.; Maj, M. Hedonic eating is associated with increased peripheral levels of ghrelin and the endocannabinoid 2-arachidonoyl-glycerol in healthy humans: A pilot study. J. Clin. Endocrinol. Metab. 2012, 97, E917–E924. [Google Scholar] [CrossRef] [PubMed]
- Engeli, S.; Bohnke, J.; Feldpausch, M.; Gorzelniak, K.; Janke, J.; Batkai, S.; Pacher, P.; Harvey-White, J.; Luft, F.C.; Sharma, A.M.; et al. Activation of the peripheral endocannabinoid system in human obesity. Diabetes 2005, 54, 2838–2843. [Google Scholar] [CrossRef]
- Bluher, M.; Engeli, S.; Kloting, N.; Berndt, J.; Fasshauer, M.; Batkai, S.; Pacher, P.; Schon, M.R.; Jordan, J.; Stumvoll, M. Dysregulation of the peripheral and adipose tissue endocannabinoid system in human abdominal obesity. Diabetes 2006, 55, 3053–3060. [Google Scholar] [CrossRef] [PubMed]
- Cote, M.; Matias, I.; Lemieux, I.; Petrosino, S.; Almeras, N.; Despres, J.P.; Di Marzo, V. Circulating endocannabinoid levels, abdominal adiposity and related cardiometabolic risk factors in obese men. Int. J. Obes. 2007, 31, 692–699. [Google Scholar] [CrossRef]
- Di Marzo, V.; Cote, M.; Matias, I.; Lemieux, I.; Arsenault, B.J.; Cartier, A.; Piscitelli, F.; Petrosino, S.; Almeras, N.; Despres, J.P. Changes in plasma endocannabinoid levels in viscerally obese men following a 1 year lifestyle modification programme and waist circumference reduction: Associations with changes in metabolic risk factors. Diabetologia 2009, 52, 213–217. [Google Scholar] [CrossRef]
- Little, T.J.; Cvijanovic, N.; DiPatrizio, N.V.; Argueta, D.A.; Rayner, C.K.; Feinle-Bisset, C.; Young, R.L. Plasma endocannabinoid levels in lean, overweight, and obese humans: Relationships to intestinal permeability markers, inflammation, and incretin secretion. Am. J. Physiol. Endocrinol. Metab. 2018, 315, E489–E495. [Google Scholar] [CrossRef]
- Hillard, C.J. Circulating Endocannabinoids: From Whence Do They Come and Where are They Going? Neuropsychopharmacology 2017. [Google Scholar] [CrossRef]
- Lotfi Yagin, N.; Aliasgharzadeh, S.; Alizadeh, M.; Aliasgari, F.; Mahdavi, R. The association of circulating endocannabinoids with appetite regulatory substances in obese women. Obes. Res. Clin. Pract. 2020, 14, 321–325. [Google Scholar] [CrossRef] [PubMed]
- Yagin, N.L.; Hajjarzadeh, S.; Aliasgharzadeh, S.; Aliasgari, F.; Mahdavi, R. The association of dietary patterns with endocannabinoids levels in overweight and obese women. Lipids Health Dis. 2020, 19, 161. [Google Scholar] [CrossRef]
- Tagliamonte, S.; Gill, C.I.R.; Pourshahidi, L.K.; Slevin, M.M.; Price, R.K.; Ferracane, R.; Lawther, R.; O’Connor, G.; Vitaglione, P. Endocannabinoids, endocannabinoid-like molecules and their precursors in human small intestinal lumen and plasma: Does diet affect them? Eur. J. Nutr. 2020. [Google Scholar] [CrossRef] [PubMed]
- Depommier, C.; Flamand, N.; Pelicaen, R.; Maiter, D.; Thissen, J.P.; Loumaye, A.; Hermans, M.P.; Everard, A.; Delzenne, N.M.; Di Marzo, V.; et al. Linking the Endocannabinoidome with Specific Metabolic Parameters in an Overweight and Insulin-Resistant Population: From Multivariate Exploratory Analysis to Univariate Analysis and Construction of Predictive Models. Cells 2021, 10, 71. [Google Scholar] [CrossRef]
