
High-Phytate Diets Increase Amyloid β Deposition and Apoptotic Neuronal Cell Death in a Rat Model



Abstract
:1. Introduction
2. Materials and Methods
2.1. Rats
2.2. Immunohistochemistry (IHC)
2.3. Biochemical Assays
2.4. Image Quantification
2.5. qRT-PCR
2.6. Western Blotting
2.7. Transcription Assays
2.8. Chromatin Immunoprecipitation Analysis
2.9. Statistical Analysis
3. Results
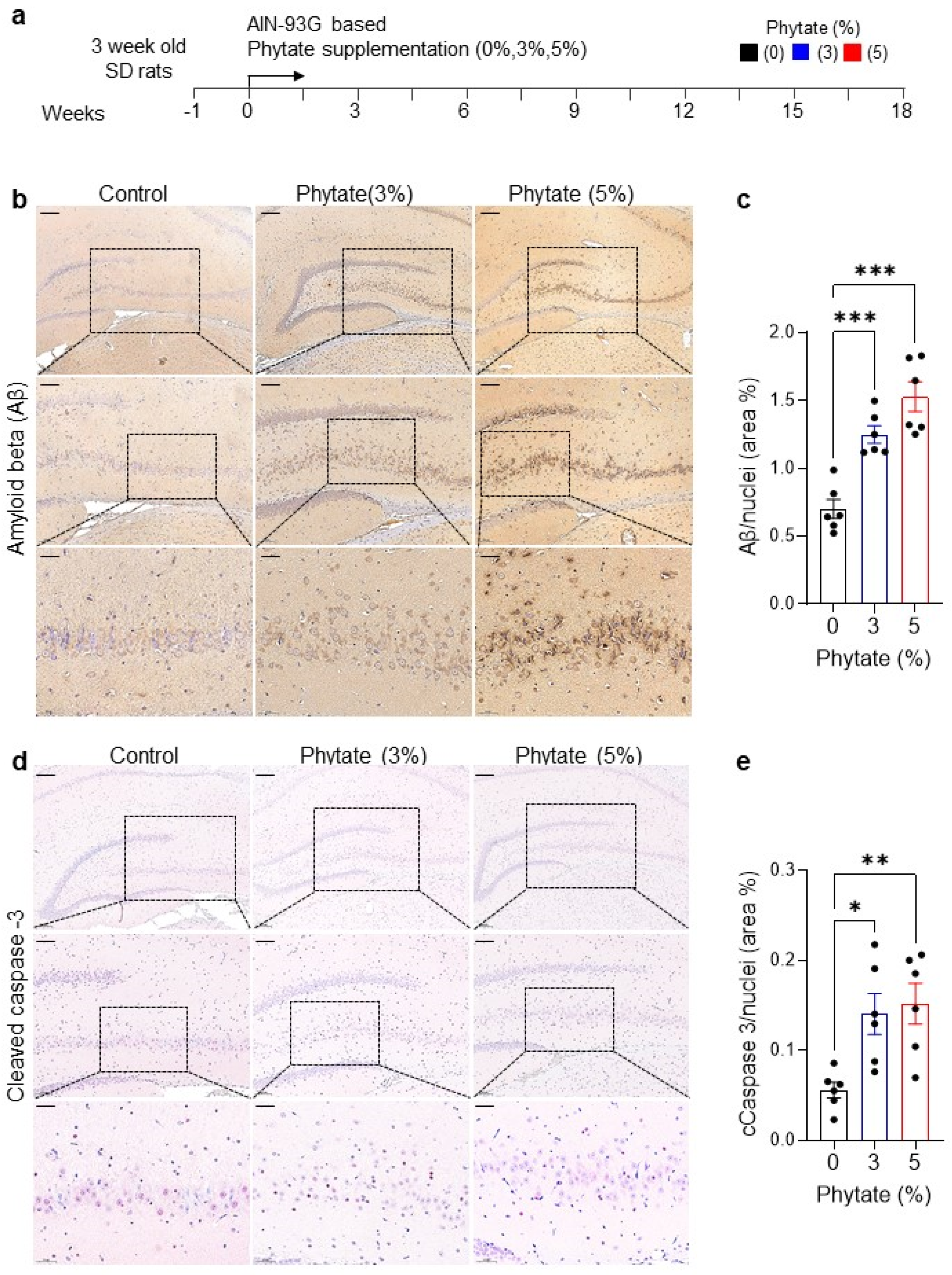
3.1. Rats Fed High-Phytate Diets Show Elevated Accumulation of Aβ Peptide and Neuronal Apoptosis in the Hippocampus
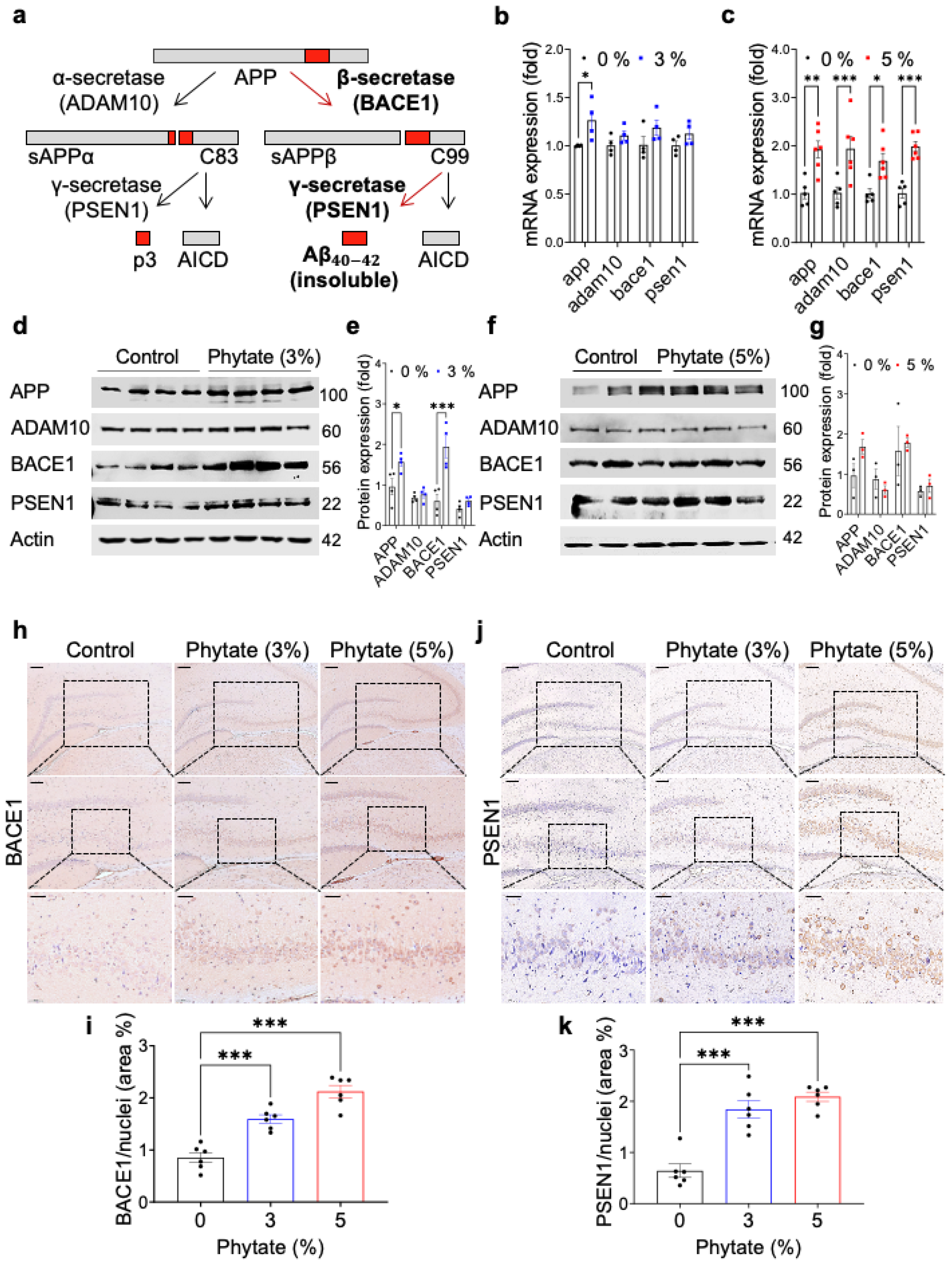
3.2. High-Phytate Diets Increase Aβ Accumulation through Activating the Amyloidogenic Pathway in the Hippocampus
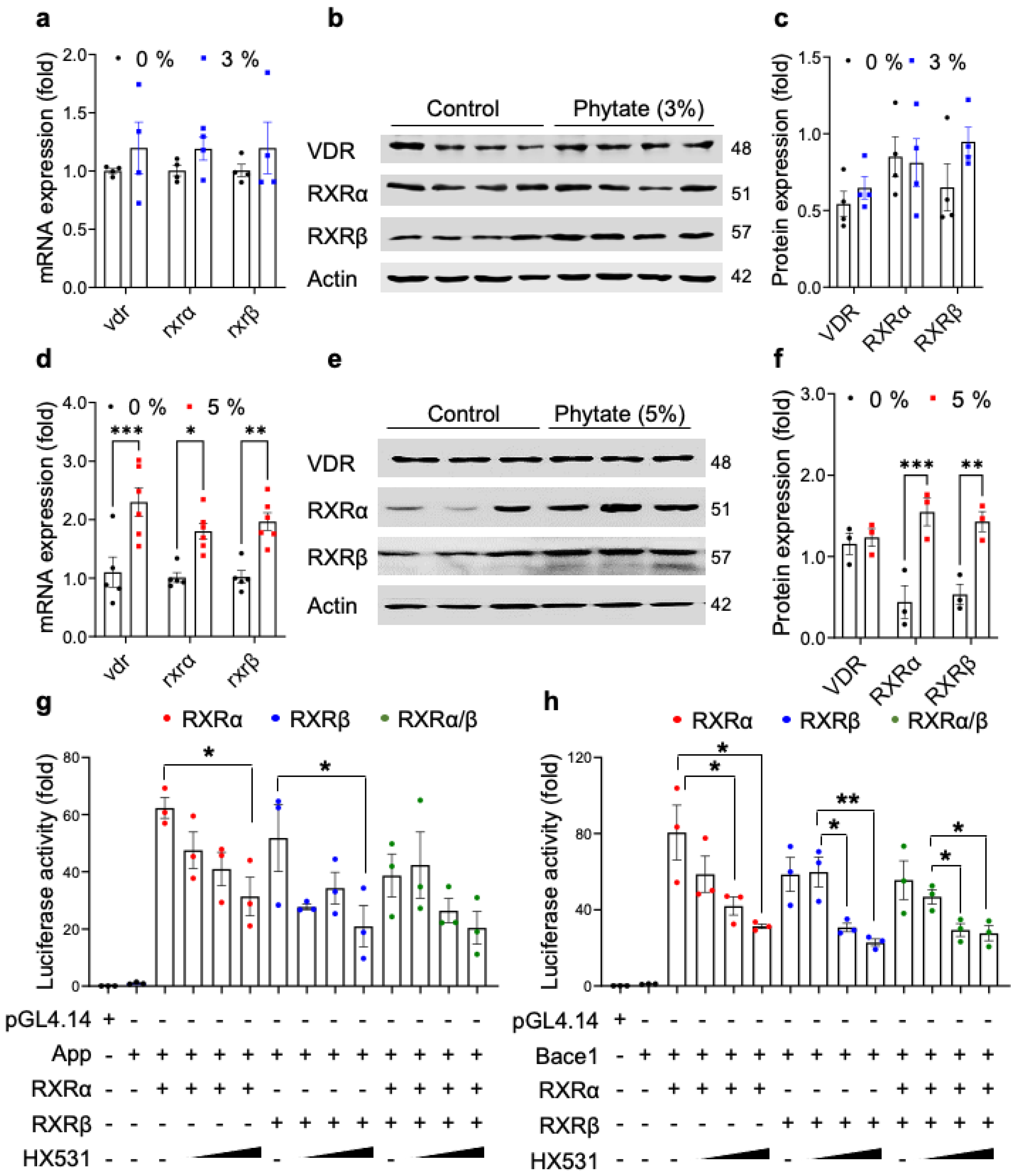
3.3. Rats Fed High-Phytate Diets Develop Secondary Hyperparathyroidism and Show Effects of PTH on the Transcriptional Activity of App and APP Processing Enzymes
3.4. High-Phytate Diets Increase the Expression of APP and BACE1 through Transcriptional Activation by RXRα/β and VDR
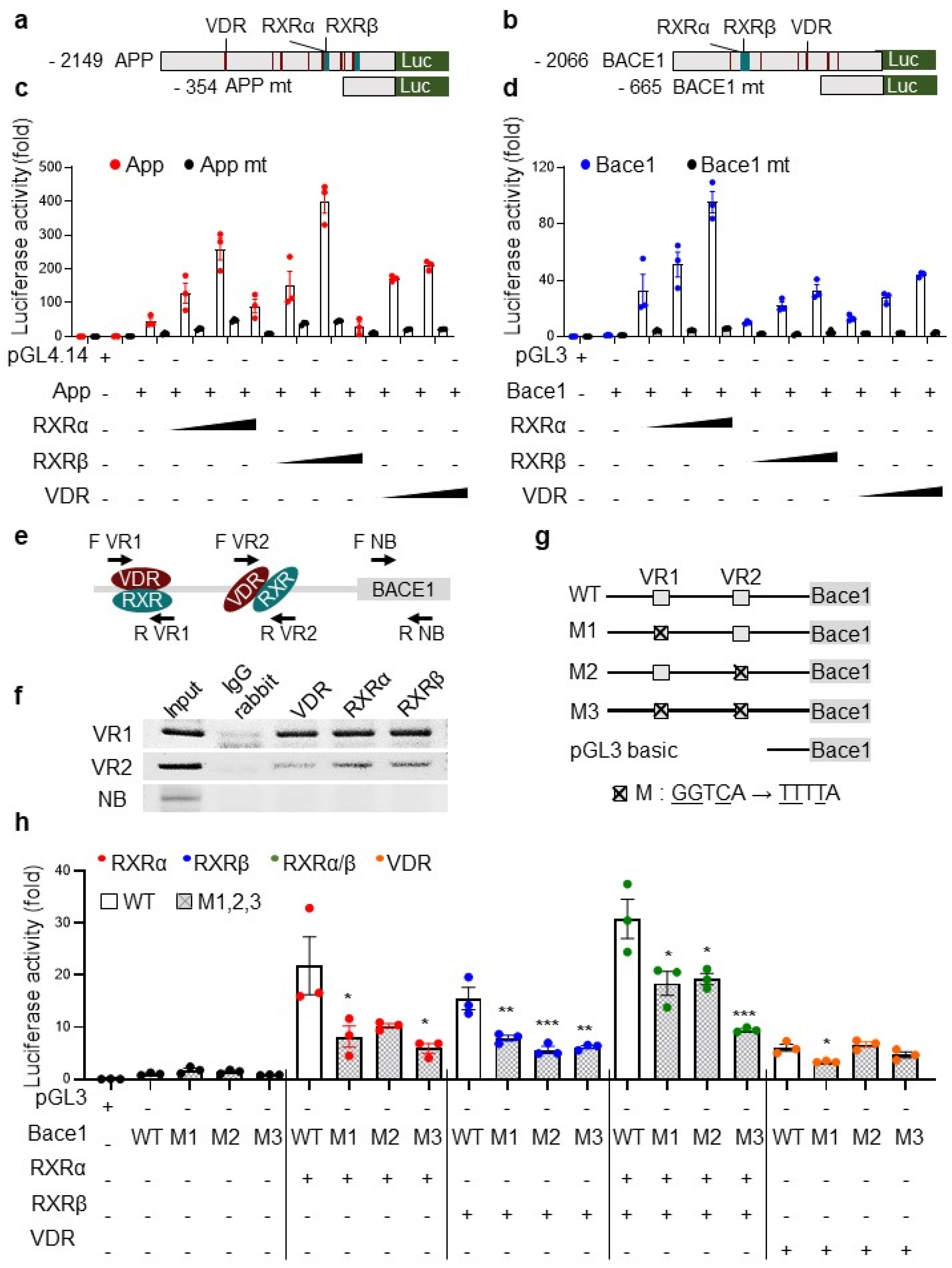
3.5. The Transcriptional Activation of App and bace1 Is Mediated by Direct Promoter Binding of RXR and VDR
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | GeneBank Accession No. | Primer | Primer Sequences | |
|---|---|---|---|---|
| 1 | App | NM_019288.2 | Forward | 5′-GATTCCTTACCGGTGCCTAGTTG |
| Reverse | 5′-TCCTGGTGTAGAAACTTGCACTTG | |||
| 2 | Adam10 | NM_019254.1 | Forward | 5′-GAGCTCAGCTGGTGGCTTACA |
| Reverse | 5′-AACGCAGAAATCAGACAATGACA | |||
| 3 | Bace1 | NM_019204.2 | Forward | 5′-TCGTTTGCCCAAGAAAGTATTTG |
| Reverse | 5′-AAGCCATCCGGGAACTTCTC | |||
| 4 | Psen1 | NM_019163.3 | Forward | 5′-CCTACTTCCAGAATGCACAGATGT |
| Reverse | 5′-GCTGCCGTTCTTGATTGTCA | |||
| 5 | RXRα | NM_012805.3 | Forward | 5′-AAACCCCCTCTAGGCCTCAA |
| Reverse | 5′-GATGTGCTTGGTGAAGGATGAC | |||
