
Obesity, Nutrients and the Immune System in the Era of COVID-19



Abstract
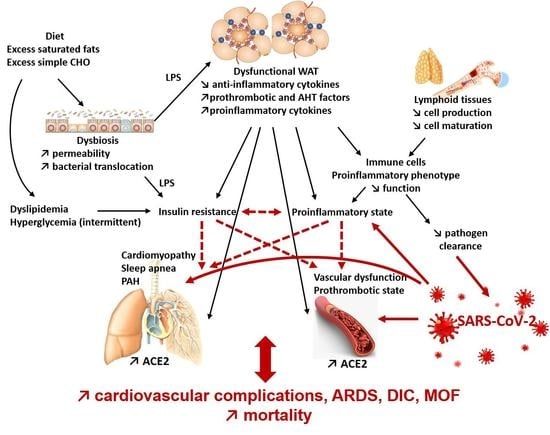
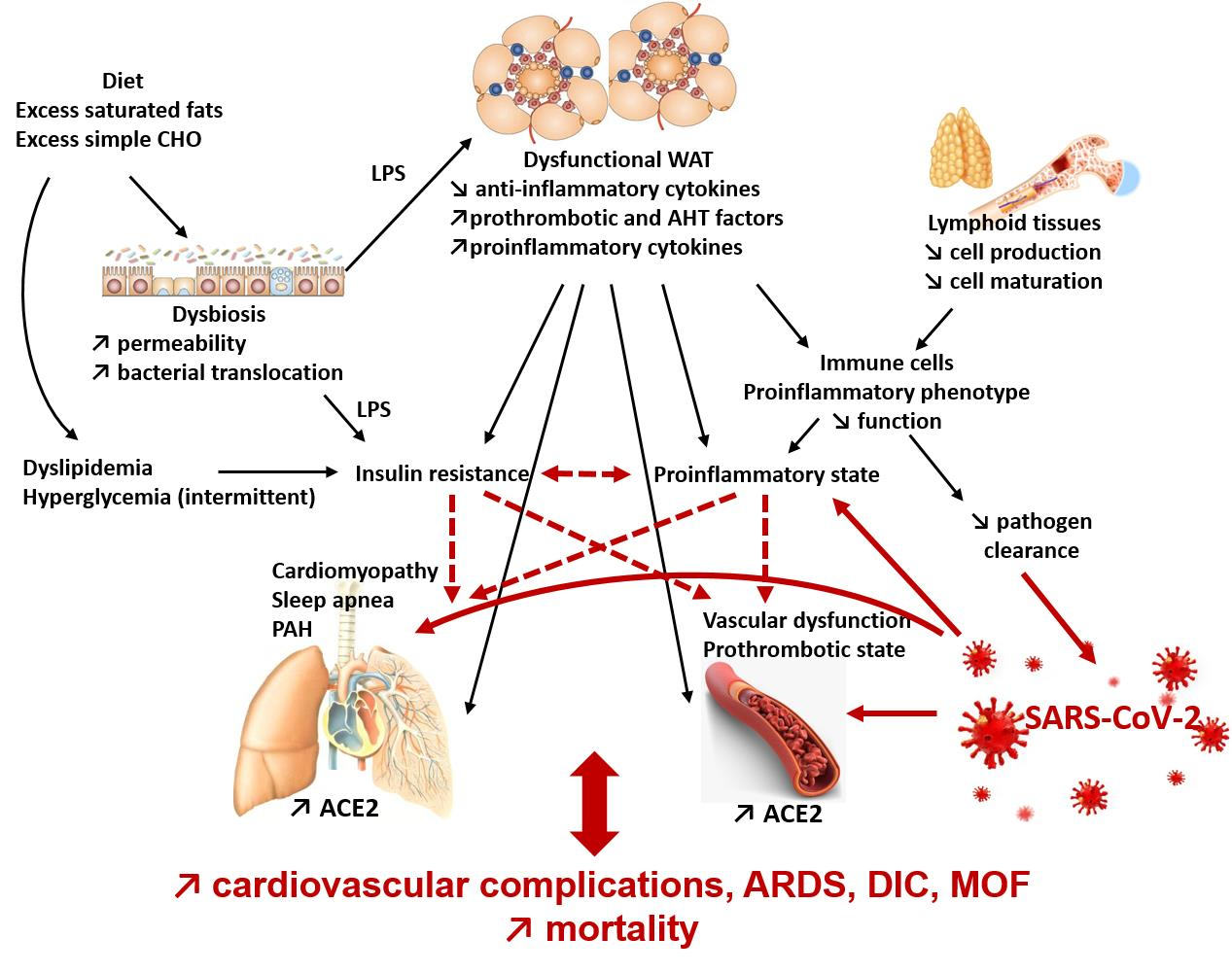{kind=link}
1. Introduction
2. Interactions between Adipose Tissue and Immunity
2.1. Leptin
2.2. Adiponectin
3. The Inflammatory State of Adipose Tissue during Obesity
3.1. Obesity and Inflammasome
3.2. Diet and Inflammation
3.3. Pro-Inflammatory Adipokines
3.4. Adipose Cell Apoptosis
4. Dysimmunity of Obesity
4.1. The Consequences of Obesity on Lymphoid Tissues
4.2. The Consequences of Obesity on Innate Immunity
4.2.1. Neutrophils
4.2.2. Macrophages
4.2.3. Dendritic Cells
4.2.4. NK Cells
4.3. The Consequences of Obesity on Adaptive Immunity
4.3.1. T Lymphocytes
4.3.2. B Lymphocytes
5. Obesity and COVID-19
5.1. Coronavirus Disease-2019
5.2. Obesity: A Risk of Severe COVID-19
5.2.1. Obesity, COVID-19, and Hyperinflammation
5.2.2. Obesity, COVID-19, and Endothelial Dysfunction
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Dhurandhar, N.V.; Bailey, D.; Thomas, D. Interaction of obesity and infections. Obes. Rev. Off. J. Int. Assoc. Study Obes. 2015, 16, 1017–1729. [Google Scholar] [CrossRef]
- Bansal, R.; Gubbi, S.; Muniyappa, R. Metabolic syndrome and COVID 19: Endocrine-immune-vascular interactions shapes clinical course. Endocrinology 2020, 161, 1–15. [Google Scholar] [CrossRef]
- Korakas, E.; Ikonomidis, I.; Kousathana, F.; Balampanis, K.; Kountouri, A.; Raptis, A.; Palaiodimou, L.; Kokkinos, A.; Lambadiari, V. Obesity and COVID-19: Immune and metabolic derangement as a possible link to adverse clinical outcomes. Am. J. Physiol. Endocrinol. Metab. 2020, 319, E105–E109. [Google Scholar] [CrossRef] [PubMed]
- Rajala, M.W.; Scherer, P.E. Minireview: The adipocyte—at the crossroads of energy homeostasis, inflammation, and atherosclerosis. Endocrinology 2003, 144, 3765–3773. [Google Scholar] [CrossRef]
- Procaccini, C.; Lourenco, E.V.; Matarese, G.; La Cava, A. Leptin signaling: A key pathway in immune responses. Curr. Signal Transduct Ther. 2009, 4, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Ahima, R.S.; Flier, J.S. Leptin. Annu. Rev. Physiol. 2000, 62, 413–437. [Google Scholar] [CrossRef]
- Vernooy, J.H.J.; Ubags, N.D.J.; Brusselle, G.G.; Tavernier, J.; Suratt, B.T.; Joos, G.F.; Wouters, E.F.; Bracke, K.R. Leptin as regulator of pulmonary immune responses: Involvement in respiratory diseases. Pulm. Pharmacol. Ther. 2013, 26, 464–472. [Google Scholar] [CrossRef] [PubMed]
- Francisco, V.; Pino, J.; Campos-Cabaleiro, V.; Ruiz-Fernández, C.; Mera, A.; Gonzalez-Gay, M.A.; Gómez, R.; Gualillo, O. Obesity, fat mass and immune system: Role for leptin. Front. Physiol. 2018, 9, 640. [Google Scholar] [CrossRef]
- Maurya, R.; Bhattacharya, P.; Dey, R.; Nakhasi, H.L. Leptin functions in infectious diseases. Front. Immunol. 2018, 9, 2741. [Google Scholar] [CrossRef] [PubMed]