- Quercioli, A.; Pataky, Z.; Vincenti, G.; Makoundou, V.; Di Marzo, V.; Montecucco, F.; Carballo, S.; Thomas, A.; Staub, C.; Steffens, S.; et al. Elevated endocannabinoid plasma levels are associated with coronary circulatory dysfunction in obesity. Eur. Heart J. 2011, 32, 1369–1378. [Google Scholar] [CrossRef]
- Lacroix, S.; Pechereau, F.; Leblanc, N.; Boubertakh, B.; Houde, A.; Martin, C.; Flamand, N.; Silvestri, C.; Raymond, F.; Di Marzo, V.; et al. Rapid and Concomitant Gut Microbiota and Endocannabinoidome Response to Diet-Induced Obesity in Mice. mSystems 2019, 4. [Google Scholar] [CrossRef]
- Kuipers, E.N.; Kantae, V.; Maarse, B.C.E.; van den Berg, S.M.; van Eenige, R.; Nahon, K.J.; Reifel-Miller, A.; Coskun, T.; de Winther, M.P.J.; Lutgens, E.; et al. High Fat Diet Increases Circulating Endocannabinoids Accompanied by Increased Synthesis Enzymes in Adipose Tissue. Front. Physiol. 2018, 9, 1913. [Google Scholar] [CrossRef] [PubMed]
- Gatta-Cherifi, B.; Matias, I.; Vallee, M.; Tabarin, A.; Marsicano, G.; Piazza, P.V.; Cota, D. Simultaneous postprandial deregulation of the orexigenic endocannabinoid anandamide and the anorexigenic peptide YY in obesity. Int. J. Obes. 2012, 36, 880–885. [Google Scholar] [CrossRef] [PubMed]
- Godlewski, G.; Cinar, R.; Coffey, N.J.; Liu, J.; Jourdan, T.; Mukhopadhyay, B.; Chedester, L.; Liu, Z.; Osei-Hyiaman, D.; Iyer, M.R.; et al. Targeting Peripheral CB1 Receptors Reduces Ethanol Intake via a Gut-Brain Axis. Cell Metab. 2019, 29, 1320–1333.e8. [Google Scholar] [CrossRef] [PubMed]
- Burdyga, G.; Varro, A.; Dimaline, R.; Thompson, D.G.; Dockray, G.J. Ghrelin receptors in rat and human nodose ganglia: Putative role in regulating CB-1 and MCH receptor abundance. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G1289–G1297. [Google Scholar] [CrossRef]
- Burdyga, G.; Lal, S.; Varro, A.; Dimaline, R.; Thompson, D.G.; Dockray, G.J. Expression of cannabinoid CB1 receptors by vagal afferent neurons is inhibited by cholecystokinin. J. Neurosci. 2004, 24, 2708–2715. [Google Scholar] [CrossRef]
- Burdyga, G.; Varro, A.; Dimaline, R.; Thompson, D.G.; Dockray, G.J. Expression of cannabinoid CB1 receptors by vagal afferent neurons: Kinetics and role in influencing neurochemical phenotype. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 299, G63–G69. [Google Scholar] [CrossRef]
- de Lartigue, G.; Barbier de la Serre, C.; Espero, E.; Lee, J.; Raybould, H.E. Leptin resistance in vagal afferent neurons inhibits cholecystokinin signaling and satiation in diet induced obese rats. PLoS ONE 2012, 7, e32967. [Google Scholar] [CrossRef] [PubMed]
- Christie, S.; O’Rielly, R.; Li, H.; Nunez-Salces, M.; Wittert, G.A.; Page, A.J. Modulatory effect of methanandamide on gastric vagal afferent satiety signals depends on nutritional status. J. Physiol. 2020, 598, 2169–2182. [Google Scholar] [CrossRef] [PubMed]