| 6 | RXRβ | NM_206849.3 | Forward | 5′-GCACAGAAACTCAGCCCATTC |
| Reverse | 5′-TCACGCATTTTGGACACTAGCT | |||
| 7 | VDR | NM_017058.2 | Forward | 5′-TCCCAGGATTCAGGGATCTCA |
| Reverse | 5′-TGAAAGACTGGTTGGAGCGT | |||
| 8 | Cyclopillin | NG_042086.1 | Forward | 5′-GCCATTCCTGGACCCAAAA |
| Reverse | 5′-GGTCTTTGGGAAGGTGAAAGAA |
Appendix B
| Gene | GeneBank Accession No. | Genecode No. | Primer | Primer Sequences | |
|---|---|---|---|---|---|
| 1 | App | NM_007471 | ENSMUST00000005406.11 | Forward | 5′-AAAGGTACCTGTATGTACATGCCTATGTG |
| Reverse | 5′-AAAAGATCTCGTGATCCTGCGTGGGCC | ||||
| 2 | Bace1 | NM_011792 | ENSMUST00000034591.10 | Forward | 5′-AAA GGT ACC CTG TCG GATCCCAGTGACT |
| Reverse | 5′-AAAAGATCTCAGGGACTCCGGGCTCCTA | ||||
| 3 | Psen1 | NM_001362271 | ENSMUST00000041806.12 | Forward | 5′-AAAGAGCTCAAGAATCTATTGGAACAC |
| Reverse | 5′-AAAGATATCAAGGCTGCTCTCAGTCTG | ||||
| 4 | App mt | NM_007471 | ENSMUST00000005406.11 | Forward | 5′-AAGGTACCACTCCCCAACTTGGTTCC |
| Reverse | 5′-AAAAGATCTCGTGATCCTGCGTGGGCC | ||||
| 5 | Bace1 mt | NM_011792 | ENSMUST00000034591.10 | Forward | 5′-AAA GGT ACC ATG GGA GAA GACCCACTG |
| Reverse | 5′-AAAAGATCTAGGCCACCATAATCCAGC | ||||
| 6 | Bace1 WT | NM_012104 | ENST00000313005.10 | Forward | 5′-AAA GGT ACC ATG GGA GAA GAC CCA CTG |
| Reverse | 5′-AAA AGA TCT AGG CCA CCA TAA TCC AGC | ||||
| 7 | Bace1 M1 (VR2) | NM_012104 | ENST00000313005.10 | Forward | 5′-GCATGAGATTTTATTGAACCCGGGAGGCGGAGGTTGC |
| Reverse | 5′-TCCCGGGTTCAATAAAATCTCATGCCTCAGCCTCTGAGTA | ||||
| 8 | Bace1 M2 (VR1) | NM_012104 | ENST00000313005.10 | Forward | 5′-GGATCACGATTTTAGGAGATCGAGACCATCTTGGC |
| Reverse | 5′-TCGATCTCCTAAAATCGTGATCCTCCCGCCTCGGCCT |
Appendix C
| Targeting Element Binding Sites | Detection Site | Primer | Primer Sequences | |
|---|---|---|---|---|
| 1 | VDR/RXR 1 (VR1) | −1538 | Forward | 5′-ATGATCGGCCGGGCGCG |
