
Berry-Derived Polyphenols in Cardiovascular Pathologies: Mechanisms of Disease and the Role of Diet and Sex





Abstract
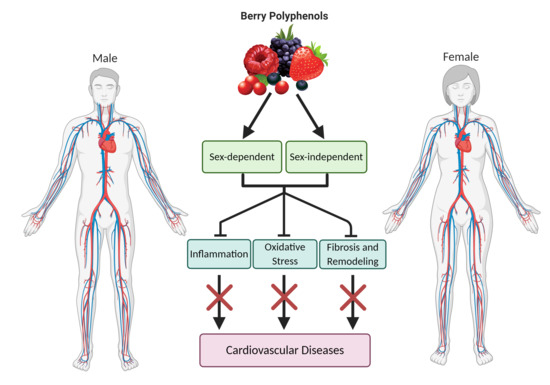
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Berry-Derived Polyphenols
2.1. Metabolism of Berry-Derived Polyphenols
2.2. Sex-Specific Effects of Polyphenols on the Microbiota
2.3. Berry Polyphenol and Estrogen Receptor Interaction
3. Inflammatory Signaling
3.1. Toll-Like Receptor-4
3.2. Tumor Necrosis Factor Receptor
3.3. Berry Polyphenols in Inflammation
4. Oxidative Stress
4.1. NADPH Oxidase and Xanthine Oxidase
4.2. Role of NOS in Oxidative Stress
4.3. Endogenous Antioxidant System
Nrf2 and Sirt1
4.4. Berry Polyphenols in Oxidative Stress
4.4.1. Berry Polyphenols in Sirt1 and Nrf2 Regulation
4.4.2. Berry Polyphenols in Endothelial Function
4.4.3. Potential ROS-Independent Effects of Berry Polyphenols
5. Cardiac and Vascular Remodeling
5.1. Apoptosis
5.2. Fibrosis
5.3. Berry Polyphenols in Cardiac and Vascular Remodeling
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Virani, S.; Alonso, A.; Benjamin, E.; Bittencourt, M.; Callaway, C.; Carson, A.; Chamberlain, A.; Chang, A.; Cheng, S.; Delling, F.; et al. Heart Disease and Stroke Statistics—2020 Update: A Report From the American Heart Association. Circulation 2020, 141, e139–e596. [Google Scholar] [CrossRef] [PubMed]
- Waheed, N.; Elias-Smale, S.; Malas, W.; Maas, A.; Sedlak, T.; Tremmel, J.; Mehta, P. Sex differences in non-obstructive coronary artery disease. Cardiovasc. Res. 2020, 116, 829–840. [Google Scholar] [CrossRef] [PubMed]
- Patel, H.; Rosengren, A.; Ekman, I. Symptoms in acute coronary syndromes: Does sex make a difference? Am. Heart J. 2004, 148, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Muller-Delp, J. The coronary microvasculation in health and disease. Physiology 2013, 2013, 1–24. [Google Scholar]
- Schwertz, D.; Penckofer, S. Sex differences and the effects of sex hormones on hemostasis and vascular reactivity. Heart Lung J. Crit. Care 2001, 30, 401–426. [Google Scholar] [CrossRef]
- Lam, C.; Arnott, C.; Beale, A.; Chandramouli, C.; Hilfiker-Kleiner, D.; Kaye, D.; Ky, B.; Santema, B.; Silwa, K.; Voors, A. Sex differences in heart failure. Eur. Heart J. 2019, 40, 3859c–3868c. [Google Scholar] [CrossRef]
- Azevedo, P.S.; Polegato, B.F.; Minicucci, M.F.; Paiva, S.A.; Zornoff, L.A. Cardiac Remodeling: Concepts, Clinical Impact, Pathophysiological Mechanisms and Pharmacologic Treatment. Arq. Bras. Cardiol. 2016, 106, 62–69. [Google Scholar] [CrossRef]
- Steven, S.; Frenis, K.; Oelze, M.; Kalinovic, S.; Kuntic, M.; Bayo Jimenez, M.T.; Vujacic-Mirski, K.; Helmstadter, J.; Kroller-Schon, S.; Munzel, T.; et al. Vascular Inflammation and Oxidative Stress: Major Triggers for Cardiovascular Disease. Oxid. Med. Cell. Longev. 2019, 2019, 7092151. [Google Scholar] [CrossRef]
- Renna, N.F.; de Las Heras, N.; Miatello, R.M. Pathophysiology of vascular remodeling in hypertension. Int. J. Hypertens. 2013, 2013, 808353. [Google Scholar] [CrossRef]
- Boese, A.; Kim, S.; Yin, K.-J.; Lee, J.-P.; Hamblin, M. Sex differences in vascular physiology and pathophysiology: Estrogen and androgen signaling in health and disease. Am. J. Physiol. Heart Circ. Physiol. 2017, 313, H524–H545. [Google Scholar] [CrossRef]
- Dinu, M.; Abbate, R.; Gensini, G.F.; Casini, A.; Sofi, F. Vegetarian, vegan diets and multiple health outcomes: A systematic review with meta-analysis of observational studies. Crit. Rev. Food Sci. Nutr. 2017, 57, 3640–3649. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Caulfield, L.E.; Garcia-Larsen, V.; Steffen, L.M.; Coresh, J.; Rebholz, C.M. Plant-Based Diets Are Associated With a Lower Risk of Incident Cardiovascular Disease, Cardiovascular Disease Mortality, and All-Cause Mortality in a General Population of Middle-Aged Adults. J. Am. Heart Assoc. 2019, 8, e012865. [Google Scholar] [CrossRef] [PubMed]
- Najjar, R.S.; Moore, C.E.; Montgomery, B.D. Consumption of a defined, plant-based diet reduces lipoprotein(a), inflammation, and other atherogenic lipoproteins and particles within 4 weeks. Clin. Cardiol. 2018, 41, 1062–1068. [Google Scholar] [CrossRef] [PubMed]
- Najjar, R.S.; Montgomery, B.D. A defined, plant-based diet as a potential therapeutic approach in the treatment of heart failure: A clinical case series. Complement. Ther. Med. 2019, 45, 211–214. [Google Scholar] [CrossRef]
- Esselstyn, C.B., Jr.; Gendy, G.; Doyle, J.; Golubic, M.; Roizen, M.F. A way to reverse CAD? J. Fam. Pract. 2014, 63, 356b–364b. [Google Scholar]
- Lin, P.; Leslie, D.; Levine, M.; Davis, G.; Esselstyn, C. Plant-Based Diet Reverses Vascular Endothelial Dysfunction in Patients with Peripheral Arterial Disease. Int. J. Dis. Reversal Prev. 2020, 2, 15. [Google Scholar]
- Upadhyay, S.; Dixit, M. Role of Polyphenols and Other Phytochemicals on Molecular Signaling. Oxid. Med. Cell. Longev. 2015, 2015, 504253. [Google Scholar] [CrossRef]
- Olas, B. Berry Phenolic Antioxidant-Implications for Human Health? Front. Pharmacol. 2018, 9, 78. [Google Scholar] [CrossRef]
- Kimble, R.; Keane, K.M.; Lodge, J.K.; Howatson, G. Dietary intake of anthocyanins and risk of cardiovascular disease: A systematic review and meta-analysis of prospective cohort studies. Crit. Rev. Food Sci. Nutr. 2019, 59, 3032–3043. [Google Scholar] [CrossRef]
- McCullough, M.L.; Peterson, J.J.; Patel, R.; Jacques, P.F.; Shah, R.; Dwyer, J.T. Flavonoid intake and cardiovascular disease mortality in a prospective cohort of US adults. Am. J. Clin. Nutr. 2012, 95, 454–464. [Google Scholar] [CrossRef]
- Huang, H.; Chen, G.; Liao, D.; Zhu, Y.; Xue, X. Effects of Berries Consumption on Cardiovascular Risk Factors: A Meta-analysis with Trial Sequential Analysis of Randomized Controlled Trials. Sci. Rep. 2016, 6, 23625. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gallegos, J.L.; Haskell-Ramsay, C.; Lodge, J.K. Effects of chronic consumption of specific fruit (berries, citrus and cherries) on CVD risk factors: A systematic review and meta-analysis of randomised controlled trials. Eur. J. Nutr. 2020. [Google Scholar] [CrossRef] [PubMed]
- Del Bo’, C.; Bernardi, S.; Marino, M.; Porrini, M.; Tucci, M.; Guglielmetti, S.; Cherubini, A.; Carrieri, B.; Kirkup, B.; Kroon, P.; et al. Systematic Review on Polyphenol Intake and Health Outcomes: Is there Sufficient Evidence to Define a Health-Promoting Polyphenol-Rich Dietary Pattern? Nutrients 2019, 11, 1355. [Google Scholar]
- Zamora-Ros, R.; Rabassa, M.; Cherubini, A.; Urpí-Sardà, M.; Bandinelli, S.; Ferrucci, L.; Andres-Lacueva, C. High Concentrations of a Urinary Biomarker of Polyphenol Intake Are Associated with Decreased Mortality in Older Adults. J. Nutr. 2013, 143, 1445–1450. [Google Scholar] [CrossRef]
- Rodriguez-Garcia, C.; Sanchez-Quesada, C.; Toledo, E.; Delgado-Rodriguez, M.; Gaforio, J.J. Naturally Lignan-Rich Foods: A Dietary Tool for Health Promotion? Molecules 2019, 24, 917. [Google Scholar] [CrossRef]
- Blaszczyk, A.; Sady, S.; Sielicka, M. The stilbene profile in edible berries. Phytochem. Rev. 2019, 18, 37–67. [Google Scholar] [CrossRef]
- Panche, A.N.; Diwan, A.D.; Chandra, S.R. Flavonoids: An overview. J. Nutr. Sci. 2016, 5, e47. [Google Scholar] [CrossRef]
- Pathak, S.B.A.; Banerjee, A.; Celep, G.S.; Bissi, L.; Marotta, F. Metabolism of Dietary Polyphenols by Human Gut Microbiota and Their Health Benefits; Academic Press: New York, NY, USA, 2018. [Google Scholar]
- Sandhu, A.K.; Miller, M.G.; Thangthaeng, N.; Scott, T.M.; Shukitt-Hale, B.; Edirisinghe, I.; Burton-Freeman, B. Metabolic fate of strawberry polyphenols after chronic intake in healthy older adults. Food Funct. 2018, 9, 96–106. [Google Scholar] [CrossRef]
- de Ferrars, R.M.; Czank, C.; Zhang, Q.; Botting, N.P.; Kroon, P.A.; Cassidy, A.; Kay, C.D. The pharmacokinetics of anthocyanins and their metabolites in humans. Br. J. Pharmacol. 2014, 171, 3268–3282. [Google Scholar] [CrossRef]
- Manach, C.; Scalbert, A.; Morand, C.; Remesy, C.; Jimenez, L. Polyphenols: Food sources and bioavailability. Am. J. Clin. Nutr. 2004, 79, 727–747. [Google Scholar] [CrossRef]
- Williamson, G.; Clifford, M.N. Colonic metabolites of berry polyphenols: The missing link to biological activity? Br. J. Nutr. 2010, 104 (Suppl. 3), S48–S66. [Google Scholar] [CrossRef] [PubMed]
- Clifford, M.N.; Scalbert, A. Ellagitannins-nature, occurrence and dietary burden. J. Sci. Food Agric. 2000, 80, 1118–1125. [Google Scholar] [CrossRef]
- Koponen, J.M.; Happonen, A.M.; Mattila, P.H.; Torronen, A.R. Contents of anthocyanins and ellagitannins in selected foods consumed in Finland. J. Agric. Food Chem. 2007, 55, 1612–1619. [Google Scholar] [CrossRef] [PubMed]
- Lipinska, L.; Klewicka, E.; Sojka, M. The structure, occurrence and biological activity of ellagitannins: A general review. Acta Sci. Pol. Technol. Aliment. 2014, 13, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Basheer, L.; Kerem, Z. Interactions between CYP3A4 and Dietary Polyphenols. Oxid. Med. Cell. Longev. 2015, 2015, 854015. [Google Scholar] [CrossRef]
- Campesi, I.; Marino, M.; Cipolletti, M.; Romani, A.; Franconi, F. Put “gender glasses” on the effects of phenolic compounds on cardiovascular function and diseases. Eur. J. Nutr. 2018, 57, 2677–2691. [Google Scholar] [CrossRef]
- Ostlund, J.; Zlabek, V.; Zamaratskaia, G. In vitro inhibition of human CYP2E1 and CYP3A by quercetin and myricetin in hepatic microsomes is not gender dependent. Toxicology 2017, 381, 10–18. [Google Scholar] [CrossRef]
- Erlund, I.; Kosonen, T.; Alfthan, G.; Mäenpää, J.; Perttunen, K.; Kenraali, J.; Parantainen, J.; Aro, A. Pharmacokinetics of quercetin from quercetin aglycone and rutin in healthy volunteers. Pharmacokinet. Dispos. 2000, 56, 545–553. [Google Scholar] [CrossRef]
- Dufour, C.; Dangles, O. Flavonoid-serum albumin complexation: Determination of binding constants and binding sites by fluorescence spectroscopy. Biochim. Biophys. Acta 2005, 1721, 164–173. [Google Scholar] [CrossRef]
- Kitson, T.M. Spectrophotometric and kinetic studies on the binding of the bioflavonoid quercetin to bovine serum albumin. Biosci. Biotechnol. Biochem. 2004, 68, 2165–2170. [Google Scholar] [CrossRef] [PubMed]
- Ziberna, L.; Lunder, M.; Moze, S.; Vanzo, A.; Tramer, F.; Passamonti, S.; Drevensek, G. Acute cardioprotective and cardiotoxic effects of bilberry anthocyanins in ischemia-reperfusion injury: Beyond concentration-dependent antioxidant activity. Cardiovasc. Toxicol. 2010, 10, 283–294. [Google Scholar] [CrossRef] [PubMed]
- van Dijk, C.; Driessen, A.J.; Recourt, K. The uncoupling efficiency and affinity of flavonoids for vesicles. Biochem. Pharmacol. 2000, 60, 1593–1600. [Google Scholar] [CrossRef]
- Goodwin, B.L.; Ruthven, C.R.; Sandler, M. Gut flora and the origin of some urinary aromatic phenolic compounds. Biochem. Pharmacol. 1994, 47, 2294–2297. [Google Scholar] [CrossRef]
- Phipps, A.N.; Stewart, J.; Wright, B.; Wilson, I.D. Effect of diet on the urinary excretion of hippuric acid and other dietary-derived aromatics in rat. A complex interaction between diet, gut microflora and substrate specificity. Xenobiotica 1998, 28, 527–537. [Google Scholar] [CrossRef]