- Cluny, N.L.; Baraboi, E.D.; Mackie, K.; Burdyga, G.; Richard, D.; Dockray, G.J.; Sharkey, K.A. High fat diet and body weight have different effects on cannabinoid CB(1) receptor expression in rat nodose ganglia. Auton. Neurosci. 2013, 179, 122–130. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Christie, S.; O’Rielly, R.; Li, H.; Wittert, G.A.; Page, A.J. Biphasic effects of methanandamide on murine gastric vagal afferent mechanosensitivity. J. Physiol. 2020, 598, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Christie, S.; O’Rielly, R.; Li, H.; Wittert, G.; Page, A. High fat diet induced obesity alters endocannabinoid and ghrelin mediated regulation of components of the endocannabinoid system in nodose ganglia. Peptides 2020, 170371. [Google Scholar] [CrossRef]
- Ripken, D.; van der Wielen, N.; van der Meulen, J.; Schuurman, T.; Witkamp, R.F.; Hendriks, H.F.; Koopmans, S.J. Cholecystokinin regulates satiation independently of the abdominal vagal nerve in a pig model of total subdiaphragmatic vagotomy. Physiol. Behav. 2015, 139, 167–176. [Google Scholar] [CrossRef]
- Reidelberger, R.D.; Hernandez, J.; Fritzsch, B.; Hulce, M. Abdominal vagal mediation of the satiety effects of CCK in rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004, 286, R1005–R1012. [Google Scholar] [CrossRef] [PubMed]
- Sykaras, A.G.; Demenis, C.; Case, R.M.; McLaughlin, J.T.; Smith, C.P. Duodenal enteroendocrine I-cells contain mRNA transcripts encoding key endocannabinoid and fatty acid receptors. PLoS ONE 2012, 7, e42373. [Google Scholar] [CrossRef] [PubMed]
- Morales, I.; Berridge, K.C. ‘Liking’ and ‘wanting’ in eating and food reward: Brain mechanisms and clinical implications. Physiol. Behav. 2020, 227, 113152. [Google Scholar] [CrossRef]
- Date, Y.; Murakami, N.; Toshinai, K.; Matsukura, S.; Niijima, A.; Matsuo, H.; Kangawa, K.; Nakazato, M. The role of the gastric afferent vagal nerve in ghrelin-induced feeding and growth hormone secretion in rats. Gastroenterology 2002, 123, 1120–1128. [Google Scholar] [CrossRef]
- Zigman, J.M.; Jones, J.E.; Lee, C.E.; Saper, C.B.; Elmquist, J.K. Expression of ghrelin receptor mRNA in the rat and the mouse brain. J. Comp. Neurol. 2006, 494, 528–548. [Google Scholar] [CrossRef] [PubMed]
- Bello, N.T.; Coughlin, J.W.; Redgrave, G.W.; Ladenheim, E.E.; Moran, T.H.; Guarda, A.S. Dietary conditions and highly palatable food access alter rat cannabinoid receptor expression and binding density. Physiol. Behav. 2012, 105, 720–726. [Google Scholar] [CrossRef][Green Version]
- Vianna, C.R.; Donato, J., Jr.; Rossi, J.; Scott, M.; Economides, K.; Gautron, L.; Pierpont, S.; Elias, C.F.; Elmquist, J.K. Cannabinoid receptor 1 in the vagus nerve is dispensable for body weight homeostasis but required for normal gastrointestinal motility. J. Neurosci. 2012, 32, 10331–10337. [Google Scholar] [CrossRef] [PubMed]
- Page, A.J.; Blackshaw, L.A. An in vitro study of the properties of vagal afferent fibres innervating the ferret oesophagus and stomach. J. Physiol. 1998, 512, 907–916. [Google Scholar] [CrossRef]
- Berthoud, H.R.; Patterson, L.M.; Zheng, H. Vagal-enteric interface: Vagal activation-induced expression of c-Fos and p-CREB in neurons of the upper gastrointestinal tract and pancreas. Anat. Rec. 2001, 262, 29–40. [Google Scholar] [CrossRef]
- Zheng, H.; Berthoud, H.R. Functional vagal input to gastric myenteric plexus as assessed by vagal stimulation-induced Fos expression. Am. J. Physiol. Gastrointest. Liver Physiol. 2000, 279, G73–G81. [Google Scholar] [CrossRef]