- Alwarawrah, Y.; Kiernan, K.; MacIver, N.J. Changes in nutritional status impact immune cell metabolism and function. Front. Immunol. 2018, 9, 1055. [Google Scholar] [CrossRef]
- Wang, Z.V.; Scherer, P.E. Adiponectin, the past two decades. J. Mol. Cell Biol. 2016, 8, 93–100. [Google Scholar] [CrossRef]
- Bastard, J.-P.; Maachi, M.; Lagathu, C.; Kim, M.J.; Caron, M.; Vidal, H.; Capeau, J.; Feve, B. Recent advances in the relationship between obesity, inflammation, and insulin resistance. Eur. Cytokine Netw. 2006, 17, 4–12. [Google Scholar]
- Ouchi, N.; Parker, J.L.; Lugus, J.J.; Walsh, K. Adipokines in inflammation and metabolic disease. Nat. Rev. Immunol. 2011, 11, 85–97. [Google Scholar] [CrossRef]
- Claycombe-Larson, K.J.; Alvine, T.; Wu, D.; Kalupahana, N.S.; Moustaid-Moussa, N.; Roemmich, J.N. Nutrients and immunometabolism: Role of macrophage NLRP3. J. Nutr. 2020, 150, 1693–1704. [Google Scholar] [CrossRef]
- López-Reyes, A.; Martinez-Armenta, C.; Espinosa-Velázquez, R.; Vázquez-Cárdenas, P.; Cruz-Ramos, M.; Palacios-Gonzalez, B.; Gomez-Quiroz, L.E.; Martínez-Nava, G.A. NLRP3 inflammasome: The stormy link between obesity and COVID-19. Front. Immunol. 2020, 11, 570251. [Google Scholar] [CrossRef]
- Aguilera, M.; Darby, T.; Melgar, S. The complex role of inflammasomes in the pathogenesis of Inflammatory Bowel Diseases—lessons learned from experimental models. Cytokine Growth Factor Rev. 2014, 25, 715–730. [Google Scholar] [CrossRef] [PubMed]
- Ta, A.; Vanaja, S.K. Inflammasome activation and evasion by bacterial pathogens. Curr. Opin. Immunol. 2020, 68, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Freeman, T.L.; Swartz, T.H. Targeting the NLRP3 inflammasome in severe COVID-19. Front. Immunol. 2020, 11, 1518. [Google Scholar] [CrossRef] [PubMed]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, A.R.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Cariou, B.; Byrne, C.D.; Loomba, R.; Sanyal, A.J. NAFLD as a metabolic disease in humans: A literature review. Diabetes Obes. Metab. 2021. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef]
- Bergheim, I.; Weber, S.; Vos, M.; Krämer, S.; Volynets, V.; Kaserouni, S.; McClain, C.J.; Bischoff, S.C. Antibiotics protect against fructose-induced hepatic lipid accumulation in mice: Role of endotoxin. J. Hepatol. 2008, 48, 983–992. [Google Scholar] [CrossRef]
- Tilg, H.; Adolph, T.E.; Moschen, A.R. Multiple parallel hits hypothesis in NAFLD—revisited after a decade. Hepatology 2020. [Google Scholar] [CrossRef] [PubMed]
- Brun, P.; Castagliuolo, I.; Di Leo, V.; Buda, A.; Pinzani, M.; Palù, G.; Martines, D. Increased intestinal permeability in obese mice: New evidence in the pathogenesis of nonalcoholic steatohepatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 292, G518–G525. [Google Scholar] [CrossRef] [PubMed]