| −1379 | Reverse | 5′-GCCTGCAACCACGCCCCGCT | ||
| 2 | VDR/RXR 2 (VR2) | −541 | Forward | 5′-ACAAACAGGTTCAGATGG |
| −414 | Reverse | 5′-TTTTCCAGGCTGCAAAC | ||
| 3 | Non binding (NB) | 101 | Forward | 5′-CGGGAGCTGCGAGCCGCG |
| 230 | Reverse | 5′-GGCGGGCCGGTGGCGGC |
References
- Zhang, Y.W.; Thompson, R.; Zhang, H.; Xu, H. App processing in Alzheimer’s disease. Mol. Brain 2011, 4, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, M.P.; LeVine, H., 3rd. Alzheimer’s disease and the amyloid-beta peptide. J. Alzheimer’s Dis. JAD 2010, 19, 311–323. [Google Scholar] [CrossRef] [Green Version]
- Cole, S.L.; Vassar, R. The Alzheimer’s disease beta-secretase enzyme, bace1. Mol. Neurodegener. 2007, 2, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, L.; McClatchy, D.B.; Maher, P.; Liang, Z.; Diedrich, J.K.; Soriano-Castell, D.; Goldberg, J.; Shokhirev, M.; Yates, J.R.; Schubert, D.; et al. Intracellular amyloid toxicity induces oxytosis/ferroptosis regulated cell death. Cell Death Dis. 2020, 11, 828. [Google Scholar] [CrossRef] [PubMed]
- Karran, E.; Mercken, M.; Strooper, B.D. The amyloid cascade hypothesis for Alzheimer’s disease: An appraisal for the development of therapeutics. Nat. Rev. Drug Discov. 2011, 10, 698–712. [Google Scholar] [CrossRef]
- Mawuenyega, K.G.; Sigurdson, W.; Ovod, V.; Munsell, L.; Kasten, T.; Morris, J.C.; Yarasheski, K.E.; Bateman, R.J. Decreased clearance of cns beta-amyloid in Alzheimer’s disease. Sciences 2010, 330, 1774. [Google Scholar] [CrossRef] [Green Version]
- Raboy, V. Myo-inositol-1,2,3,4,5,6-hexakisphosphate. Phytochemistry 2003, 64, 1033–1043. [Google Scholar] [CrossRef]