- Pradhan, S.N.; Roth, T. Comparative behavioral effects of several anticholinergic agents in rats. Psychopharmacologia 1968, 12, 358–366. [Google Scholar] [CrossRef]
- Lorenz, D.; Nardi, P.; Smith, G.P. Atropine methyl nitrate inhibits sham feeding in the rat. Pharmcol. Biochem. Behav. 1978, 8, 405–407. [Google Scholar] [CrossRef]
- Muise, E.D.; Gandotra, N.; Tackett, J.J.; Bamdad, M.C.; Cowles, R.A. Distribution of muscarinic acetylcholine receptor subtypes in the murine small intestine. Life Sci. 2017, 169, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Rinaldo, L.; Hansel, C. Muscarinic acetylcholine receptor activation blocks long-term potentiation at cerebellar parallel fiber-Purkinje cell synapses via cannabinoid signaling. Proc. Natl. Acad. Sci. USA 2013, 110, 11181–11186. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Tzounopoulos, T. Physiological activation of cholinergic inputs controls associative synaptic plasticity via modulation of endocannabinoid signaling. J. Neurosci. 2011, 31, 3158–3168. [Google Scholar] [CrossRef]
- Kim, J.; Isokawa, M.; Ledent, C.; Alger, B.E. Activation of muscarinic acetylcholine receptors enhances the release of endogenous cannabinoids in the hippocampus. J. Neurosci. 2002, 22, 10182–10191. [Google Scholar] [CrossRef]
- Ramikie, T.S.; Nyilas, R.; Bluett, R.J.; Gamble-George, J.C.; Hartley, N.D.; Mackie, K.; Watanabe, M.; Katona, I.; Patel, S. Multiple mechanistically distinct modes of endocannabinoid mobilization at central amygdala glutamatergic synapses. Neuron 2014, 81, 1111–1125. [Google Scholar] [CrossRef] [PubMed]
- Straiker, A.; Mackie, K. Metabotropic suppression of excitation in murine autaptic hippocampal neurons. J. Physiol. 2007, 578, 773–785. [Google Scholar] [CrossRef]
- Stella, N.; Schweitzer, P.; Piomelli, D. A second endogenous cannabinoid that modulates long-term potentiation. Nature 1997, 388, 773–778. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.M.; Mangieri, R.; Stapleton, C.; Kim, J.; Fegley, D.; Wallace, M.; Mackie, K.; Piomelli, D. Stimulation of endocannabinoid formation in brain slice cultures through activation of group I metabotropic glutamate receptors. Mol. Pharmcol. 2005, 68, 1196–1202. [Google Scholar] [CrossRef]
- Jung, K.M.; Astarita, G.; Zhu, C.; Wallace, M.; Mackie, K.; Piomelli, D. A key role for diacylglycerol lipase-alpha in metabotropic glutamate receptor-dependent endocannabinoid mobilization. Mol. Pharmcol. 2007, 72, 612–621. [Google Scholar] [CrossRef] [PubMed]
- Bellocchio, L.; Soria-Gomez, E.; Quarta, C.; Metna-Laurent, M.; Cardinal, P.; Binder, E.; Cannich, A.; Delamarre, A.; Haring, M.; Martin-Fontecha, M.; et al. Activation of the sympathetic nervous system mediates hypophagic and anxiety-like effects of CB1 receptor blockade. Proc. Natl. Acad. Sci. USA 2013, 110, 4786–4791. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Abu El Haija, M.; Morgan, D.A.; Guo, D.; Song, Y.; Frank, A.; Tian, L.; Riedl, R.A.; Burnett, C.M.L.; Gao, Z.; et al. Endocannabinoid Receptor-1 and Sympathetic Nervous System Mediate the Beneficial Metabolic Effects of Gastric Bypass. Cell Rep. 2020, 33, 108270. [Google Scholar] [CrossRef]