- Gulhane, M.; Murray, L.; Lourie, R.; Tong, H.; Sheng, Y.H.; Wang, R.; Kang, A.; Schreiber, V.; Wong, K.Y.; Magor, G.; et al. High fat diets induce colonic epithelial cell stress and inflammation that is reversed by IL-22. Sci. Rep. 2016, 6, 28990. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Nemoto, Y.; Takei, Y.; Morikawa, R.; Oshima, S.; Nagaishi, T.; Okamoto, R.; Tsuchiya, K.; Nakamura, T.; Stutte, S.; et al. High-fat diet-derived free fatty acids impair the intestinal immune system and increase sensitivity to intestinal epithelial damage. Biochem. Biophys. Res. Commun. 2020, 522, 971–977. [Google Scholar] [CrossRef]
- De Bandt, J.-P.; Waligora-Dupriet, A.-J.; Butel, M.-J. Intestinal microbiota in inflammation and insulin resistance: Relevance to humans. Curr. Opin. Clin. Nutr. Metab. Care 2011, 14, 334–340. [Google Scholar] [CrossRef] [PubMed]
- Jegatheesan, P.; De Bandt, J.-P. Fructose and NAFLD: The multifaceted aspects of fructose metabolism. Nutrients 2017, 9, 230. [Google Scholar] [CrossRef]
- Ghanim, H.; Abuaysheh, S.; Sia, C.L.; Korzeniewski, K.; Chaudhuri, A.; Fernandez-Real, J.M.; Dandona, P. Increase in plasma endotoxin concentrations and the expression of Toll-like receptors and suppressor of cytokine signaling-3 in mononuclear cells after a high-fat, high-carbohydrate meal: Implications for insulin resistance. Diabetes Care 2009, 32, 2281–2287. [Google Scholar] [CrossRef]
- Nagareddy, P.R.; Murphy, A.J.; Stirzaker, R.A.; Hu, Y.; Yu, S.; Miller, R.G.; Ramkhelawon, B.; Distel, E.; Westerterp, M.; Huang, L.-S.; et al. Hyperglycemia promotes myelopoiesis and impairs the resolution of atherosclerosis. Cell Metab. 2013, 17, 695–708. [Google Scholar] [CrossRef] [PubMed]
- Flynn, M.C.; Kraakman, M.J.; Tikellis, C.; Lee, M.K.S.; Hanssen, N.M.J.; Kammoun, H.L.; Pickering, R.J.; Dragoljevic, D.; Al-Sharea, A.; Barrett, T.J.; et al. Transient intermittent hyperglycemia accelerates atherosclerosis by promoting myelopoiesis. Circ. Res. 2020, 127, 877–892. [Google Scholar] [CrossRef]
- Wu, N.; Shen, H.; Liu, H.; Wang, Y.; Bai, Y.; Han, P. Acute blood glucose fluctuation enhances rat aorta endothelial cell apoptosis, oxidative stress and pro-inflammatory cytokine expression in vivo. Cardiovasc. Diabetol. 2016, 15, 109. [Google Scholar] [CrossRef]
- Al-Rashed, F.; Sindhu, S.; Arefanian, H.; Al Madhoun, A.; Kochumon, S.; Thomas, R.; Al-Kandari, S.; Alghaith, A.; Jacob, T.; Al-Mulla, F.; et al. Repetitive intermittent hyperglycemia drives the M1 polarization and inflammatory responses in THP-1 macrophages through the mechanism involving the TLR4-IRF5 pathway. Cells 2020, 9, 1892. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Luo, H.-Y.; Liu, Q.; Xiao, Y.; Tang, L.; Zhong, F.; Huang, G.; Xu, J.