- Kim, O.-H.; Kim, Y.-O.; Shim, J.-H.; Jung, Y.-S.; Jung, W.-J.; Choi, W.-C.; Lee, H.; Lee, S.-J.; Kim, K.-K.; Auh, J.-H.; et al. Β-propeller phytase hydrolyzes insoluble ca2+-phytate salts and completely abrogates the ability of phytate to chelate metal ions. Biochemistry 2010, 49, 10216–10227. [Google Scholar] [CrossRef] [PubMed]
- Reddy, N.R.; Sathe, S.K.; Salunkhe, D.K. Phytates in legumes and cereals. In Advances in Food Research; Chichester, C.O., Mrak, E.M., Stewart, G.F., Eds.; Academic Press: Cambridge, MA, USA, 1982; Volume 28, pp. 1–92. [Google Scholar]
- Moe, S.M.; Zidehsarai, M.P.; Chambers, M.A.; Jackman, L.A.; Radcliffe, J.S.; Trevino, L.L.; Donahue, S.E.; Asplin, J.R. Vegetarian compared with meat dietary protein source and phosphorus homeostasis in chronic kidney disease. Clin. J. Am. Soc. Nephrol. CJASN 2011, 6, 257–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, O.-H.; Booth, C.J.; Choi, H.S.; Lee, J.; Kang, J.; Hur, J.; Jung, W.J.; Jung, Y.-S.; Choi, H.J.; Kim, H.; et al. High-phytate/low-calcium diet is a risk factor for crystal nephropathies, renal phosphate wasting, and bone loss. eLife 2020, 9, e52709. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.H.; Pike, J.W. Human vitamin d receptor-dependent transactivation in saccharomyces cerevisiae requires retinoid x receptor. Mol. Endocrinol. 1996, 10, 196–205. [Google Scholar] [PubMed] [Green Version]
- Dusso, A.S. Vitamin d receptor: Mechanisms for vitamin d resistance in renal failure. Kidney Int. 2003, 63, S6–S9. [Google Scholar] [CrossRef] [Green Version]
- Kang, J.K.; Kim, O.-H.; Hur, J.; Yu, S.H.; Lamichhane, S.; Lee, J.W.; Ojha, U.; Hong, J.H.; Lee, C.S.; Cha, J.-Y.; et al. Increased intracellular Ca2+ concentrations prevent membrane localization of ph domains through the formation of Ca2+-phosphoinositides. Proc. Natl. Acad. Sci. USA 2017, 114, 11926–11931. [Google Scholar] [CrossRef] [Green Version]