- Ziberna, L.; Tramer, F.; Moze, S.; Vrhovsek, U.; Mattivi, F.; Passamonti, S. Transport and bioactivity of cyanidin 3-glucoside into the vascular endothelium. Free Radic. Biol. Med. 2012, 52, 1750–1759. [Google Scholar] [CrossRef] [PubMed]
- Maestro, A.; Terdoslavich, M.; Vanzo, A.; Kuku, A.; Tramer, F.; Nicolin, V.; Micali, F.; Decorti, G.; Passamonti, S. Expression of bilitranslocase in the vascular endothelium and its function as a flavonoid transporter. Cardiovasc. Res. 2010, 85, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Ziberna, L.; Kim, J.H.; Auger, C.; Passamonti, S.; Schini-Kerth, V. Role of endothelial cell membrane transport in red wine polyphenols-induced coronary vasorelaxation: Involvement of bilitranslocase. Food Funct. 2013, 4, 1452–1456. [Google Scholar] [CrossRef]
- Ziberna, L.; Lunder, M.; Tramer, F.; Drevensek, G.; Passamonti, S. The endothelial plasma membrane transporter bilitranslocase mediates rat aortic vasodilation induced by anthocyanins. Nutr. Metab. Cardiovasc. Dis. 2013, 23, 68–74. [Google Scholar] [CrossRef]
- Ziberna, L.; Fornasaro, S.; Čvorović, J.; Tramer, F.; Passamonti, S. Bioavailability of Flavonoids: The Role of Cell Membrane Transporters. In Polyphenols in Human Health and Disease; Academic Press: New York, NY, USA, 2014; pp. 489–511. [Google Scholar] [CrossRef]
- Morris, M.E.; Lee, H.J.; Predko, L.M. Gender differences in the membrane transport of endogenous and exogenous compounds. Pharmacol. Rev. 2003, 55, 229–240. [Google Scholar] [CrossRef]
- Soukup, S.T.; Helppi, J.; Muller, D.R.; Zierau, O.; Watzl, B.; Vollmer, G.; Diel, P.; Bub, A.; Kulling, S.E. Phase II metabolism of the soy isoflavones genistein and daidzein in humans, rats and mice: A cross-species and sex comparison. Arch. Toxicol. 2016, 90, 1335–1347. [Google Scholar] [CrossRef]
- Battson, M.L.; Lee, D.M.; Weir, T.L.; Gentile, C.L. The gut microbiota as a novel regulator of cardiovascular function and disease. J. Nutr. Biochem. 2018, 56, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Witkowski, M.; Weeks, T.L.; Hazen, S.L. Gut Microbiota and Cardiovascular Disease. Circ. Res. 2020, 127, 553–570. [Google Scholar] [CrossRef] [PubMed]
- Santos-Marcos, J.; Rangel-Zuñiga, O.; Jimenez-Lucena, R.; Quintana-Navarro, G.; Garcia-Carpintero, S.; Malagon, M.; Landa, B.; Tena-Sempere, M.; Perez-Martinez, P.; Lopez-Miranda, J.; et al. Influence of gender and menopausal status on gut microbiota. Maturitas 2018, 116, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Edogawa, S.; Peters, S.; Jenkins, G.; Gurunathan, S.; Sundt, W.; Johnson, S.; Lennon, R.; Dyer, R.; Camilleri, M.; Kashyap, P.; et al. Sex differences in NSAID-induced perturbation of human intestinal barrier function and microbiota. FASEB J. 2018, 32. [Google Scholar] [CrossRef] [PubMed]
- Haro, C.; Rangel-Zuniga, O.A.; Alcala-Diaz, J.F.; Gomez-Delgado, F.; Perez-Martinez, P.; Delgado-Lista, J.; Quintana-Navarro, G.M.; Landa, B.B.; Navas-Cortes, J.A.; Tena-Sempere, M.; et al. Intestinal Microbiota Is Influenced by Gender and Body Mass Index. PLoS ONE 2016, 11, e0154090. [Google Scholar] [CrossRef] [PubMed]
- Sinha, T.; Vila, A.; Garmaeva, S.; Jankipersadsing, S.; Imhann, F.; Collij, V.; Bonder, M.; Jiang, X.; Gurry, T.; Alm, E.; et al. Analysis of 1135 gut metagenomes identifies sex-specific resistome profiles. Gut Microbes 2018, 10, 358–366. [Google Scholar] [CrossRef]
- Santos-Marcos, J.A.; Haro, C.; Vega-Rojas, A.; Alcala-Diaz, J.F.; Molina-Abril, H.; Leon-Acuna, A.; Lopez-Moreno, J.; Landa, B.B.; Tena-Sempere, M.; Perez-Martinez, P.; et al. Sex Differences in the Gut Microbiota as Potential Determinants of Gender Predisposition to Disease. Mol. Nutr. Food Res. 2019, 63, e1800870. [Google Scholar] [CrossRef]
- Dominianni, C.; Sinha, R.; Goedert, J.J.; Pei, Z.; Yang, L.; Hayes, R.B.; Ahn, J. Sex, body mass index, and dietary fiber intake influence the human gut microbiome. PLoS ONE 2015, 10, e0124599. [Google Scholar] [CrossRef]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef]
- Richards, L.B.; Li, M.; van Esch, B.C.; Garssen, J.; Folkerts, G. The effects of short-chain fatty acids on the cardiovascular system. PharmaNutrition 2016, 4, 68–111. [Google Scholar] [CrossRef]
- Jiao, X.; Wang, Y.; Lin, Y.; Lang, Y.; Li, E.; Zhang, X.; Zhang, Q.; Feng, Y.; Meng, X.; Li, B. Blueberry polyphenols extract as a potential prebiotic with anti-obesity effects on C57BL/6 J mice by modulating the gut microbiota. J. Nutr. Biochem. 2019, 64, 88–100. [Google Scholar] [CrossRef] [PubMed]
- Etxeberria, U.; Arias, N.; Boque, N.; Macarulla, M.T.; Portillo, M.P.; Martinez, J.A.; Milagro, F.I. Reshaping faecal gut microbiota composition by the intake of trans-resveratrol and quercetin in high-fat sucrose diet-fed rats. J. Nutr. Biochem. 2015, 26, 651–660. [Google Scholar] [CrossRef] [PubMed]
- Larrosa, M.; Yanez-Gascon, M.J.; Selma, M.V.; Gonzalez-Sarrias, A.; Toti, S.; Ceron, J.J.; Tomas-Barberan, F.; Dolara, P.; Espin, J.C. Effect of a low dose of dietary resveratrol on colon microbiota, inflammation and tissue damage in a DSS-induced colitis rat model. J. Agric. Food Chem. 2009, 57, 2211–2220. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Bose, S.; Seo, J.G.; Chung, W.S.; Lim, C.Y.; Kim, H. The effects of co-administration of probiotics with herbal medicine on obesity, metabolic endotoxemia and dysbiosis: A randomized double-blind controlled clinical trial. Clin. Nutr. 2014, 33, 973–981. [Google Scholar] [CrossRef] [PubMed]
- DiRienzo, D.B. Effect of probiotics on biomarkers of cardiovascular disease: Implications for heart-healthy diets. Nutr. Rev. 2014, 72, 18–29. [Google Scholar] [CrossRef]
- Gonzalez-Sarrias, A.; Romo-Vaquero, M.; Garcia-Villalba, R.; Cortes-Martin, A.; Selma, M.V.; Espin, J.C. The Endotoxemia Marker Lipopolysaccharide-Binding Protein is Reduced in Overweight-Obese Subjects Consuming Pomegranate Extract by Modulating the Gut Microbiota: A Randomized Clinical Trial. Mol. Nutr. Food Res. 2018, 62, e1800160. [Google Scholar] [CrossRef]
- Tzounis, X.; Rodriguez-Mateos, A.; Vulevic, J.; Gibson, G.R.; Kwik-Uribe, C.; Spencer, J.P. Prebiotic evaluation of cocoa-derived flavanols in healthy humans by using a randomized, controlled, double-blind, crossover intervention study. Am. J. Clin. Nutr. 2011, 93, 62–72. [Google Scholar] [CrossRef]
- Moreno-Indias, I.; Sanchez-Alcoholado, L.; Perez-Martinez, P.; Andres-Lacueva, C.; Cardona, F.; Tinahones, F.; Queipo-Ortuno, M.I. Red wine polyphenols modulate fecal microbiota and reduce markers of the metabolic syndrome in obese patients. Food Funct. 2016, 7, 1775–1787. [Google Scholar] [CrossRef]
- Ntemiri, A.; Ghosh, T.; Gheller, M.; Tran, T.; Blum, J.; Pellanda, P.; Vlckova, K.; Neto, M.; Howell, A.; Thalacker-Mercer, A.; et al. Whole Blueberry and Isolated Polyphenol-Rich Fractions Modulate Specific Gut Microbes in an In Vitro Colon Model and in a Pilot Study in Human Consumers. Nutrients 2020, 12, 2800. [Google Scholar] [CrossRef]
- Stanhewicz, A.; Wenner, M.; Stachenfeld, N. Sex differences in endothelial function important to vascular health and overall cardiovascular disease risk across the lifespan. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H1569–H1588. [Google Scholar] [CrossRef]
- Villablanca, A.; Jayachandran, M.; Banka, C. Atherosclerosis and sex hormones: Current concepts. Clin. Sci. 2010, 119, 493–513. [Google Scholar] [CrossRef] [PubMed]
- Gilliver, S. Sex steroids as inflammatory regulators. J. Steroid Biochem. Mol. Biol. 2010, 120, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Fairweather, D. Sex Differences in Inflammation During Atherosclerosis. Clin. Med. Insights Cardiol. 2014, 8, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Shen, Y.; Li, R. Estrogen synthesis and signaling pathways during ageing: From periphery to brain. Trends Mol. Med. 2013, 19, 197–209. [Google Scholar] [CrossRef]
- Gaskins, A.; Wilchesky, M.; Mumford, S.; Whitcomb, B.; Browne, R.; Wactawski-Wende, J.; Perkins, N.; Schisterman, E. Endogenous Reproductive Hormones and C-reactive Protein Across the Menstrual Cycle: The BioCycle Study. Am. J. Epidemiol. 2012, 175, 423–431. [Google Scholar] [CrossRef]
- Karowicz-Bilinska, A.; Plodzidym, M.; Krol, J.; Lewinska, A.; Bartosz, G. Changes of markers of oxidative stress during menstrual cycle. Redox Rep. 2008, 13, 237–240. [Google Scholar] [CrossRef]
- Palan, P.; Magneson, A.; Castillo, M.; Dunne, J.; Mikhail, M. Effects of menstrual cycle and oral contraceptive use on serum levels of lipid-soluble antioxidants. Am. J. Obstet. Gynecol. 2006, 194, e35–e38. [Google Scholar] [CrossRef]
- Hutson, D.D.; Gurrala, R.; Ogola, B.O.; Zimmerman, M.A.; Mostany, R.; Satou, R.; Lindsey, S.H. Estrogen receptor profiles across tissues from male and female Rattus norvegicus. Biol. Sex Differ. 2019, 10, 4. [Google Scholar] [CrossRef]
- Nordmeyer, J.; Eder, S.; Mahmoodzadeh, S.; Martus, P.; Fielitz, J.; Bass, J.; Bethke, N.; Zurbrügg, H.; Pregla, R.; Hetzer, R.; et al. Upregulation of Myocardial Estrogen Receptors in Human Aortic Stenosis. Circulation 2004, 110, 3270–3275. [Google Scholar] [CrossRef]
- Gavin, K.; Seals, D.; Silver, A.; Moreau, K. Vascular Endothelial Estrogen Receptor Alpha Is Modulated by Estrogen Status and Related to Endothelial Function and Endothelial Nitric Oxide Synthase in Healthy Women. J. Clin. Endocrinol. Metab. 2009, 94, 3513–3520. [Google Scholar] [CrossRef]
- Cooke, P.; Nanjappa, M.; Ko, C.; Prins, G.; Hess, R. Estrogen in Male Physiology. Physiol. Rev. 2017, 97, 995–1043. [Google Scholar] [CrossRef] [PubMed]
- Coburn, S.; Stanczyk, F.; Falk, R.; McGlynn, K.; Brinton, L.; Sampson, J.; Bradwin, G.; Xu, X.; Trabert, B. Comparability of serum, plasma, and urinary estrogen and estrogen metabolite measurements by sex and menopausal status. Cancer Causes Control 2019, 30, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Brzozowski, A.M.; Pike, A.C.; Dauter, Z.; Hubbard, R.E.; Bonn, T.; Engstrom, O.; Ohman, L.; Greene, G.L.; Gustafsson, J.A.; Carlquist, M. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature 1997, 389, 753–758. [Google Scholar] [CrossRef] [PubMed]
- Jung, B.; Kim, M.; Kim, H.; Kim, D.; Yang, J.; Her, S.; Song, Y. Caffeic acid phenethyl ester, a component of beehive propolis, is a novel selective estrogen receptor modulator. Phytother. Res. 2010, 24, 295–300. [Google Scholar] [CrossRef] [PubMed]
- van der Woude, H.; Ter Veld, M.G.; Jacobs, N.; van der Saag, P.T.; Murk, A.J.; Rietjens, I.M. The stimulation of cell proliferation by quercetin is mediated by the estrogen receptor. Mol. Nutr. Food Res. 2005, 49, 763–771. [Google Scholar] [CrossRef] [PubMed]
- Papoutsi, Z.; Kassi, E.; Tsiapara, A.; Fokialakis, N.; Chrousos, G.P.; Moutsatsou, P. Evaluation of estrogenic/antiestrogenic activity of ellagic acid via the estrogen receptor subtypes ERalpha and ERbeta. J. Agric. Food Chem. 2005, 53, 7715–7720. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Sun, B. Toll-like receptor 4 in atherosclerosis. J. Cell. Mol. Med. 2007, 11, 88–95. [Google Scholar] [CrossRef]
- Nunes, K.P.; de Oliveira, A.A.; Lima, V.V.; Webb, R.C. Toll-Like Receptor 4 and Blood Pressure: Lessons From Animal Studies. Front. Physiol. 2019, 10, 655. [Google Scholar] [CrossRef]
- Yu, L.; Feng, Z. The Role of Toll-Like Receptor Signaling in the Progression of Heart Failure. Mediat. Inflamm. 2018, 2018, 9874109. [Google Scholar] [CrossRef]