-M.; Xu, A.-M.; Zhou, Z.-G.; et al. Intermittent high glucose exacerbates A-FABP activation and inflammatory response through TLR4-JNK signaling in THP-1 Cells. J. Immunol. Res. 2018, 2018, 1319272. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Kokoeva, M.V.; Inouye, K.; Tzameli, I.; Yin, H.; Flier, J.S. TLR4 links innate immunity and fatty acid-induced insulin resistance. J. Clin. Investig. 2006, 116, 3015–3025. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.T.A.; Favelyukis, S.; Nguyen, A.-K.; Reichart, D.; Scott, P.A.; Jenn, A.; Liu-Bryan, R.; Glass, C.K.; Neels, J.G.; Olefsky, J.M. A subpopulation of macrophages infiltrates hypertrophic adipose tissue and is activated by free fatty acids via Toll-like receptors 2 and 4 and JNK-dependent pathways. J. Biol. Chem. 2007, 282, 35279–35292. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, C.M.; McGillicuddy, F.C.; Harford, K.A.; Finucane, O.M.; Mills, K.H.G.; Roche, H.M. Dietary saturated fatty acids prime the NLRP3 inflammasome via TLR4 in dendritic cells-implications for diet-induced insulin resistance. Mol. Nutr. Food Res. 2012, 56, 1212–1222. [Google Scholar] [CrossRef]
- Di Conza, G.; Ho, P.-C. ER stress responses: An emerging modulator for innate immunity. Cells 2020, 9, 695. [Google Scholar] [CrossRef]
- Lancaster, G.I.; Langley, K.G.; Berglund, N.A.; Kammoun, H.L.; Reibe, S.; Estevez, E.; Weir, J.; Mellett, N.A.; Pernes, G.; Conway, J.R.; et al. Evidence that TLR4 is not a receptor for saturated fatty acids but mediates lipid-induced inflammation by reprogramming macrophage metabolism. Cell Metab. 2018, 27, 1096–1110.e5. [Google Scholar] [CrossRef]
- McKernan, K.; Varghese, M.; Patel, R.; Singer, K. Role of TLR4 in the induction of inflammatory changes in adipocytes and macrophages. Adipocyte 2020, 9, 212–222. [Google Scholar] [CrossRef]
- Andersen, C.J.; Murphy, K.E.; Fernandez, M.L. Impact of obesity and metabolic syndrome on immunity. Adv. Nutr. 2016, 7, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Glass, C.K.; Witztum, J.L. Atherosclerosis the road ahead. Cell 2001, 104, 503–516. [Google Scholar] [CrossRef]
- Calder, P.C.; Bosco, N.; Bourdet-Sicard, R.; Capuron, L.; Delzenne, N.; Doré, J.; Franceschi, C.; Lehtinen, M.J.; Recker, T.; Salvioli, S.; et al. Health relevance of the modification of low grade inflammation in ageing (inflammageing) and the role of nutrition. Ageing Res. Rev. 2017, 40, 95–119. [Google Scholar] [CrossRef]
- Guilherme, A.; Virbasius, J.V.; Puri, V.; Czech, M.P. Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nat. Rev. Mol. Cell. Biol. 2008, 9, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Bray, G.A.; Heisel, W.E.; Afshin, A.; Jensen, M.D.; Dietz, W.H.; Long, M.; Kushner, R.F.; Daniels, S.R.; Wadden, T.A.; Tsai, A.G.; et al. The science of obesity management: An endocrine society scientific statement. Endocr. Rev. 2018, 39, 79–132. [Google Scholar] [CrossRef]