- Palmer, S.C.; Hayen, A.; Macaskill, P.; Pellegrini, F.; Craig, J.C.; Elder, G.J.; Strippoli, G.F. Serum levels of phosphorus, parathyroid hormone, and calcium and risks of death and cardiovascular disease in individuals with chronic kidney disease: A systematic review and meta-analysis. JAMA 2011, 305, 1119–1127. [Google Scholar] [CrossRef]
- Reynolds, J.L.; Joannides, A.J.; Skepper, J.N.; McNair, R.; Schurgers, L.J.; Proudfoot, D.; Jahnen-Dechent, W.; Weissberg, P.L.; Shanahan, C.M. Human vascular smooth muscle cells undergo vesicle-mediated calcification in response to changes in extracellular calcium and phosphate concentrations: A potential mechanism for accelerated vascular calcification in esrd. J. Am. Soc. Nephrol. JASN 2004, 15, 2857–2867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jono, S.; McKee, M.D.; Murry, C.E.; Shioi, A.; Nishizawa, Y.; Mori, K.; Morii, H.; Giachelli, C.M. Phosphate regulation of vascular smooth muscle cell calcification. Circ. Res. 2000, 87, E10–E17. [Google Scholar] [CrossRef]
- LaFerla, F.M. Calcium dyshomeostasis and intracellular signalling in Alzheimer’s disease. Nat. Rev. Neurosci. 2002, 3, 862–872. [Google Scholar] [CrossRef] [PubMed]
- Bojarski, L.; Herms, J.; Kuznicki, J. Calcium dysregulation in Alzheimer’s disease. Neurochem. Int. 2008, 52, 621–633. [Google Scholar] [CrossRef]
- Berridge, M.J. Calcium hypothesis of Alzheimer’s disease. Pflug. Arch. Eur. J. Physiol. 2010, 459, 441–449. [Google Scholar] [CrossRef]
- Brzyska, M.; Elbaum, D. Dysregulation of calcium in Alzheimer’s disease. Acta Neurobiol. Exp. 2003, 63, 171–183. [Google Scholar]
- Corona, C.; Pensalfini, A.; Frazzini, V.; Sensi, S.L. New therapeutic targets in Alzheimer’s disease: Brain deregulation of calcium and zinc. Cell Death Dis. 2011, 2, e176. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Xie, Y.; Bowe, B.; Xian, H.; Al-Aly, Z. Serum phosphorus levels and risk of incident dementia. PLoS ONE 2017, 12, e0171377. [Google Scholar] [CrossRef] [Green Version]