- Zhang, L.; Peppel, K.; Sivashanmugam, P.; Orman, E.S.; Brian, L.; Exum, S.T.; Freedman, N.J. Expression of tumor necrosis factor receptor-1 in arterial wall cells promotes atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1087–1094. [Google Scholar] [CrossRef]
- Mehaffey, E.; Majid, D.S.A. Tumor necrosis factor-alpha, kidney function, and hypertension. Am. J. Physiol. Renal Physiol. 2017, 313, F1005–F1008. [Google Scholar] [CrossRef] [PubMed]
- Duerrschmid, C.; Trial, J.; Wang, Y.; Entman, M.L.; Haudek, S.B. Tumor necrosis factor: A mechanistic link between angiotensin-II-induced cardiac inflammation and fibrosis. Circ. Heart Fail. 2015, 8, 352–361. [Google Scholar] [CrossRef] [PubMed]
- Jacob, T.; Ascher, E.; Alapat, D.; Olevskaia, Y.; Hingorani, A. Activation of p38MAPK signaling cascade in a VSMC injury model: Role of p38MAPK inhibitors in limiting VSMC proliferation. Eur. J. Vasc. Endovasc. Surg. 2005, 29, 470–478. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ushio-Fukai, M.; Alexander, R.W.; Akers, M.; Griendling, K.K. p38 Mitogen-activated protein kinase is a critical component of the redox-sensitive signaling pathways activated by angiotensin II. Role in vascular smooth muscle cell hypertrophy. J. Biol. Chem. 1998, 273, 15022–15029. [Google Scholar] [CrossRef] [PubMed]
- Yoshizumi, M.; Tsuchiya, K.; Kirima, K.; Kyaw, M.; Suzaki, Y.; Tamaki, T. Quercetin inhibits Shc- and phosphatidylinositol 3-kinase-mediated c-Jun N-terminal kinase activation by angiotensin II in cultured rat aortic smooth muscle cells. Mol. Pharmacol. 2001, 60, 656–665. [Google Scholar]
- Liu, Q.; Chen, Y.; Auger-Messier, M.; Molkentin, J.D. Interaction between NFkappaB and NFAT coordinates cardiac hypertrophy and pathological remodeling. Circ. Res. 2012, 110, 1077–1086. [Google Scholar] [CrossRef] [PubMed]
- Schaub, M.C.; Hefti, M.A.; Harder, B.A.; Eppenberger, H.M. Various hypertrophic stimuli induce distinct phenotypes in cardiomyocytes. J. Mol. Med. 1997, 75, 901–920. [Google Scholar] [CrossRef]
- Liao, P.; Georgakopoulos, D.; Kovacs, A.; Zheng, M.; Lerner, D.; Pu, H.; Saffitz, J.; Chien, K.; Xiao, R.P.; Kass, D.A.; et al. The in vivo role of p38 MAP kinases in cardiac remodeling and restrictive cardiomyopathy. Proc. Natl. Acad. Sci. USA 2001, 98, 12283–12288. [Google Scholar] [CrossRef]
- Petrich, B.G.; Molkentin, J.D.; Wang, Y. Temporal activation of c-Jun N-terminal kinase in adult transgenic heart via cre-loxP-mediated DNA recombination. FASEB J. 2003, 17, 749–751. [Google Scholar] [CrossRef]
- Deopurkar, R.; Ghanim, H.; Friedman, J.; Abuaysheh, S.; Sia, C.L.; Mohanty, P.; Viswanathan, P.; Chaudhuri, A.; Dandona, P. Differential effects of cream, glucose, and orange juice on inflammation, endotoxin, and the expression of Toll-like receptor-4 and suppressor of cytokine signaling-3. Diabetes Care 2010, 33, 991–997. [Google Scholar] [CrossRef]
- Zhang, Y.; Xia, G.; Zhang, Y.; Liu, J.; Liu, X.; Li, W.; Lv, Y.; Wei, S.; Liu, J.; Quan, J. Palmitate induces VSMC apoptosis via toll like receptor (TLR)4/ROS/p53 pathway. Atherosclerosis 2017, 263, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Chavez-Sanchez, L.; Madrid-Miller, A.; Chavez-Rueda, K.; Legorreta-Haquet, M.V.; Tesoro-Cruz, E.; Blanco-Favela, F. Activation of TLR2 and TLR4 by minimally modified low-density lipoprotein in human macrophages and monocytes triggers the inflammatory response. Hum. Immunol. 2010, 71, 737–744. [Google Scholar] [CrossRef] [PubMed]
- Smiley, S.T.; King, J.A.; Hancock, W.W. Fibrinogen stimulates macrophage chemokine secretion through toll-like receptor 4. J. Immunol. 2001, 167, 2887–2894. [Google Scholar] [CrossRef]
- Zhang, P.; Cox, C.J.; Alvarez, K.M.; Cunningham, M.W. Cutting edge: Cardiac myosin activates innate immune responses through TLRs. J. Immunol. 2009, 183, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Dauphinee, S.M.; Karsan, A. Lipopolysaccharide signaling in endothelial cells. Lab. Investig. 2006, 86, 9–22. [Google Scholar] [CrossRef]
- Deshpande, R.; Khalili, H.; Pergolizzi, R.; Michael, S.; Change, M. Estradiol down-regulates LPS-induced cytokine production and NFκB activation in murine macrophages. Am. J. Reprod. Immunol. 1997, 38, 46–64. [Google Scholar] [CrossRef]
- Michelsen, K.S.; Wong, M.H.; Shah, P.K.; Zhang, W.; Yano, J.; Doherty, T.M.; Akira, S.; Rajavashisth, T.B.; Arditi, M. Lack of Toll-like receptor 4 or myeloid differentiation factor 88 reduces atherosclerosis and alters plaque phenotype in mice deficient in apolipoprotein E. Proc. Natl. Acad. Sci. USA 2004, 101, 10679–10684. [Google Scholar] [CrossRef]
- Riad, A.; Jager, S.; Sobirey, M.; Escher, F.; Yaulema-Riss, A.; Westermann, D.; Karatas, A.; Heimesaat, M.M.; Bereswill, S.; Dragun, D.; et al. Toll-like receptor-4 modulates survival by induction of left ventricular remodeling after myocardial infarction in mice. J. Immunol. 2008, 180, 6954–6961. [Google Scholar] [CrossRef]
- Menghini, R.; Campia, U.; Tesauro, M.; Marino, A.; Rovella, V.; Rodia, G.; Schinzari, F.; Tolusso, B.; di Daniele, N.; Federici, M.; et al. Toll-like receptor 4 mediates endothelial cell activation through NF-kappaB but is not associated with endothelial dysfunction in patients with rheumatoid arthritis. PLoS ONE 2014, 9, e99053. [Google Scholar] [CrossRef]
- Han, J.; Zou, C.; Mei, L.; Zhang, Y.; Qian, Y.; You, S.; Pan, Y.; Xu, Z.; Bai, B.; Huang, W.; et al. MD2 mediates angiotensin II-induced cardiac inflammation and remodeling via directly binding to Ang II and activating TLR4/NF-kappaB signaling pathway. Basic Res. Cardiol. 2017, 112, 9. [Google Scholar] [CrossRef]
- Dange, R.B.; Agarwal, D.; Masson, G.S.; Vila, J.; Wilson, B.; Nair, A.; Francis, J. Central blockade of TLR4 improves cardiac function and attenuates myocardial inflammation in angiotensin II-induced hypertension. Cardiovasc. Res. 2014, 103, 17–27. [Google Scholar] [CrossRef]
- Sullivan, J.; Rodriguez-Miguelez, P.; Zimmerman, M.; Harris, R. Differences in angiotensin (1–7) between men and women. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H1171–H1176. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Engel, A.; Morcillo, L.; Aranda, F.; Ruiz, M.; Gaitan, M.; Mayor-Olea, A.; Aranda, P.; Ferrario, C. Influence of gender and genetic variability on plasma angiotensin peptides. J. Renin Angiotensin Aldosterone Syst. 2006, 7, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Rifai, N.; Pfeffer, M.; Sacks, F.; Lepage, S.; Braunwald, E. Elevation of tumor necrosis factor-alpha and increased risk of recurrent coronary events after myocardial infarction. Circulation 2000, 101, 2149–2153. [Google Scholar] [CrossRef] [PubMed]
- Haudek, S.B.; Taffet, G.E.; Schneider, M.D.; Mann, D.L. TNF provokes cardiomyocyte apoptosis and cardiac remodeling through activation of multiple cell death pathways. J. Clin. Investig. 2007, 117, 2692–2701. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Xu, X.; Potter, B.J.; Wang, W.; Kuo, L.; Michael, L.; Bagby, G.J.; Chilian, W.M. TNF-alpha contributes to endothelial dysfunction in ischemia/reperfusion injury. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 475–480. [Google Scholar] [CrossRef]
- Picchi, A.; Gao, X.; Belmadani, S.; Potter, B.J.; Focardi, M.; Chilian, W.M.; Zhang, C. Tumor necrosis factor-alpha induces endothelial dysfunction in the prediabetic metabolic syndrome. Circ. Res. 2006, 99, 69–77. [Google Scholar] [CrossRef]
- Kroetsch, J.T.; Levy, A.S.; Zhang, H.; Aschar-Sobbi, R.; Lidington, D.; Offermanns, S.; Nedospasov, S.A.; Backx, P.H.; Heximer, S.P.; Bolz, S.S. Constitutive smooth muscle tumour necrosis factor regulates microvascular myogenic responsiveness and systemic blood pressure. Nat. Commun. 2017, 8, 14805. [Google Scholar] [CrossRef]
- Bozkurt, B.; Kribbs, S.B.; Clubb, F.J., Jr.; Michael, L.H.; Didenko, V.V.; Hornsby, P.J.; Seta, Y.; Oral, H.; Spinale, F.G.; Mann, D.L. Pathophysiologically relevant concentrations of tumor necrosis factor-alpha promote progressive left ventricular dysfunction and remodeling in rats. Circulation 1998, 97, 1382–1391. [Google Scholar] [CrossRef]
- Moe, K.T.; Khairunnisa, K.; Yin, N.O.; Chin-Dusting, J.; Wong, P.; Wong, M.C. Tumor necrosis factor-alpha-induced nuclear factor-kappaB activation in human cardiomyocytes is mediated by NADPH oxidase. J. Physiol. Biochem. 2014, 70, 769–779. [Google Scholar] [CrossRef]
- Defer, N.; Azroyan, A.; Pecker, F.; Pavoine, C. TNFR1 and TNFR2 signaling interplay in cardiac myocytes. J. Biol. Chem. 2007, 282, 35564–35573. [Google Scholar] [CrossRef]
- Hamid, T.; Gu, Y.; Ortines, R.V.; Bhattacharya, C.; Wang, G.; Xuan, Y.T.; Prabhu, S.D. Divergent tumor necrosis factor receptor-related remodeling responses in heart failure: Role of nuclear factor-kappaB and inflammatory activation. Circulation 2009, 119, 1386–1397. [Google Scholar] [CrossRef] [PubMed]
- Kadokami, T.; McTiernan, C.F.; Kubota, T.; Frye, C.S.; Feldman, A.M. Sex-related survival differences in murine cardiomyopathy are associated with differences in TNF-receptor expression. J. Clin. Investig. 2000, 106, 589–597. [Google Scholar] [CrossRef]
- Wang, M.; Crisostomo, P.R.; Markel, T.A.; Wang, Y.; Meldrum, D.R. Mechanisms of sex differences in TNFR2-mediated cardioprotection. Circulation 2008, 118, S38–S45. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Weitzmann, M.; Cenci, S.; Ross, F.; Adler, S.; Pacifici, R. Estrogen decreases TNF gene expression by blocking JNK activity and the resulting production of c-Jun and JunD. J. Clin. Investig. 1999, 104, 503–513. [Google Scholar] [CrossRef]
- Yoshizumi, M.; Perrella, M.A.; Burnett, J.C., Jr.; Lee, M.E. Tumor necrosis factor downregulates an endothelial nitric oxide synthase mRNA by shortening its half-life. Circ. Res. 1993, 73, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.; Liu, M.; Wu, Y.; Sharma, V.; Luo, T.; Ouyang, J.; McNeill, J.H. N-acetylcysteine attenuates TNF-alpha-induced human vascular endothelial cell apoptosis and restores eNOS expression. Eur. J. Pharmacol. 2006, 550, 134–142. [Google Scholar] [CrossRef]
- Grumbach, I.; Chen, W.; Mertens, S.; Harrison, D. A negative feedback mechanism involving nitric oxide and nuclear factor kappa-B modulates endothelial nitric oxide synthase transcription. J. Mol. Cell. Cardiol. 2005, 39, 595–603. [Google Scholar] [CrossRef]
- Revankar, C.; Cimino, D.; Sklar, L.; Arterburn, J.; Prossnitz, E. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science 2005, 307, 1625–1630. [Google Scholar] [CrossRef]
- Stirone, C.; Boroujerdi, A.; Duckles, S.; Krause, D. Estrogen receptor activation of phosphoinositide-3 kinase, akt, and nitric oxide signaling in cerebral blood vessels: Rapid and long-term effects. Mol. Pharmacol. 2005, 67, 105–113. [Google Scholar] [CrossRef]
- Sharma, G.; Prossnitz, E. Mechanisms of estradiol-induced insulin secretion by the G protein-coupled estrogen receptor GPR30/GPER in pancreatic β-cells. Endocrinology 2011, 152, 3030–3039. [Google Scholar] [CrossRef] [PubMed]
- Peluso, A.A.; Bertelsen, J.B.; Andersen, K.; Mortsensen, T.P.; Hansen, P.B.; Sumners, C.; Bader, M.; Santos, R.A.; Steckelings, U.M. Identification of protein phosphatase involvement in the AT2 receptor-induced activation of endothelial nitric oxide synthase. Clin. Sci. 2018, 132, 777–790. [Google Scholar] [CrossRef] [PubMed]