- Wu, D.; Molofsky, A.B.; Liang, H.-E.; Ricardo-Gonzalez, R.R.; Jouihan, H.A.; Bando, J.K.; Chawla, A.; Locksley, R.M. Eosinophils sustain adipose alternatively activated macrophages associated with glucose homeostasis. Science 2011, 332, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Li, C. Leptin: A multifunctional hormone. Cell Res. 2000, 10, 81–92. [Google Scholar] [CrossRef]
- Frydrych, L.M.; Bian, G.; O’Lone, D.E.; Ward, P.A.; Delano, M.J. Obesity and type 2 diabetes mellitus drive immune dysfunction, infection development, and sepsis mortality. J. Leukoc. Biol. 2018, 104, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Pangrazzi, L.; Naismith, E.; Miggitsch, C.; Carmona, A.J.A.; Keller, M.; Grubeck-Loebenstein, B.; Weinberger, B. The impact of body mass index on adaptive immune cells in the human bone marrow. Immun. Ageing 2020, 17, 15. [Google Scholar] [CrossRef]
- Brotfain, E.; Hadad, N.; Shapira, Y.; Avinoah, E.; Zlotnik, A.; Raichel, L.; Levy, R. Neutrophil functions in morbidly obese subjects. Clin. Exp. Immunol. 2015, 181, 156–163. [Google Scholar] [CrossRef]
- O’Shea, D.; Cawood, T.J.; O’Farrelly, C.; Lynch, L. Natural killer cells in obesity: Impaired function and increased susceptibility to the effects of cigarette smoke. PLoS ONE 2010, 5, e8660. [Google Scholar] [CrossRef]
- Arias-Loste, M.T.; Iruzubieta, P.; Puente, Á.; Ramos, D.; Santa Cruz, C.; Estébanez, Á.; Llerena, S.; Alonso-Martín, C.; San Segundo, D.; Álvarez, L.; et al. Increased expression profile and functionality of TLR6 in peripheral blood mononuclear cells and hepatocytes of morbidly obese patients with non-alcoholic fatty liver disease. Int. J. Mol. Sci. 2016, 17, 1878. [Google Scholar] [CrossRef]
- Bernotiene, E.; Palmer, G.; Gabay, C. The role of leptin in innate and adaptive immune responses. Arthritis Res. Ther. 2006, 8, 217. [Google Scholar] [CrossRef][Green Version]
- Richard, C.; Wadowski, M.; Goruk, S.; Cameron, L.; Sharma, A.M.; Field, C.J. Individuals with obesity and type 2 diabetes have additional immune dysfunction compared with obese individuals who are metabolically healthy. BMJ Open Diabetes Res. Care 2017, 5, e000379. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, D.; Corrigan, M.; Dunne, M.R.; Jackson, R.; Woods, C.; Gaoatswe, G.; Moynagh, P.N.; O’Connell, J.; Hogan, A.E. Changes in human dendritic cell number and function in severe obesity may contribute to increased susceptibility to viral infection. Int. J. Obes. 2005, 2013, 1510–1513. [Google Scholar] [CrossRef]
- Bähr, I.; Spielmann, J.; Quandt, D.; Kielstein, H. Obesity-associated alterations of Natural Killer cells and immunosurveillance of cancer. Front. Immunol. 2020, 11, 245. [Google Scholar] [CrossRef]