- Ashraf, A.; Stosnach, H.; Parkes, H.G.; Hye, A.; Powell, J.; So, P.-W.; Soinine, H.; Tsolaki, M.; Vellas, B.; Lovestone, S.; et al. Pattern of altered plasma elemental phosphorus, calcium, zinc, and iron in Alzheimer’s disease. Sci. Rep. 2019, 9, 3147. [Google Scholar] [CrossRef] [PubMed]
- Landfield, P.W.; Applegate, M.D.; Schmitzer-Osborne, S.E.; Naylor, C.E. Phosphate/calcium alterations in the first stages of Alzheimer’s disease: Implications for etiology and pathogenesis. J. Neurol. Sci. 1991, 106, 221–229. [Google Scholar] [CrossRef]
- Brown, J.; de Boer, I.H.; Robinson-Cohen, C.; Siscovick, D.S.; Kestenbaum, B.; Allison, M.; Vaidya, A. Aldosterone, parathyroid hormone, and the use of renin-angiotensin-aldosterone system inhibitors: The multi-ethnic study of atherosclerosis. J. Clin. Endocrinol. Metab. 2015, 100, 490–499. [Google Scholar] [CrossRef] [PubMed]
- Björkman, M.P.; Sorva, A.J.; Tilvis, R.S. Does elevated parathyroid hormone concentration predict cognitive decline in older people? Aging Clin. Exp. Res. 2010, 22, 164–169. [Google Scholar] [CrossRef]
- Chou, F.-F.; Chen, J.-B.; Hsieh, K.-C.; Liou, C.-W. Cognitive changes after parathyroidectomy in patients with secondary hyperparathyroidism. Surgery 2008, 143, 526–532. [Google Scholar] [CrossRef] [PubMed]
- Papageorgiou, S.G.; Christou, Y.; Kontaxis, T.; Bonakis, A.; Anagnostouli, M.; Potagas, C.; Kalfakis, N. Dementia as presenting symptom of primary hyperparathyroidism: Favourable outcome after surgery. Clin. Neurol. Neurosurg. 2008, 110, 1038–1040. [Google Scholar] [CrossRef]