- Simoncini, T.; Maffei, S.; Basta, G.; Barsacchi, G.; Genazzani, A.; Liao, J.; De Caterina, R. Estrogens and glucocorticoids inhibit endothelial vascular cell adhesion molecule-1 expression by different transcriptional mechanisms. Circ. Res. 2000, 87, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Torres-Estay, V.; Carreño, D.; Francisco, I.S.; Sotomayor, P.; Godoy, A.; Smith, G. Androgen receptor in human endothelial cells. J. Endocrinol. 2015, 224, R131–R137. [Google Scholar] [CrossRef]
- Liu, P.; Death, A.; Handelsman, D. Androgens and cardiovascular disease. Endocr. Rev. 2003, 24, 313–340. [Google Scholar] [CrossRef]
- Diamanti-Kandarakis, E. Polycystic ovarian syndrome: Pathophysiology, molecular aspects and clinical implications. Expert Rev. Mol. Med. 2008, 10, e3. [Google Scholar] [CrossRef]
- Goodwin, B.L.; Pendleton, L.C.; Levy, M.M.; Solomonson, L.P.; Eichler, D.C. Tumor necrosis factor-alpha reduces argininosuccinate synthase expression and nitric oxide production in aortic endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H1115–H1121. [Google Scholar] [CrossRef]
- Karlsen, A.; Paur, I.; Bohn, S.K.; Sakhi, A.K.; Borge, G.I.; Serafini, M.; Erlund, I.; Laake, P.; Tonstad, S.; Blomhoff, R. Bilberry juice modulates plasma concentration of NF-kappaB related inflammatory markers in subjects at increased risk of CVD. Eur. J. Nutr. 2010, 49, 345–355. [Google Scholar] [CrossRef]
- Lehtonen, H.M.; Suomela, J.P.; Tahvonen, R.; Yang, B.; Venojarvi, M.; Viikari, J.; Kallio, H. Different berries and berry fractions have various but slightly positive effects on the associated variables of metabolic diseases on overweight and obese women. Eur. J. Clin. Nutr. 2011, 65, 394–401. [Google Scholar] [CrossRef]
- Basu, A.; Wilkinson, M.; Penugonda, K.; Simmons, B.; Betts, N.M.; Lyons, T.J. Freeze-dried strawberry powder improves lipid profile and lipid peroxidation in women with metabolic syndrome: Baseline and post intervention effects. Nutr. J. 2009, 8, 43. [Google Scholar] [CrossRef]
- Basu, A.; Fu, D.X.; Wilkinson, M.; Simmons, B.; Wu, M.; Betts, N.M.; Du, M.; Lyons, T.J. Strawberries decrease atherosclerotic markers in subjects with metabolic syndrome. Nutr. Res. 2010, 30, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.; Betts, N.M.; Nguyen, A.; Newman, E.D.; Fu, D.; Lyons, T.J. Freeze-dried strawberries lower serum cholesterol and lipid peroxidation in adults with abdominal adiposity and elevated serum lipids. J. Nutr. 2014, 144, 830–837. [Google Scholar] [CrossRef] [PubMed]
- Stanoeva, J.P.; Stefova, M.; Andonovska, K.B.; Vankova, A.; Stafilov, T. Phenolics and mineral content in bilberry and bog bilberry from Macedonia. Int. J. Food Prop. 2017, 20, S863–S883. [Google Scholar] [CrossRef]
- Aaby, K.; Wrolstad, R.E.; Ekeberg, D.; Skrede, G. Polyphenol composition and antioxidant activity in strawberry purees; impact of achene level and storage. J. Agric. Food Chem. 2007, 55, 5156–5166. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.S.; Kim, S.; Hong, S.J.; Choi, S.C.; Choi, J.H.; Kim, J.H.; Park, C.Y.; Cho, J.Y.; Lee, T.B.; Kwon, J.W.; et al. Black Raspberry Extract Increased Circulating Endothelial Progenitor Cells and Improved Arterial Stiffness in Patients with Metabolic Syndrome: A Randomized Controlled Trial. J. Med. Food. 2016, 19, 346–352. [Google Scholar] [CrossRef]
- Aghababaee, S.K.; Vafa, M.; Shidfar, F.; Tahavorgar, A.; Gohari, M.; Katebi, D.; Mohammadi, V. Effects of blackberry (Morus nigra L.) consumption on serum concentration of lipoproteins, apo A-I, apo B, and high-sensitivity-C-reactive protein and blood pressure in dyslipidemic patients. J. Res. Med. Sci. 2015, 20, 684–691. [Google Scholar] [CrossRef]
- Schell, J.; Betts, N.M.; Lyons, T.J.; Basu, A. Raspberries Improve Postprandial Glucose and Acute and Chronic Inflammation in Adults with Type 2 Diabetes. Ann. Nutr. Metab. 2019, 74, 165–174. [Google Scholar] [CrossRef]
- Nair, A.R.; Mariappan, N.; Stull, A.J.; Francis, J. Blueberry supplementation attenuates oxidative stress within monocytes and modulates immune cell levels in adults with metabolic syndrome: A randomized, double-blind, placebo-controlled trial. Food Funct. 2017, 8, 4118–4128. [Google Scholar] [CrossRef]
- Li, T.; Li, F.; Liu, X.; Liu, J.; Li, D. Synergistic anti-inflammatory effects of quercetin and catechin via inhibiting activation of TLR4-MyD88-mediated NF-kappaB and MAPK signaling pathways. Phytother. Res. 2019, 33, 756–767. [Google Scholar] [CrossRef]
- Bhaskar, S.; Sudhakaran, P.R.; Helen, A. Quercetin attenuates atherosclerotic inflammation and adhesion molecule expression by modulating TLR-NF-kappaB signaling pathway. Cell. Immunol. 2016, 310, 131–140. [Google Scholar] [CrossRef]
- Zhang, M.; Xue, Y.; Chen, H.; Meng, L.; Chen, B.; Gong, H.; Zhao, Y.; Qi, R. Resveratrol Inhibits MMP3 and MMP9 Expression and Secretion by Suppressing TLR4/NF-kappaB/STAT3 Activation in Ox-LDL-Treated HUVECs. Oxid. Med. Cell. Longev. 2019, 2019, 9013169. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Lin, G.; Wan, W.; Li, X.; Zeng, B.; Yang, B.; Huang, C. Resveratrol, a polyphenol phytoalexin, protects cardiomyocytes against anoxia/reoxygenation injury via the TLR4/NF-kappaB signaling pathway. Int. J. Mol. Med. 2012, 29, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Sivasinprasasn, S.; Pantan, R.; Thummayot, S.; Tocharus, J.; Suksamrarn, A.; Tocharus, C. Cyanidin-3-glucoside attenuates angiotensin II-induced oxidative stress and inflammation in vascular endothelial cells. Chem. Biol. Interact. 2016, 260, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.Y.; Liu, Y.M.; Wang, J.; Wang, X.N.; Li, C.Y. Anti-inflammatory effect of the blueberry anthocyanins malvidin-3-glucoside and malvidin-3-galactoside in endothelial cells. Molecules 2014, 19, 12827–12841. [Google Scholar] [CrossRef]
- Liao, H.H.; Zhang, N.; Meng, Y.Y.; Feng, H.; Yang, J.J.; Li, W.J.; Chen, S.; Wu, H.M.; Deng, W.; Tang, Q.Z. Myricetin Alleviates Pathological Cardiac Hypertrophy via TRAF6/TAK1/MAPK and Nrf2 Signaling Pathway. Oxid. Med. Cell. Longev. 2019, 2019, 6304058. [Google Scholar] [CrossRef]
- Nwachukwu, J.; Srinivasan, S.; Bruno, N.; Parent, A.; Hughes, T.; Pollock, J.; Gjyshi, O.; Cavett, V.; Nowak, J.; Garcia-Ordonez, R.; et al. Resveratrol modulates the inflammatory response via an estrogen receptor-signal integration network. eLife 2014, 3. [Google Scholar] [CrossRef]
- Han, D.; Ybanez, M.D.; Ahmadi, S.; Yeh, K.; Kaplowitz, N. Redox regulation of tumor necrosis factor signaling. Antioxid. Redox Signal. 2009, 11, 2245–2263. [Google Scholar] [CrossRef]
- Dikalov, S.I.; Ungvari, Z. Role of mitochondrial oxidative stress in hypertension. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H1417–H1427. [Google Scholar] [CrossRef]
- Dey, S.; DeMazumder, D.; Sidor, A.; Foster, D.B.; O’Rourke, B. Mitochondrial ROS Drive Sudden Cardiac Death and Chronic Proteome Remodeling in Heart Failure. Circ. Res. 2018, 123, 356–371. [Google Scholar] [CrossRef]
- Kornfeld, O.S.; Hwang, S.; Disatnik, M.H.; Chen, C.H.; Qvit, N.; Mochly-Rosen, D. Mitochondrial reactive oxygen species at the heart of the matter: New therapeutic approaches for cardiovascular diseases. Circ. Res. 2015, 116, 1783–1799. [Google Scholar] [CrossRef]
- Hajjar, R.J.; Leopold, J.A. Xanthine oxidase inhibition and heart failure: Novel therapeutic strategy for ventricular dysfunction? Circ. Res. 2006, 98, 169–171. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; DeLano, F.A.; Parks, D.A.; Jamshidi, N.; Granger, D.N.; Ishii, H.; Suematsu, M.; Zweifach, B.W.; Schmid-Schonbein, G.W. Xanthine oxidase activity associated with arterial blood pressure in spontaneously hypertensive rats. Proc. Natl. Acad. Sci. USA 1998, 95, 4754–4759. [Google Scholar] [CrossRef] [PubMed]
- Murdoch, C.E.; Alom-Ruiz, S.P.; Wang, M.; Zhang, M.; Walker, S.; Yu, B.; Brewer, A.; Shah, A.M. Role of endothelial Nox2 NADPH oxidase in angiotensin II-induced hypertension and vasomotor dysfunction. Basic Res. Cardiol. 2011, 106, 527–538. [Google Scholar] [CrossRef] [PubMed]
- Bouabout, G.; Ayme-Dietrich, E.; Jacob, H.; Champy, M.F.; Birling, M.C.; Pavlovic, G.; Madeira, L.; Fertak, L.E.; Petit-Demouliere, B.; Sorg, T.; et al. Nox4 genetic inhibition in experimental hypertension and metabolic syndrome. Arch. Cardiovasc. Dis. 2018, 111, 41–52. [Google Scholar] [CrossRef]
- Langbein, H.; Brunssen, C.; Hofmann, A.; Cimalla, P.; Brux, M.; Bornstein, S.R.; Deussen, A.; Koch, E.; Morawietz, H. NADPH oxidase 4 protects against development of endothelial dysfunction and atherosclerosis in LDL receptor deficient mice. Eur. Heart J. 2016, 37, 1753–1761. [Google Scholar] [CrossRef] [PubMed]
- D’Oria, R.; Schipani, R.; Leonardini, A.; Natalicchio, A.; Perrini, S.; Cignarelli, A.; Laviola, L.; Giorgino, F. The Role of Oxidative Stress in Cardiac Disease: From Physiological Response to Injury Factor. Oxid. Med. Cell. Longev. 2020, 2020, 5732956. [Google Scholar] [CrossRef]
- Ide, T.; Tsutsui, H.; Ohashi, N.; Hayashidani, S.; Suematsu, N.; Tsuchihashi, M.; Tamai, H.; Takeshita, A. Greater oxidative stress in healthy young men compared with premenopausal women. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 438–442. [Google Scholar] [CrossRef]
- Barp, J.; Araújo, A.; Fernandes, T.G.; Rigatto, K.; Llesuy, S.; Belló-Klein, A.; Singal, P. Myocardial antioxidant and oxidative stress changes due to sex hormones. Braz. J. Med. Biol. Res. 2002, 35, 1075–1081. [Google Scholar] [CrossRef]
- Khalifa, A.R.; Abdel-Rahman, E.A.; Mahmoud, A.M.; Ali, M.H.; Noureldin, M.; Saber, S.H.; Mohsen, M.; Ali, S.S. Sex-specific differences in mitochondria biogenesis, morphology, respiratory function, and ROS homeostasis in young mouse heart and brain. Physiol. Rep. 2017, 5. [Google Scholar] [CrossRef]
- Konior, A.; Schramm, A.; Czesnikiewicz-Guzik, M.; Guzik, T.J. NADPH oxidases in vascular pathology. Antioxid. Redox Signal. 2014, 20, 2794–2814. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Cordeiro, R.M. Reactive oxygen species at phospholipid bilayers: Distribution, mobility and permeation. Biochim. Biophys. Acta 2014, 1838, 438–444. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.; Ing, M.H.; Salazar, A.; Lassegue, B.; Griendling, K.; Navab, M.; Sevanian, A.; Hsiai, T.K. Pulsatile versus oscillatory shear stress regulates NADPH oxidase subunit expression: Implication for native LDL oxidation. Circ. Res. 2003, 93, 1225–1232. [Google Scholar] [CrossRef] [PubMed]
- Li, J.M.; Shah, A.M. Mechanism of endothelial cell NADPH oxidase activation by angiotensin II. Role of the p47phox subunit. J. Biol. Chem. 2003, 278, 12094–12100. [Google Scholar] [CrossRef] [PubMed]
- Duerrschmidt, N.; Wippich, N.; Goettsch, W.; Broemme, H.J.; Morawietz, H. Endothelin-1 induces NAD(P)H oxidase in human endothelial cells. Biochem. Biophys. Res. Commun. 2000, 269, 713–717. [Google Scholar] [CrossRef]