- Papadopoulou, M.; Sanchez, G.; Vermijlen, D. Innate and adaptive γδ T cells: How, when, and why. Immunol. Rev. 2020, 298, 99–116. [Google Scholar] [CrossRef] [PubMed]
- Honce, R.; Schultz-Cherry, S. Impact of obesity on influenza A virus pathogenesis, immune response, and evolution. Front. Immunol. 2019, 10, 1071. [Google Scholar] [CrossRef]
- Collins, N. Dietary Regulation of memory T cells. Int. J. Mol. Sci. 2020, 21, 4363. [Google Scholar] [CrossRef] [PubMed]
- Rebeles, J.; Green, W.D.; Alwarawrah, Y.; Nichols, A.G.; Eisner, W.; Danzaki, K.; MacIver, N.J.; A Beck, M. Obesity-induced changes in T-cell metabolism are associated with impaired memory T-cell response to influenza and are not reversed with weight loss. J. Infect. Dis. 2019, 219, 1652–1661. [Google Scholar] [CrossRef] [PubMed]
- Farnsworth, C.W.; Schott, E.M.; Benvie, A.; Kates, S.L.; Schwarz, E.M.; Gill, S.R.; Zuscik, M.J.; Mooney, R.A. Exacerbated Staphylococcus aureus foot infections in obese/diabetic mice are associated with impaired germinal center ceactions, Ig class switching, and humoral immunity. J. Immunol. 2018, 201, 560–572. [Google Scholar] [CrossRef] [PubMed]
- Frasca, D.; Ferracci, F.; Diaz, A.; Romero, M.; Lechner, S.; Blomberg, B.B. Obesity decreases B cell responses in young and elderly individuals. Obesity 2016, 24, 615–625. [Google Scholar] [CrossRef]
- Rodrigues, T.S.; De Sá, K.S.G.; Ishimoto, A.Y.; Becerra, A.; Oliveira, S.; Almeida, L.; Gonçalves, A.V.; Perucello, D.B.; Andrade, W.A.; Castro, R.; et al. Inflammasomes are activated in response to SARS-CoV-2 infection and are associated with COVID-19 severity in patients. J. Exp. Med. 2021, 218. [Google Scholar] [CrossRef] [PubMed]
- Luzi, L.; Radaelli, M.G. Influenza and obesity: Its odd relationship and the lessons for COVID-19 pandemic. Acta Diabetol. 2020, 57, 759–764. [Google Scholar] [CrossRef]
- Stefan, N.; Birkenfeld, A.L.; Schulze, M.B.; Ludwig, D.S. Obesity and impaired metabolic health in patients with COVID-19. Nat. Rev. Endocrinol. 2020, 16, 341–342. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Bandt, J.-P.; Monin, C. Obesity, Nutrients and the Immune System in the Era of COVID-19. Nutrients 2021, 13, 610. https://doi.org/10.3390/nu13020610
De Bandt J-P, Monin C. Obesity, Nutrients and the Immune System in the Era of COVID-19. Nutrients. 2021; 13(2):610. https://doi.org/10.3390/nu13020610
Chicago/Turabian StyleDe Bandt, Jean-Pascal, and Charlotte Monin. 2021. "Obesity, Nutrients and the Immune System in the Era of COVID-19" Nutrients 13, no. 2: 610. https://doi.org/10.3390/nu13020610
APA StyleDe Bandt, J.-P., & Monin, C. (2021). Obesity, Nutrients and the Immune System in the Era of COVID-19. Nutrients, 13(2), 610. https://doi.org/10.3390/nu13020610