- Hagström, E.; Kilander, L.; Nylander, R.; Larsson, E.M.; Michaëlsson, K.; Melhus, H.; Ahlström, H.; Johansson, L.; Lind, L.; Arnlöv, J. Plasma parathyroid hormone is associated with vascular dementia and cerebral hyperintensities in two community-based cohorts. J. Clin. Endocrinol. Metab. 2014, 99, 4181–4189. [Google Scholar] [CrossRef]
- Zhao, Y.; Shen, L.; Ji, H.F. Alzheimer’s disease and risk of hip fracture: A meta-analysis study. TheScientificWorldJournal 2012, 2012, 872173. [Google Scholar] [CrossRef] [Green Version]
- Timmons, J.G.; Manners, R.; Bailey, M.; McDougall, C. Cognitive impairment reversed by cinacalcet administration in primary hyperparathyroidism. Hormones 2021, 20, 587–589. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brouillette, J.; Caillierez, R.; Zommer, N.; Alves-Pires, C.; Benilova, I.; Blum, D.; De Strooper, B.; Buée, L. Neurotoxicity and memory deficits induced by soluble low-molecular-weight amyloid-β1–42 oligomers are revealed in vivo by using a novel animal model. J. Neurosci. 2012, 32, 7852–7861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mucke, L.; Selkoe, D.J. Neurotoxicity of amyloid β-protein: Synaptic and network dysfunction. Cold Spring Harb. Perspect. Med. 2012, 2, a006338. [Google Scholar] [CrossRef] [Green Version]
- Vassar, R. Caspase-3 cleavage of gga3 stabilizes bace: Implications for Alzheimer’s disease. Neuron 2007, 54, 671–673. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Lee, X.; Shao, Z.; Apicco, D.; Huang, G.; Gong, B.J.; Pepinsky, R.B.; Mi, S. A dr6/p75ntr complex is responsible for β-amyloid-induced cortical neuron death. Cell Death Dis. 2013, 4, e579. [Google Scholar] [CrossRef] [PubMed]
- Kadowaki, H.; Nishitoh, H.; Urano, F.; Sadamitsu, C.; Matsuzawa, A.; Takeda, K.; Masutani, H.; Yodoi, J.; Urano, Y.; Nagano, T.; et al. Amyloid β induces neuronal cell death through ros-mediated ask1 activation. Cell Death Differ. 2005, 12, 19–24. [Google Scholar] [CrossRef] [Green Version]