- Frey, R.S.; Rahman, A.; Kefer, J.C.; Minshall, R.D.; Malik, A.B. PKCzeta regulates TNF-alpha-induced activation of NADPH oxidase in endothelial cells. Circ. Res. 2002, 90, 1012–1019. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Pimentel, D.R.; Wang, J.; Singh, K.; Colucci, W.S.; Sawyer, D.B. Role of reactive oxygen species and NAD(P)H oxidase in alpha(1)-adrenoceptor signaling in adult rat cardiac myocytes. Am. J. Physiol. Cell Physiol. 2002, 282, C926–C934. [Google Scholar] [CrossRef]
- Miller, A.A.; Drummond, G.R.; Mast, A.E.; Schmidt, H.H.; Sobey, C.G. Effect of gender on NADPH-oxidase activity, expression, and function in the cerebral circulation: Role of estrogen. Stroke 2007, 38, 2142–2149. [Google Scholar] [CrossRef]
- Wong, P.S.; Randall, M.D.; Roberts, R.E. Sex differences in the role of NADPH oxidases in endothelium-dependent vasorelaxation in porcine isolated coronary arteries. Vasc. Pharmacol. 2015, 72, 83–92. [Google Scholar] [CrossRef]
- De Silva, T.M.; Broughton, B.R.; Drummond, G.R.; Sobey, C.G.; Miller, A.A. Gender influences cerebral vascular responses to angiotensin II through Nox2-derived reactive oxygen species. Stroke 2009, 40, 1091–1097. [Google Scholar] [CrossRef]
- Zhang, R.; Thor, D.; Han, X.; Anderson, L.; Rahimian, R. Sex differences in mesenteric endothelial function of streptozotocin-induced diabetic rats: A shift in the relative importance of EDRFs. Am. J. Physiol. Heart Circ. Physiol. 2012, 303, H1183–H1198. [Google Scholar] [CrossRef] [PubMed]
- Pacher, P.; Nivorozhkin, A.; Szabo, C. Therapeutic effects of xanthine oxidase inhibitors: Renaissance half a century after the discovery of allopurinol. Pharmacol. Rev. 2006, 58, 87–114. [Google Scholar] [CrossRef] [PubMed]
- Linder, N.; Rapola, J.; Raivio, K.O. Cellular expression of xanthine oxidoreductase protein in normal human tissues. Lab. Investig. 1999, 79, 967–974. [Google Scholar] [PubMed]
- Spiekermann, S.; Landmesser, U.; Dikalov, S.; Bredt, M.; Gamez, G.; Tatge, H.; Reepschlager, N.; Hornig, B.; Drexler, H.; Harrison, D.G. Electron spin resonance characterization of vascular xanthine and NAD(P)H oxidase activity in patients with coronary artery disease: Relation to endothelium-dependent vasodilation. Circulation 2003, 107, 1383–1389. [Google Scholar] [CrossRef] [PubMed]
- Budhiraja, R.; Kayyali, U.; Karamsetty, M.; Fogel, M.; Hill, N.; Chalkley, R.; Finlay, G.; Hassoun, P. Estrogen modulates xanthine dehydrogenase/xanthine oxidase activity by a receptor-independent mechanism. Antioxid. Redox Signal. 2003, 5, 705–711. [Google Scholar] [CrossRef] [PubMed]
- Seyrek, M.; Yildiz, O.; Ulusoy, H.B.; Yildirim, V. Testosterone relaxes isolated human radial artery by potassium channel opening action. J. Pharmacol. Sci. 2007, 103, 309–316. [Google Scholar] [CrossRef]
- Puttabyatappa, Y.; Stallone, J.; Ergul, A.; El-Remessy, A.; Kumar, S.; Black, S.; Johnson, M.; Owen, M.; White, R. Peroxynitrite mediates testosterone-induced vasodilation of microvascular resistance vessels. J. Pharmacol. Exp. Ther. 2013, 345, 7–14. [Google Scholar] [CrossRef]
- Berry, C.E.; Hare, J.M. Xanthine oxidoreductase and cardiovascular disease: Molecular mechanisms and pathophysiological implications. J. Physiol. 2004, 555, 589–606. [Google Scholar] [CrossRef]
- McNally, J.; Davis, M.; Giddens, D.; Saha, A.; Hwang, J.; Dikalov, S.; Jo, H.; Harrison, D. Role of xanthine oxidoreductase and NAD(P)H oxidase in endothelial superoxide production in response to oscillatory shear stress. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H2290–H2297. [Google Scholar] [CrossRef]
- Bredemeier, M.; Lopes, L.; Eisenreich, M.; Hickmann, S.; Bongiorno, G.; d’Avila, R.; Morsch, A.; da Silva Stein, F.; Campos, G. Xanthine oxidase inhibitors for prevention of cardiovascular events: A systematic review and meta-analysis of randomized controlled trials. BMC Cardiovasc. Disord. 2018, 18, 24. [Google Scholar] [CrossRef]
- Guzik, T.; Sadowski, J.; Guzik, B.; Jopek, A.; Kapelak, B.; Przybylowski, P.; Wierzbicki, K.; Korbut, R.; Harrison, D.; Channon, K. Coronary artery superoxide production and nox isoform expression in human coronary artery disease. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Cicero, A.; Cosentino, E.; Kuwabara, M.; Esposti, D.; Borghi, C. Effects of allopurinol and febuxostat on cardiovascular mortality in elderly heart failure patients. Intern. Emerg. Med. 2019, 14, 949–956. [Google Scholar] [CrossRef]
- Zhang, Y.; Murugesan, P.; Huang, K.; Cai, H. NADPH oxidases and oxidase crosstalk in cardiovascular diseases: Novel therapeutic targets. Nat. Rev. Cardiol. 2020, 17, 170–194. [Google Scholar] [CrossRef] [PubMed]
- Bryk, D.; Olejarz, W.; Zapolska-Downar, D. The role of oxidative stress and NADPH oxidase in the pathogenesis of atherosclerosis. Postepy Hig. Med. Dosw. (Online) 2017, 71, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Tsutsui, H.; Kinugawa, S.; Matsushima, S. Oxidative stress and heart failure. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H2181–H2190. [Google Scholar] [CrossRef]
- Vallance, P.; Chan, N. Endothelial function and nitric oxide: Clinical relevance. Heart 2001, 85, 342–350. [Google Scholar] [CrossRef]
- Tousoulis, D.; Kampoli, A.-M.; Papageorgiou, C.; Stefanadis, C. The Role of Nitric Oxide on Endothelial Function. Curr. Vasc. Pharmacol. 2012, 10, 4–18. [Google Scholar] [CrossRef]
- Jones, S.P.; Greer, J.J.; van Haperen, R.; Duncker, D.J.; de Crom, R.; Lefer, D.J. Endothelial nitric oxide synthase overexpression attenuates congestive heart failure in mice. Proc. Natl. Acad. Sci. USA 2003, 100, 4891–4896. [Google Scholar] [CrossRef]
- Widmer, R.; Lerman, A. Endothelial dysfunction and cardiovascular disease. Glob. Cardiol. Sci. Pract. 2014, 2014, 291–308. [Google Scholar] [CrossRef]
- Li, Q.; Yon, J.-Y.; Cai, H. Mechanisms and Consequences of eNOS Dysfunction in Hypertension. J. Hypertens. 2015, 33, 1128–1136. [Google Scholar] [CrossRef]
- Yang, Y.-M.; Huang, A.; Kaley, G.; Sun, D. eNOS uncoupling and endothelial dysfunction in aged vessels. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H1829–H1836. [Google Scholar] [CrossRef] [PubMed]
- Landmesser, U.; Dikalov, S.; PRice, S.; McCann, L.; Fukai, T.; Holland, S.; Mitch, W.; Harrison, D. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J. Clin. Investig. 2003, 111, 1201–1209. [Google Scholar] [CrossRef] [PubMed]
- White, A.; Ryoo, S.; Li, D.; Champion, H.; Steppan, J.; Wang, D.; Nyhan, D.; Shoukas, A.; Hare, J.; Berkowitz, D. Knockdown of arginase I restores NO signaling in the vasculature of old rats. Hypertension 2006, 47, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Holowatz, L.; Thompson, C.; Kenney, W. l-Arginine supplementation or arginase inhibition augments reflex cutaneous vasodilatation in aged human skin. J. Physiol. 2006, 574, 573–581. [Google Scholar] [CrossRef]
- Katusic, Z. Vascular endothelial dysfunction: Does tetrahydrobiopterin play a role? Am. J. Physiol. Heart Circ. Physiol. 2001, 281, H981–H986. [Google Scholar] [CrossRef]
- Stroes, E.; Hijmering, M.; van Zandvoost, M.; Wever, R.; Rabelink, T.; van Faassen, E. Origin of superoxide production by endothelial nitric oxide synthase. FEBS Lett. 1998, 438, 161–164. [Google Scholar] [CrossRef]
- Holowatz, L.; Kenney, W. Up-regulation of arginase activity contributes to attenuated reflex cutaneous vasodilatation in hypertensive humans. J. Physiol. 2007, 581, 863–872. [Google Scholar] [CrossRef]
- Antoniades, C.; Shirodaria, C.; Leeson, P.; Antonopoulos, A.; Warrick, N.; Van-Assche, T.; Cunnington, C.; Tousoulis, D.; Pillai, R.; Ratnatunga, C.; et al. Association of plasma asymmetrical dimethylarginine (ADMA) with elevated vascular superoxide production and endothelial nitric oxide synthase uncoupling: Implications for endothelial function in human atherosclerosis. Eur. Heart J. 2009, 30, 1142–1150. [Google Scholar] [CrossRef]
- Böger, R. Asymmetric dimethylarginine, an endogenous inhibitor of nitric oxide synthase, explains the “l-arginine paradox” and acts as a novel cardiovascular risk factor. J Nutr. 2004, 134, 2842S–2847S. [Google Scholar] [CrossRef]
- Pastore, M.; Talwar, S.; Conley, M.; Magness, R. Identification of Differential ER-Alpha Versus ER-Beta Mediated Activation of eNOS in Ovine Uterine Artery Endothelial Cells. Biol. Reprod. 2016, 94, 1–9. [Google Scholar] [CrossRef]
- Klawitter, J.; Hildreth, K.; Christians, U.; Kohrt, W.; Moreau, K. A relative l-arginine deficiency contributes to endothelial dysfunction across the stages of the menopausal transition. Physiol. Rep. 2017, 5, e13409. [Google Scholar] [CrossRef] [PubMed]
- Moreau, K.; Meditz, A.; Deane, K.; Kohrt, W. Tetrahydrobiopterin improves endothelial function and decreases arterial stiffness in estrogen-deficient postmenopausal women. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H1211–H1218. [Google Scholar] [CrossRef] [PubMed]
- Sandoo, A.; Zanten, J.V.V.; Metsios, G.; Carroll, D.; Kitas, G. The Endothelium and Its Role in Regulating Vascular Tone. Open Cardiovasc. Med. J. 2010, 4, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Tyml, K.; Wilson, J.X. iNOS expression requires NADPH oxidase-dependent redox signaling in microvascular endothelial cells. J. Cell. Physiol. 2008, 217, 207–214. [Google Scholar] [CrossRef]
- Kuo, P.; Schroeder, R. The emerging multifaceted roles of nitric oxide. Ann. Surg. 1995, 221, 220–235. [Google Scholar] [CrossRef]
- Hong, H.-J.; Loh, S.-H.; Yen, M.-H. Suppression of the development of hypertension by the inhibition of inducible nitric oxide synthase. Br. J. Pharmacol. 2000, 131, 631–637. [Google Scholar] [CrossRef]
- Smith, C.; Santhanam, L.; Bruning, R.; Stanhewicz, A.; Berkowitz, D.; Holowatz, L. Upregulation of inducible nitric oxide synthase contributes to attenuated cutaneous vasodilation in essential hypertensive humans. Hypertension 2011, 58, 935–942. [Google Scholar] [CrossRef]
- Fukai, T.; Ushio-Fukai, M. Superoxide dismutases: Role in redox signaling, vascular function, and diseases. Antioxid. Redox Signal. 2011, 15, 1583–1606. [Google Scholar] [CrossRef]
- Carr, A.; McCall, M.; Frei, B. Oxidation of LDL by Myeloperoxidase and Reactive Nitrogen Species: Reaction Pathways and Antioxidant Protection. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1716–1723. [Google Scholar] [CrossRef]
- Cignarella, A.; Bolego, C.; Pelosi, V.; Meda, C.; Krust, A.; Pinna, C.; Gaion, R.; Vegeto, E.; Maggi, A. Distinct Roles of Estrogen Receptor-a and B in the Modulation of Vascular Inducible Nitric-Oxide Synthase in Diabetes. J. Pharmacol. Exp. Ther. 2009, 328, 174–182. [Google Scholar] [CrossRef]
- Juliet, P.; Hayashi, T.; Daigo, S.; Matsui-Hirai, H.; Miyazaki, A.; Fukatsu, A.; Funami, J.; Iguchi, A.; Ignarro, L. Combined effect of testosterone and apocynin on nitric oxide and superoxide production in PMA-differentiated THP-1 cells. Biochim. Biophys. Acta 2004, 1693, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Flores-Mateo, G.; Carrillo-Santisteve, P.; Elosua, R.; Guallar, E.; Marrugat, J.; Bleys, J.; Covas, M.-I. Antioxidant Enzyme Activity and Coronary Heart Disease: Meta-analyses of Observational Studies. Am. J. Epidemiol. 2009, 170, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Espinola-Klein, C.; Rupprecht, H.; Bickel, C.; Schnabel, R.; Genth-Zotz, S.; Torzewski, M.; Lackner, K.; Munzel, T.; Blankenberg, S. Glutathione Peroxidase-1 Activity, Atherosclerotic Burden, and Cardiovascular Prognosis. Am. J. Cardiol. 2007, 99, 808–812. [Google Scholar] [CrossRef] [PubMed]