- Monteleone, A.M.; Di Marzo, V.; Monteleone, P.; Dalle Grave, R.; Aveta, T.; Ghoch, M.E.; Piscitelli, F.; Volpe, U.; Calugi, S.; Maj, M. Responses of peripheral endocannabinoids and endocannabinoid-related compounds to hedonic eating in obesity. Eur. J. Nutr. 2016, 55, 1799–1805. [Google Scholar] [CrossRef] [PubMed]
- Pi-Sunyer, F.X.; Aronne, L.J.; Heshmati, H.M.; Devin, J.; Rosenstock, J. Effect of rimonabant, a cannabinoid-1 receptor blocker, on weight and cardiometabolic risk factors in overweight or obese patients: RIO-North America: A randomized controlled trial. JAMA 2006, 295, 761–775. [Google Scholar] [CrossRef]
- Manca, C.; Boubertakh, B.; Leblanc, N.; Deschenes, T.; Lacroix, S.; Martin, C.; Houde, A.; Veilleux, A.; Flamand, N.; Muccioli, G.G.; et al. Germ-free mice exhibit profound gut microbiota-dependent alterations of intestinal endocannabinoidome signaling. J. Lipid Res. 2020, 61, 70–85. [Google Scholar] [CrossRef] [PubMed]
- Markey, L.; Hooper, A.; Melon, L.C.; Baglot, S.; Hill, M.N.; Maguire, J.; Kumamoto, C.A. Colonization with the commensal fungus Candida albicans perturbs the gut-brain axis through dysregulation of endocannabinoid signaling. Psychoneuroendocrinology 2020, 121, 104808. [Google Scholar] [CrossRef]
- Forte, N.; Fernandez-Rilo, A.C.; Palomba, L.; Di Marzo, V.; Cristino, L. Obesity Affects the Microbiota-Gut-Brain Axis and the Regulation Thereof by Endocannabinoids and Related Mediators. Int J. Mol. Sci. 2020, 21, 1554. [Google Scholar] [CrossRef]
- Mehrpouya-Bahrami, P.; Chitrala, K.N.; Ganewatta, M.S.; Tang, C.; Murphy, E.A.; Enos, R.T.; Velazquez, K.T.; McCellan, J.; Nagarkatti, M.; Nagarkatti, P. Blockade of CB1 cannabinoid receptor alters gut microbiota and attenuates inflammation and diet-induced obesity. Sci. Rep. 2017, 7, 15645. [Google Scholar] [CrossRef]
- Muccioli, G.G.; Naslain, D.; Backhed, F.; Reigstad, C.S.; Lambert, D.M.; Delzenne, N.M.; Cani, P.D. The endocannabinoid system links gut microbiota to adipogenesis. Mol. Syst. Biol. 2010, 6, 392. [Google Scholar] [CrossRef]
- Ellermann, M.; Pacheco, A.R.; Jimenez, A.G.; Russell, R.M.; Cuesta, S.; Kumar, A.; Zhu, W.; Vale, G.; Martin, S.A.; Raj, P.; et al. Endocannabinoids Inhibit the Induction of Virulence in Enteric Pathogens. Cell 2020. [Google Scholar] [CrossRef]
- Sclafani, A. Gut-brain nutrient signaling. Appetition vs. satiation. Appetite 2013, 71, 454–458. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
DiPatrizio, N.V. Endocannabinoids and the Gut-Brain Control of Food Intake and Obesity. Nutrients 2021, 13, 1214. https://doi.org/10.3390/nu13041214
DiPatrizio NV. Endocannabinoids and the Gut-Brain Control of Food Intake and Obesity. Nutrients. 2021; 13(4):1214. https://doi.org/10.3390/nu13041214
Chicago/Turabian StyleDiPatrizio, Nicholas V. 2021. "Endocannabinoids and the Gut-Brain Control of Food Intake and Obesity" Nutrients 13, no. 4: 1214. https://doi.org/10.3390/nu13041214
APA StyleDiPatrizio, N. V. (2021). Endocannabinoids and the Gut-Brain Control of Food Intake and Obesity. Nutrients, 13(4), 1214. https://doi.org/10.3390/nu13041214