- Buijsse, B.; Lee, D.-H.; Erickson, R.; Luepker, R.; Jacobs, D.; Holtzman, J. Low Serum Glutathione Peroxidase Activity Is Associated with Increased Cardiovascular Mortality in Individuals with Low HDLc’s. PLoS ONE 2012, 7, e38901. [Google Scholar] [CrossRef] [PubMed]
- Fukai, T.; Galis, Z.; Meng, X.; Parthasarathy, S.; Harrison, D. Vascular expression of extracellular superoxide dismutase in atherosclerosis. J. Clin. Investig. 1998, 101, 2101–2111. [Google Scholar] [CrossRef]
- Tanaka, M.; Mokhtari, G.K.; Terry, R.D.; Balsam, L.B.; Lee, K.H.; Kofidis, T.; Tsao, P.S.; Robbins, R.C. Overexpression of human copper/zinc superoxide dismutase (SOD1) suppresses ischemia-reperfusion injury and subsequent development of graft coronary artery disease in murine cardiac grafts. Circulation 2004, 110, II200–II206. [Google Scholar] [CrossRef]
- Kang, P.T.; Chen, C.L.; Ohanyan, V.; Luther, D.J.; Meszaros, J.G.; Chilian, W.M.; Chen, Y.R. Overexpressing superoxide dismutase 2 induces a supernormal cardiac function by enhancing redox-dependent mitochondrial function and metabolic dilation. J. Mol. Cell. Cardiol. 2015, 88, 14–28. [Google Scholar] [CrossRef]
- Fukai, T.; Siegfried, M.; Ushio-Fukai, M.; Cheng, Y.; Kojda, G.; Harrison, D. Regulation of the vascular extracellular superoxide dismutase by nitric oxide and exercise training. J. Clin. Investig. 2000, 105, 1631–1639. [Google Scholar] [CrossRef]
- Duckles, S.; Krause, D.; Stirone, C.; Procaccio, V. Estrogen and Mitochondria: A New Paradigm for Vascular Protection? Mol. Interv. 2006, 6, 26–35. [Google Scholar] [CrossRef]
- Stirone, C.; Duckles, S.; Krause, D.; Procaccio, V. Estrogen increases mitochondrial efficiency and reduces oxidative stress in cerebral blood vessels. Mol. Pharmacol. 2005, 68, 959–965. [Google Scholar] [CrossRef]
- Skogastierna, C.; Hotzen, M.; Rane, A.; Ekstrom, L. A supraphysiological dose of testosterone induces nitric oxide production and oxidative stress. Eur. J. Prev. Cardiol. 2014, 21, 1049–1054. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wu, S.; Ruan, Y.; Hong, L.; Xing, X.; Lai, W. Testosterone suppresses oxidative stress via androgen receptor-independent pathway in murine cardiomyocytes. Mol. Med. Rep. 2011, 4, 1183–1188. [Google Scholar] [PubMed]
- Jeney, V.; Balla, J.; Yachie, A.; Varga, Z.; Vercellotti, G.M.; Eaton, J.W.; Balla, G. Pro-oxidant and cytotoxic effects of circulating heme. Blood 2002, 100, 879–887. [Google Scholar] [CrossRef] [PubMed]
- Fredenburgh, L.E.; Merz, A.A.; Cheng, S. Haeme oxygenase signalling pathway: Implications for cardiovascular disease. Eur. Heart J. 2015, 36, 1512–1518. [Google Scholar] [CrossRef] [PubMed]
- Otterbein, L.E.; Bach, F.H.; Alam, J.; Soares, M.; Tao Lu, H.; Wysk, M.; Davis, R.J.; Flavell, R.A.; Choi, A.M. Carbon monoxide has anti-inflammatory effects involving the mitogen-activated protein kinase pathway. Nat. Med. 2000, 6, 422–428. [Google Scholar] [CrossRef] [PubMed]
- Christova, T.; Diankova, Z.; Setchenska, M. Heme oxygenase--carbon monoxide signalling pathway as a physiological regulator of vascular smooth muscle cells. Acta Physiol. Pharmacol. Bulg. 2000, 25, 9–17. [Google Scholar] [PubMed]
- Pósa, A.; Kupai, K.; Ménesi, R.; Szalai, Z.; Szabó, R.; Pintér, Z.; Pálfi, G.; Gyöngyösi, M.; Berkó, A.; Pávó, I.; et al. Sexual Dimorphism of Cardiovascular Ischemia Susceptibility Is Mediated by Heme Oxygenase. Oxid. Med. Cell. Longev. 2013, 2013, 521563. [Google Scholar] [CrossRef] [PubMed]
- Chong, Z.Z.; Wang, S.; Shang, Y.C.; Maiese, K. Targeting cardiovascular disease with novel SIRT1 pathways. Future Cardiol. 2012, 8, 89–100. [Google Scholar] [CrossRef]
- Augustine, L.; Fisher, C.; Lickteig, A.; Aleksunes, L.; Slitt, A.; Cherrington, N. Gender divergent expression of Nqo1 in Sprague Dawley and August Copenhagen x Irish rats. J. Biochem. Mol. Toxicol. 2008, 22, 93–100. [Google Scholar] [CrossRef]
- Rougée, L.; Riches, A.; Berman, J.; Collier, A. The Ontogeny and Population Variability of Human Hepatic NADPH Dehydrogenase Quinone Oxido-Reductase 1 (NQO1). Drug Metab. Dispos. 2016, 44, 967–974. [Google Scholar] [CrossRef]
- Chen, Q.M.; Maltagliati, A.J. Nrf2 at the heart of oxidative stress and cardiac protection. Physiol. Genom. 2018, 50, 77–97. [Google Scholar] [CrossRef] [PubMed]
- Dinkova-Kostova, A.T.; Kostov, R.V.; Canning, P. Keap1, the cysteine-based mammalian intracellular sensor for electrophiles and oxidants. Arch. Biochem. Biophys. 2017, 617, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Katoh, Y.; Itoh, K.; Yoshida, E.; Miyagishi, M.; Fukamizu, A.; Yamamoto, M. Two domains of Nrf2 cooperatively bind CBP, a CREB binding protein, and synergistically activate transcription. Genes Cells 2001, 6, 857–868. [Google Scholar] [CrossRef] [PubMed]
- Wardyn, J.D.; Ponsford, A.H.; Sanderson, C.M. Dissecting molecular cross-talk between Nrf2 and NF-kappaB response pathways. Biochem. Soc. Trans. 2015, 43, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.H.; Qu, J.; Shen, X. NF-kappaB/p65 antagonizes Nrf2-ARE pathway by depriving CBP from Nrf2 and facilitating recruitment of HDAC3 to MafK. Biochim. Biophys. Acta 2008, 1783, 713–727. [Google Scholar] [CrossRef]
- Kerns, M.; Hakim, K.; Zieman, A.; Lu, R.; Coulombe, P. Sexual dimorphism in response to a NRF2 inducer in a model for pachyonychia congenita. J. Investig. Dermatol. 2018, 138, 1094–1100. [Google Scholar] [CrossRef]
- Yu, J.; Zhao, Y.; Li, B.; Sun, L.; Huo, H. 17beta-estradiol regulates the expression of antioxidant enzymes in myocardial cells by increasing Nrf2 translocation. J. Biochem. Mol. Toxicol. 2012, 26, 264–269. [Google Scholar] [CrossRef]
- D’Onofrio, N.; Servillo, L.; Balestrieri, M.L. SIRT1 and SIRT6 Signaling Pathways in Cardiovascular Disease Protection. Antioxid. Redox Signal. 2018, 28, 711–732. [Google Scholar] [CrossRef]
- Li, J.; Ichikawa, T.; Villacorta, L.; Janicki, J.S.; Brower, G.L.; Yamamoto, M.; Cui, T. Nrf2 protects against maladaptive cardiac responses to hemodynamic stress. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1843–1850. [Google Scholar] [CrossRef]
- Wang, C.; Luo, Z.; Carter, G.; Wellstein, A.; Jose, P.A.; Tomlinson, J.; Leiper, J.; Welch, W.J.; Wilcox, C.S.; Wang, D. NRF2 prevents hypertension, increased ADMA, microvascular oxidative stress, and dysfunction in mice with two weeks of ANG II infusion. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2018, 314, R399–R406. [Google Scholar] [CrossRef]
- Sanz, M.N.; Grimbert, L.; Moulin, M.; Gressette, M.; Rucker-Martin, C.; Lemaire, C.; Mericskay, M.; Veksler, V.; Ventura-Clapier, R.; Garnier, A.; et al. Inducible Cardiac-Specific Deletion of Sirt1 in Male Mice Reveals Progressive Cardiac Dysfunction and Sensitization of the Heart to Pressure Overload. Int. J. Mol. Sci. 2019, 20, 5. [Google Scholar] [CrossRef]
- Fry, J.L.; Shiraishi, Y.; Turcotte, R.; Yu, X.; Gao, Y.Z.; Akiki, R.; Bachschmid, M.; Zhang, Y.; Morgan, K.G.; Cohen, R.A.; et al. Vascular Smooth Muscle Sirtuin-1 Protects Against Aortic Dissection During Angiotensin II-Induced Hypertension. J. Am. Heart Assoc. 2015, 4, e002384. [Google Scholar] [CrossRef]
- Alcendor, R.R.; Gao, S.; Zhai, P.; Zablocki, D.; Holle, E.; Yu, X.; Tian, B.; Wagner, T.; Vatner, S.F.; Sadoshima, J. Sirt1 regulates aging and resistance to oxidative stress in the heart. Circ. Res. 2007, 100, 1512–1521. [Google Scholar] [CrossRef]
- Gao, P.; Xu, T.T.; Lu, J.; Li, L.; Xu, J.; Hao, D.L.; Chen, H.Z.; Liu, D.P. Overexpression of SIRT1 in vascular smooth muscle cells attenuates angiotensin II-induced vascular remodeling and hypertension in mice. J. Mol. Med. 2014, 92, 347–357. [Google Scholar] [CrossRef]
- Wang, W.J.; Li, S.Y.; Xing, Y.F.; Horvath, G.; Wang, Q.; Cui, T.X. Cardiac Specific Overexpression of Nrf2 Protects Against Pressure Overload-Induced Heart Failure Via Enhancing Autophagic Clearance of Ubiquitinated Proteins. J. Cardiac. Fail. 2012, 18, S1. [Google Scholar] [CrossRef]
- Shimabukuro, M. SIRT1 and Gender Differences in Atherosclerotic Cardiovascular Disease. J. Atheroscler. 2020, 27, 8–10. [Google Scholar] [CrossRef]
- Del Bo’, C.; Martini, D.; Porrini, M.; Klimis-Zacas, D.; Riso, P. Berries and oxidative stress markers: An overview of human intervention studies. Food Funct. 2015, 6. [Google Scholar] [CrossRef]
- Marniemi, J.; Hakala, P.; Mäki, J.; Ahotupa, M. Partial resistance of low-density lipoprotein to oxidation in vivo after increased intake of berries. Nutr. Metab. Cardiovasc. Dis. 2000, 10, 331–337. [Google Scholar]
- Mazza, G.; Kay, C.; Cottrell, T.; Holub, B. Absorption of anthocyanins from blueberries and serum antioxidant status in human subjects. J. Agric. Food Chem. 2002, 50, 7731–7737. [Google Scholar] [CrossRef]
- Kay, C.; Holub, B. The effect of wild blueberry (Vaccinium angustifolium) consumption on postprandial serum antioxidant status in human subjects. Br. J. Nutr. 2002, 88, 389–397. [Google Scholar] [CrossRef]
- Netzel, M.; Strass, G.; Kaul, C.; Bitsch, I.; Dietrich, H.; Bitsch, R. In vivo antioxidative capacity of a composite berry juice. Food Res. Int. 2002, 35, 213–216. [Google Scholar] [CrossRef]
- Netzel, M.; Strass, G.; Herbst, M.; Dietrich, H.; Bitsch, R.; Bitsch, I.; Frank, T. The excretion and biological antioxidant activity of elderberry antioxidants in healthy humans. Food Res. Int. 2005, 38, 905–910. [Google Scholar] [CrossRef]
- Tulipani, S.; Romandini, S.; Busco, F.; Bompadre, S.; Mezzetti, B.; Battino, M. Ascorbate, not urate, modulates the plasma antioxidant capacity after strawberry intake. Food Chem. 2009, 117, 181–188. [Google Scholar] [CrossRef]
- McLeay, Y.; Barnes, M.; Mundel, T.; Hurst, S.; Hurst, R.; Stannard, S. Effect of New Zealand blueberry consumption on recovery from eccentric exercise-induced muscle damage. J. Int. Soc. Sports Nutr. 2012, 9. [Google Scholar] [CrossRef]
- Mathison, B.; Kimble, L.; Kaspar, K.; Khoo, C.; Chew, B. Consumption of cranberry beverage improved endogenous antioxidant status and protected against bacteria adhesion in healthy humans: A randomized controlled trial. Nutr. Res. 2014, 34, 420–427. [Google Scholar] [CrossRef]
- Johnson, S.A.; Figueroa, A.; Navaei, N.; Wong, A.; Kalfon, R.; Ormsbee, L.T.; Feresin, R.G.; Elam, M.L.; Hooshmand, S.; Payton, M.E.; et al. Daily blueberry consumption improves blood pressure and arterial stiffness in postmenopausal women with pre- and stage 1-hypertension: A randomized, double-blind, placebo-controlled clinical trial. J. Acad. Nutr. Diet 2015, 115, 369–377. [Google Scholar] [CrossRef]
- Basu, A.; Du, M.; Leyva, M.J.; Sanchez, K.; Betts, N.M.; Wu, M.; Aston, C.E.; Lyons, T.J. Blueberries decrease cardiovascular risk factors in obese men and women with metabolic syndrome. J. Nutr. 2010, 140, 1582–1587. [Google Scholar] [CrossRef]
- Feresin, R.G.; Johnson, S.A.; Pourafshar, S.; Campbell, J.C.; Jaime, S.J.; Navaei, N.; Elam, M.L.; Akhavan, N.S.; Alvarez-Alvarado, S.; Tenenbaum, G.; et al. Impact of daily strawberry consumption on blood pressure and arterial stiffness in pre- and stage 1-hypertensive postmenopausal women: A randomized controlled trial. Food Funct. 2017, 8, 4139–4149. [Google Scholar] [CrossRef]
- Burton-Freeman, B.; Linares, A.; Hyson, D.; Kappagoda, T. Strawberry modulates LDL oxidation and postprandial lipemia in response to high-fat meal in overweight hyperlipidemic men and women. J. Am. Coll. Nutr. 2010, 29, 46–54. [Google Scholar] [CrossRef]
- An, J.H.; Kim, D.L.; Lee, T.B.; Kim, K.J.; Kim, S.H.; Kim, N.H.; Kim, H.Y.; Choi, D.S.; Kim, S.G. Effect of Rubus Occidentalis Extract on Metabolic Parameters in Subjects with Prediabetes: A Proof-of-concept, Randomized, Double-blind, Placebo-controlled Clinical Trial. Phytother. Res. 2016, 30, 1634–1640. [Google Scholar] [CrossRef]
- Hung, C.H.; Chan, S.H.; Chu, P.M.; Tsai, K.L. Quercetin is a potent anti-atherosclerotic compound by activation of SIRT1 signaling under oxLDL stimulation. Mol. Nutr. Food Res. 2015, 59, 1905–1917. [Google Scholar] [CrossRef]
- Zhou, Y.; Jiang, Z.; Lu, H.; Xu, Z.; Tong, R.; Shi, J.; Jia, G. Recent Advances of Natural Polyphenols Activators for Keap1-Nrf2 Signaling Pathway. Chem. Biodivers. 2019, 16, e1900400. [Google Scholar] [CrossRef]
- Bello, M.; Morales-Gonzalez, J.A. Molecular recognition between potential natural inhibitors of the Keap1-Nrf2 complex. Int. J. Biol. Macromol. 2017, 105, 981–992. [Google Scholar] [CrossRef]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef]
- Silva-Islas, C.A.; Maldonado, P.D. Canonical and non-canonical mechanisms of Nrf2 activation. Pharmacol. Res. 2018, 134, 92–99. [Google Scholar] [CrossRef]
- Zheng, D.; Liu, Z.; Zhou, Y.; Hou, N.; Yan, W.; Qin, Y.; Ye, Q.; Cheng, X.; Xiao, Q.; Bao, Y.; et al. Urolithin B, a gut microbiota metabolite, protects against myocardial ischemia/reperfusion injury via p62/Keap1/Nrf2 signaling pathway. Pharmacol. Res. 2020, 153, 104655. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, Y.; Wang, X.; Liu, Y.; Xia, M. Supplementation with cyanidin-3-O-beta-glucoside protects against hypercholesterolemia-mediated endothelial dysfunction and attenuates atherosclerosis in apolipoprotein E-deficient mice. J. Nutr. 2012, 142, 1033–1037. [Google Scholar] [CrossRef]
- Ding, Y.; Zhang, B.; Zhou, K.; Chen, M.; Wang, M.; Jia, Y.; Song, Y.; Li, Y.; Wen, A. Dietary ellagic acid improves oxidant-induced endothelial dysfunction and atherosclerosis: Role of Nrf2 activation. Int. J. Cardiol. 2014, 175, 508–514. [Google Scholar] [CrossRef]
- Xu, J.W.; Ikeda, K.; Yamori, Y. Inhibitory effect of polyphenol cyanidin on TNF-alpha-induced apoptosis through multiple signaling pathways in endothelial cells. Atherosclerosis 2007, 193, 299–308. [Google Scholar] [CrossRef]
- Jin, L.; Piao, Z.H.; Sun, S.; Liu, B.; Kim, G.R.; Seok, Y.M.; Lin, M.Q.; Ryu, Y.; Choi, S.Y.; Kee, H.J.; et al. Gallic Acid Reduces Blood Pressure and Attenuates Oxidative Stress and Cardiac Hypertrophy in Spontaneously Hypertensive Rats. Sci. Rep. 2017, 7, 15607. [Google Scholar] [CrossRef]
- Yan, X.; Zhang, Q.Y.; Zhang, Y.L.; Han, X.; Guo, S.B.; Li, H.H. Gallic Acid Attenuates Angiotensin II-Induced Hypertension and Vascular Dysfunction by Inhibiting the Degradation of Endothelial Nitric Oxide Synthase. Front. Pharmacol. 2020, 11, 1121. [Google Scholar] [CrossRef]
- Kalea, A.Z.; Clark, K.; Schuschke, D.A.; Kristo, A.S.; Klimis-Zacas, D.J. Dietary enrichment with wild blueberries (Vaccinium angustifolium) affects the vascular reactivity in the aorta of young spontaneously hypertensive rats. J. Nutr. Biochem. 2010, 21, 14–22. [Google Scholar] [CrossRef]
- Rodriguez-Mateos, A.; Rendeiro, C.; Bergillos-Meca, T.; Tabatabaee, S.; George, T.W.; Heiss, C.; Spencer, J.P. Intake and time dependence of blueberry flavonoid-induced improvements in vascular function: A randomized, controlled, double-blind, crossover intervention study with mechanistic insights into biological activity. Am. J. Clin. Nutr. 2013, 98, 1179–1191. [Google Scholar] [CrossRef]
- Istas, G.; Feliciano, R.P.; Weber, T.; Garcia-Villalba, R.; Tomas-Barberan, F.; Heiss, C.; Rodriguez-Mateos, A. Plasma urolithin metabolites correlate with improvements in endothelial function after red raspberry consumption: A double-blind randomized controlled trial. Arch. Biochem. Biophys. 2018, 651, 43–51. [Google Scholar] [CrossRef]
- Djurica, D.; Holt, R.; Ren, J.; Shindel, A.; Hackman, R.; Keen, C. Effects of a dietary strawberry powder on parameters of vascular health in adolescent males. Br. J. Nutr. 2016, 116, 639–647. [Google Scholar] [CrossRef]
- Jeong, H.S.; Hong, S.J.; Lee, T.B.; Kwon, J.W.; Jeong, J.T.; Joo, H.J.; Park, J.H.; Ahn, C.M.; Yu, C.W.; Lim, D.S. Effects of black raspberry on lipid profiles and vascular endothelial function in patients with metabolic syndrome. Phytother. Res. 2014, 28, 1492–1498. [Google Scholar] [CrossRef]
- Jeong, H.S.; Hong, S.J.; Cho, J.Y.; Lee, T.B.; Kwon, J.W.; Joo, H.J.; Park, J.H.; Yu, C.W.; Lim, D.S. Effects of Rubus occidentalis extract on blood pressure in patients with prehypertension: Randomized, double-blinded, placebo-controlled clinical trial. Nutrition 2016, 32, 461–467. [Google Scholar] [CrossRef]
- Kane, M.; Anselm, E.; Rattmann, Y.; Auger, C.; Schini-Kerth, V. Role of gender and estrogen receptors in the rat aorta endothelium-dependent relaxation to red wine polyphenols. Vasc. Pharmacol. 2009, 51, 140–146. [Google Scholar] [CrossRef]
- Yao, F.; Liu, Y.; Liu, D.; Wu, S.; Lin, H.; Fan, R.; Li, C. Sex differences between vascular endothelial function and carotid intima-media thickness by Framingham Risk Score. J. Ultrasound Med. 2014, 33, 281–286. [Google Scholar] [CrossRef]
- Son, Y.; Cheong, Y.K.; Kim, N.H.; Chung, H.T.; Kang, D.G.; Pae, H.O. Mitogen-Activated Protein Kinases and Reactive Oxygen Species: How Can ROS Activate MAPK Pathways? J. Signal Transduct. 2011, 2011, 792639. [Google Scholar] [CrossRef]
- Narula, J.; Haider, N.; Arbustini, E.; Chandrashekhar, Y. Mechanisms of disease: Apoptosis in heart failure--seeing hope in death. Nat. Clin. Pract. Cardiovasc. Med. 2006, 3, 681–688. [Google Scholar] [CrossRef]
- Bennett, M.R. Apoptosis of vascular smooth muscle cells in vascular remodelling and atherosclerotic plaque rupture. Cardiovasc. Res. 1999, 41, 361–368. [Google Scholar] [CrossRef]
- Kim, R. Unknotting the roles of Bcl-2 and Bcl-xL in cell death. Biochem. Biophys. Res. Commun. 2005, 333, 336–343. [Google Scholar] [CrossRef]
- Zorov, D.B.; Filburn, C.R.; Klotz, L.O.; Zweier, J.L.; Sollott, S.J. Reactive oxygen species (ROS)-induced ROS release: A new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J. Exp. Med. 2000, 192, 1001–1014. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Nijhawan, D.; Budihardjo, I.; Srinivasula, S.M.; Ahmad, M.; Alnemri, E.S.; Wang, X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 1997, 91, 479–489. [Google Scholar] [CrossRef]
- Porter, A.G.; Janicke, R.U. Emerging roles of caspase-3 in apoptosis. Cell. Death Differ. 1999, 6, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Chandrashekhar, Y.; Sen, S.; Anway, R.; Shuros, A.; Anand, I. Long-term caspase inhibition ameliorates apoptosis, reduces myocardial troponin-I cleavage, protects left ventricular function, and attenuates remodeling in rats with myocardial infarction. J. Am. Coll. Cardiol. 2004, 43, 295–301. [Google Scholar] [CrossRef]
- Miyashita, T.; Reed, J.C. Tumor-Suppressor P53 Is a Direct Transcriptional Activator of the Human Bax Gene. Cell 1995, 80, 293–299. [Google Scholar]
- Graupner, V.; Alexander, E.; Overkamp, T.; Rothfuss, O.; De Laurenzi, V.; Gillissen, B.F.; Daniel, P.T.; Schulze-Osthoff, K.; Essmann, F. Differential regulation of the proapoptotic multidomain protein Bak by p53 and p73 at the promoter level. Cell Death Differ. 2011, 18, 1130–1139. [Google Scholar] [CrossRef]
- Cesselli, D.; Jakoniuk, I.; Barlucchi, L.; Beltrami, A.P.; Hintze, T.H.; Nadal-Ginard, B.; Kajstura, J.; Leri, A.; Anversa, P. Oxidative stress-mediated cardiac cell death is a major determinant of ventricular dysfunction and failure in dog dilated cardiomyopathy. Circ. Res. 2001, 89, 279–286. [Google Scholar] [CrossRef]
- Song, H.; Conte, J.V., Jr.; Foster, A.H.; McLaughlin, J.S.; Wei, C. Increased p53 protein expression in human failing myocardium. J. Heart Lung Transplant. 1999, 18, 744–749. [Google Scholar] [CrossRef]
- Fan, M.; Goodwin, M.; Vu, T.; Brantley-Finley, C.; Gaarde, W.A.; Chambers, T.C. Vinblastine-induced phosphorylation of Bcl-2 and Bcl-XL is mediated by JNK and occurs in parallel with inactivation of the Raf-1/MEK/ERK cascade. J. Biol. Chem. 2000, 275, 29980–29985. [Google Scholar] [CrossRef] [PubMed]
- Olivetti, G.; Giordano, G.; Corradi, D.; Melissari, M.; Lagrasta, C.; Gambert, S.; Anversa, P. Gender differences and aging: Effects on the human heart. J. Am. Coll. Cardiol. 1995, 26, 1068–1079. [Google Scholar] [CrossRef]
- Guerra, S.; Leri, A.; Wang, X.; Finato, N.; Di Loreto, C.; Beltrami, C.; Kajstura, J.; Anversa, P. Myocyte death in the failing human heart is gender dependent. Circ. Res. 1999, 85, 856–866. [Google Scholar] [CrossRef] [PubMed]
- Biondi-Zoccai, C.; Abate, A.; Bussani, R.; Camilot, D.; Giorgio, F.; Marino, M.; Silvestri, F.; Baldi, F.; Biasucci, L.; Baldi, A. Reduced post-infarction myocardial apoptosis in women: A clue to their different clinical course? Heart 2005, 91, 99–101. [Google Scholar] [CrossRef]
- Bouma, W.; Noma, M.; Kanemoto, S.; Matsubara, M.; Leshnower, B.; Hinmon, R.; Gorman, J.H., 3rd; Gorman, R. Sex-related resistance to myocardial ischemia-reperfusion injury is associated with high constitutive ARC expression. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H1510–H1517. [Google Scholar] [CrossRef]
- Gabel, S.; Walker, V.; London, R.; Steenbergen, C.; Korach, K.; Murphy, E. Estrogen receptor beta mediates gender differences in ischemia/reperfusion injury. JMCC 2005, 38, 289–297. [Google Scholar] [CrossRef]
- Wang, M.; Tsai, B.; Reiger, K.; Brown, J.; Meldrum, D. 17-beta-Estradiol decreases p38 MAPK-mediated myocardial inflammation and dysfunction following acute ischemia. J. Mol. Cell. Cardiol. 2006, 40, 205–212. [Google Scholar] [CrossRef]
- Matsui, T.; Li, L.; del Monte, F.; Fukui, Y.; Franke, T.; Hajjar, R.; Rosenzweig, A. Adenoviral gene transfer of activated phosphatidylinositol 3′-kinase and Akt inhibits apoptosis of hypoxic cardiomyocytes in vitro. Circulation 1999, 100, 2373–2379. [Google Scholar] [CrossRef]
- Fujio, Y.; Nguyen, T.; Wencker, D.; Kitsis, R.; Walsh, K. Akt promotes survival of cardiomyocytes in vitro and protects against ischemia-reperfusion injury in mouse heart. Circulation 2000, 101, 660–667. [Google Scholar] [CrossRef]
- Abeyrathna, P.; Su, Y. The Critical Role of Akt in Cardiovascular Function. Vasc. Pharmacol. 2015, 74, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Camper-Kirby, D.; Welch, S.; Walker, A.; Shiraishi, I.; Schaefer, K.; Kajstura, J.; Anversa, P.; Sussman, M. Myocardial Akt activation and gender: Increased nuclear activity in females versus males. Circ. Res. 2001, 88, 1020–1027. [Google Scholar] [CrossRef] [PubMed]
- Ogawara, Y.; Kishishita, S.; Obata, T.; Isazawa, Y.; Suzuki, T.; Tanaka, K.; Masuyama, N.; Gotoh, Y. Akt enhances Mdm2-mediated ubiquitination and degradation of p53. J. Biol. Chem. 2002, 277, 21843–21850. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, T.; Leal, J.; Seger, R.; Taya, Y.; Oren, M. Cross-talk between Akt, p53 and Mdm2: Possible implications for the regulation of apoptosis. Oncogene 2002, 21, 1299–1303. [Google Scholar] [CrossRef] [PubMed]
- Mayo, L.; Donner, D. A phosphatidylinositol 3-kinase/Akt pathway promotes translocation of Mdm2 from the cytoplasm to the nucleus. Proc. Natl. Acad. Sci. USA 2001, 98, 11598–11603. [Google Scholar] [CrossRef]
- Zhou, B.; Liao, Y.; Xia, W.; Zou, Y.; Spohn, B.; Hung, M. HER-2/neu induces p53 ubiquitination via Akt-mediated MDM2 phosphorylation. Nat. Cell Biol. 2001, 3, 973–982. [Google Scholar] [CrossRef]
- Moll, U.M.; Petrenko, O. The MDM2-p53 interaction. Mol. Cancer Res. 2003, 1, 1001–1008. [Google Scholar]
- Cardone, M.; Roy, N.; Stennicke, H.; Salvesen, G.; Franke, T.; Stanbridge, E.; Frisch, S.; Reed, J. Regulation of cell death protease caspase-9 by phosphorylation. Science 1998, 282, 1318–1321. [Google Scholar] [CrossRef]
- Patten, R.; Pourati, I.; Aronovitz, M.; Baur, J.; Celestin, F.; Chen, X.; Michael, A.; Haq, S.; Nuedling, S.; Grohe, C.; et al. 17beta-estradiol reduces cardiomyocyte apoptosis in vivo and in vitro via activation of phospho-inositide-3 kinase/Akt signaling. Circ. Res. 2004, 95, 692–699. [Google Scholar] [CrossRef]
- Ikeda, Y.; Aihara, K.; Akaike, M.; Sato, T.; Ishikawa, K.; Ise, T.; Yagi, S.; Iwase, T.; Ueda, Y.; Yoshida, S.; et al. Androgen receptor counteracts Doxorubicin-induced cardiotoxicity in male mice. Mol. Endocrinol. 2010, 24, 1338–1348. [Google Scholar] [CrossRef]
- Huang, C.; Gu, H.; Zhang, W.; Herrmann, J.; Wang, M. Testosterone-down-regulated Akt pathway during cardiac ischemia/reperfusion: A mechanism involving BAD, Bcl-2 and FOXO3a. J. Surg. Res. 2010, 164, e1–e11. [Google Scholar] [CrossRef]
- Le, T.; Ashton, A.; Mardini, M.; Stanton, P.; Funder, J.; Handelsman, D.; Mihailidou, A. Role of androgens in sex differences in cardiac damage during myocardial infarction. Endocrinology 2014, 155, 568–575. [Google Scholar] [CrossRef]
- Wang, M.; Tsai, B.; Kher, A.; Baker, L.; Wairiuko, G.; Meldrum, D. Role of endogenous testosterone in myocardial proinflammatory and proapoptotic signaling after acute ischemia-reperfusion. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H221–H226. [Google Scholar] [CrossRef]
- Kim, J.; Pedram, A.; Razandi, M.; Levin, E. Estrogen prevents cardiomyocyte apoptosis through inhibition of reactive oxygen species and differential regulation of p38 kinase isoforms. J. Biol. Chem. 2006, 281, 6760–6767. [Google Scholar] [CrossRef]
- Liu, H.; Pedram, A.; Kim, J. Oestrogen prevents cardiomyocyte apoptosis by suppressing p38α-mediated activation of p53 and by down-regulating p53 inhibition on p38β. Cardiovasc. Res. 2011, 89, 119–128. [Google Scholar] [CrossRef]
- Lee, S.-D.; Kuo, W.-W.; Ho, Y.-J.; Lin, A.-C.; Tsai, C.-H.; Wang, H.-F.; Kuo, C.-H.; Yang, A.-L.; Huang, C.-Y.; Hwang, J.-M. Cardiac Fas-dependent and mitochondria-dependent apoptosis in ovariectomized rats. Maturitas 2008, 61, 268–277. [Google Scholar] [CrossRef]
- Liou, C.-M.; Yang, A.-L.; Kuo, C.-H.; Tin, H.; Huang, C.-Y.; Lee, S.-D. Effects of 17beta-estradiol on cardiac apoptosis in ovariectomized rats. Cell Biochem. Funct. 2010, 28, 521–528. [Google Scholar] [CrossRef]
- Lin, Y.-Y.; Chen, J.-S.; Wu, X.-B.; Shyu, W.-C.; Chaunchaiyakul, R.; Zhao, X.-L.; Kuo, C.-H.; Cheng, Y.-J.; Yang, A.-L.; Lee, S.-D. Combined effects of 17β-estradiol and exercise training on cardiac apoptosis in ovariectomized rats. PLoS ONE 2018, 13. [Google Scholar] [CrossRef]
- Lin, Y.-Y.; Hong, Y.; Yu, S.-H.; Wu, X.-B.; Shyu, W.-C.; Chen, J.-S.; Ting, H.; Yang, A.-L.; Lee, S.-D. Antiapoptotic and mitochondrial biogenetic effects of exercise training on ovariectomized hypertensive rat hearts. J. Appl. Physiol. 2019, 126, 1661–1672. [Google Scholar] [CrossRef]
- Chen, C.; Hu, L.; Dong, T.; Wang, G.; Wang, L.; Zhou, X.; Jiang, Y.; Murao, K.; Lu, S.; Chen, J.; et al. Apoptosis and autophagy contribute to gender difference in cardiac ischemia-reperfusion induced injury in rats. Life Sci. 2013, 93, 265–270. [Google Scholar] [CrossRef]
- Gordon, J.W.; Shaw, J.A.; Kirshenbaum, L.A. Multiple facets of NF-kappaB in the heart: To be or not to NF-kappaB. Circ. Res. 2011, 108, 1122–1132. [Google Scholar] [CrossRef] [PubMed]
- Bujak, M.; Dobaczewski, M.; Chatila, K.; Mendoza, L.H.; Li, N.; Reddy, A.; Frangogiannis, N.G. Interleukin-1 receptor type I signaling critically regulates infarct healing and cardiac remodeling. Am. J. Pathol. 2008, 173, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Huynh, M.L.; Fadok, V.A.; Henson, P.M. Phosphatidylserine-dependent ingestion of apoptotic cells promotes TGF-beta1 secretion and the resolution of inflammation. J. Clin. Investig. 2002, 109, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Thompson, N.L.; Flanders, K.C.; Smith, J.M.; Ellingsworth, L.R.; Roberts, A.B.; Sporn, M.B. Expression of transforming growth factor-beta 1 in specific cells and tissues of adult and neonatal mice. J. Cell Biol. 1989, 108, 661–669. [Google Scholar] [CrossRef] [PubMed]
- Barcellos-Hoff, M.H.; Dix, T.A. Redox-mediated activation of latent transforming growth factor-beta 1. Mol. Endocrinol. 1996, 10, 1077–1083. [Google Scholar] [CrossRef]
- Mohamed, R.; Cao, Y.; Afroz, R.; Xu, S.; Ta, H.T.; Barras, M.; Zheng, W.; Little, P.J.; Kamato, D. ROS directly activates transforming growth factor beta type 1 receptor signalling in human vascular smooth muscle cells. Biochim. Biophys. Acta Gen. Subj. 2020, 1864, 129463. [Google Scholar] [CrossRef] [PubMed]
- Sartore, S.; Chiavegato, A.; Faggin, E.; Franch, R.; Puato, M.; Ausoni, S.; Pauletto, P. Contribution of adventitial fibroblasts to neointima formation and vascular remodeling: From innocent bystander to active participant. Circ. Res. 2001, 89, 1111–1121. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. Cardiac fibrosis: Cell biological mechanisms, molecular pathways and therapeutic opportunities. Mol. Aspects Med. 2019, 65, 70–99. [Google Scholar] [CrossRef]
- Iwanaga, Y.; Aoyama, T.; Kihara, Y.; Onozawa, Y.; Yoneda, T.; Sasayama, S. Excessive activation of matrix metalloproteinases coincides with left ventricular remodeling during transition from hypertrophy to heart failure in hypertensive rats. J. Am. Coll. Cardiol. 2002, 39, 1384–1391. [Google Scholar] [CrossRef]
- Regitz-Zagrosek, V.; Kararigas, G. Mechanistic Pathways of Sex Differences in Cardiovascular Disease. Physiol. Rev. 2016, 97, 1–37. [Google Scholar] [CrossRef]
- Venkatesh, B.; Volpe, G.; Donekal, S.; Mewton, N.; Liu, C.; Shea, S.; Liu, K.; Burke, G.; Wu, C.; Bluemke, D.; et al. Association of longitudinal changes in left ventricular structure and function with myocardial fibrosis: The Multi-Ethnic Study of Atherosclerosis study. Hypertension 2014, 64, 508–515. [Google Scholar] [CrossRef] [PubMed]
- Campbell, D.; Somaratne, J.; Jenkins, A.; Prior, D.; Yii, M.; Kenny, J.; Newcomb, A.; Kelly, D.; Black, M. Differences in myocardial structure and coronary microvasculature between men and women with coronary artery disease. Hypertension 2011, 57, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Dworatzek, E.; Mahmoodzadeh, S.; Schriever, C.; Kusumoto, K.; Kramer, L.; Santos, G.; Fliegner, D.; Leung, Y.-K.; Ho, S.-M.; Zimmermann, W.-H.; et al. Sex-specific regulation of collagen I and III expression by 17β-Estradiol in cardiac fibroblasts: Role of estrogen receptors. Cardiovasc. Res. 2019, 115, 315–327. [Google Scholar] [CrossRef] [PubMed]
- Petrov, G.; Regitz-Zagrosek, V.; Lehmkuhl, E.; Krabatsch, T.; Dunkel, A.; Dandel, M.; Dworatzek, E.; Mahmoodzadeh, S.; Schubert, C.; Becher, E.; et al. Regression of myocardial hypertrophy after aortic valve replacement: Faster in women? Circulation 2010, 122, S23–S28. [Google Scholar] [CrossRef] [PubMed]
- Mahmoodzadeh, S.; Dworatzek, E.; Fritschka, S.; Pham, T.; Regitz-Zagrosek, V. 17β-Estradiol inhibits matrix metalloproteinase-2 transcription via MAP kinase in fibroblasts. Cardiovasc. Res. 2010, 85, 719–728. [Google Scholar] [CrossRef]
- Ahmet, I.; Spangler, E.; Shukitt-Hale, B.; Juhaszova, M.; Sollott, S.J.; Joseph, J.A.; Ingram, D.K.; Talan, M. Blueberry-enriched diet protects rat heart from ischemic damage. PLoS ONE 2009, 4, e5954. [Google Scholar] [CrossRef] [PubMed]
- Isaak, C.K.; Petkau, J.C.; Blewett, H.; O, K.; Siow, Y.L. Lingonberry anthocyanins protect cardiac cells from oxidative-stress-induced apoptosis. Can. J. Physiol. Pharmacol. 2017, 95, 904–910. [Google Scholar] [CrossRef]
- Feresin, R.G.; Huang, J.; Klarich, D.S.; Zhao, Y.; Pourafshar, S.; Arjmandi, B.H.; Salazar, G. Blackberry, raspberry and black raspberry polyphenol extracts attenuate angiotensin II-induced senescence in vascular smooth muscle cells. Food Funct. 2016, 7, 4175–4187. [Google Scholar] [CrossRef]
- Choi, Y.J.; Kang, J.S.; Park, J.H.; Lee, Y.J.; Choi, J.S.; Kang, Y.H. Polyphenolic flavonoids differ in their antiapoptotic efficacy in hydrogen peroxide-treated human vascular endothelial cells. J. Nutr. 2003, 133, 985–991. [Google Scholar] [CrossRef]
- Jin, L.; Piao, Z.H.; Liu, C.P.; Sun, S.; Liu, B.; Kim, G.R.; Choi, S.Y.; Ryu, Y.; Kee, H.J.; Jeong, M.H. Gallic acid attenuates calcium calmodulin-dependent kinase II-induced apoptosis in spontaneously hypertensive rats. J. Cell Mol. Med. 2018, 22, 1517–1526. [Google Scholar] [CrossRef]
- Jin, L.; Sun, S.; Ryu, Y.; Piao, Z.H.; Liu, B.; Choi, S.Y.; Kim, G.R.; Kim, H.S.; Kee, H.J.; Jeong, M.H. Gallic acid improves cardiac dysfunction and fibrosis in pressure overload-induced heart failure. Sci. Rep. 2018, 8, 9302. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Jiang, Y.; Jing, W.; Sun, B.; Miao, C.; Ren, L. Quercetin provides greater cardioprotective effect than its glycoside derivative rutin on isoproterenol-induced cardiac fibrosis in the rat. Can. J. Physiol. Pharmacol. 2013, 91, 951–959. [Google Scholar] [CrossRef] [PubMed]
- Olson, E.R.; Naugle, J.E.; Zhang, X.; Bomser, J.A.; Meszaros, J.G. Inhibition of cardiac fibroblast proliferation and myofibroblast differentiation by resveratrol. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H1131–H1138. [Google Scholar] [CrossRef] [PubMed]
- Ruel, G.; Pomerleau, S.; Couture, P.; Lemieux, S.; Lamarche, B.; Couillard, C. Plasma matrix metalloproteinase (MMP)-9 levels are reduced following low-calorie cranberry juice supplementation in men. J. Am. Coll. Nutr. 2009, 28, 694–701. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Najjar, R.S.; Turner, C.G.; Wong, B.J.; Feresin, R.G. Berry-Derived Polyphenols in Cardiovascular Pathologies: Mechanisms of Disease and the Role of Diet and Sex. Nutrients 2021, 13, 387. https://doi.org/10.3390/nu13020387
Najjar RS, Turner CG, Wong BJ, Feresin RG. Berry-Derived Polyphenols in Cardiovascular Pathologies: Mechanisms of Disease and the Role of Diet and Sex. Nutrients. 2021; 13(2):387. https://doi.org/10.3390/nu13020387
Chicago/Turabian StyleNajjar, Rami S., Casey G. Turner, Brett J. Wong, and Rafaela G. Feresin. 2021. "Berry-Derived Polyphenols in Cardiovascular Pathologies: Mechanisms of Disease and the Role of Diet and Sex" Nutrients 13, no. 2: 387. https://doi.org/10.3390/nu13020387
APA StyleNajjar, R. S., Turner, C. G., Wong, B. J., & Feresin, R. G. (2021). Berry-Derived Polyphenols in Cardiovascular Pathologies: Mechanisms of Disease and the Role of Diet and Sex. Nutrients, 13(2), 387. https://doi.org/10.3390/nu13020387