- Nicholson, D.W.; Ali, A.; Thornberry, N.A.; Vaillancourt, J.P.; Ding, C.K.; Gallant, M.; Gareau, Y.; Griffin, P.R.; Labelle, M.; Lazebnik, Y.A.; et al. Identification and inhibition of the ice/ced-3 protease necessary for mammalian apoptosis. Nature 1995, 376, 37–43. [Google Scholar] [CrossRef]
- Nunan, J.; Small, D.H. Regulation of app cleavage by α-, β- and γ-secretases. FEBS Lett. 2000, 483, 6–10. [Google Scholar] [CrossRef] [Green Version]
- Roberds, S.L.; Anderson, J.; Basi, G.; Bienkowski, M.J.; Branstetter, D.G.; Chen, K.S.; Freedman, S.B.; Frigon, N.L.; Games, D.; Hu, K.; et al. Bace knockout mice are healthy despite lacking the primary beta-secretase activity in brain: Implications for Alzheimer’s disease therapeutics. Hum. Mol. Genet. 2001, 10, 1317–1324. [Google Scholar] [CrossRef] [Green Version]
- Luo, Y.; Bolon, B.; Kahn, S.; Bennett, B.D.; Babu-Khan, S.; Denis, P.; Fan, W.; Kha, H.; Zhang, J.; Gong, Y.; et al. Mice deficient in bace1, the Alzheimer’s beta-secretase, have normal phenotype and abolished beta-amyloid generation. Nat. Neurosci. 2001, 4, 231–232. [Google Scholar] [CrossRef] [PubMed]
- Dovey, H.F.; John, V.; Anderson, J.P.; Chen, L.Z.; de Saint Andrieu, P.; Fang, L.Y.; Freedman, S.B.; Folmer, B.; Goldbach, E.; Holsztynska, E.J.; et al. Functional gamma-secretase inhibitors reduce beta-amyloid peptide levels in brain. J. Neurochem. 2001, 76, 173–181. [Google Scholar] [CrossRef]
- Fishilevich, S.; Nudel, R.; Rappaport, N.; Hadar, R.; Plaschkes, I.; Iny Stein, T.; Rosen, N.; Kohn, A.; Twik, M.; Safran, M.; et al. Genehancer: Genome-wide integration of enhancers and target genes in genecards. Database J. Biol. Databases Curation 2017, 2017. [Google Scholar] [CrossRef] [Green Version]
- Ebisawa, M.; Umemiya, H.; Ohta, K.; Fukasawa, H.; Kawachi, E.; Christoffel, G.; Gronemeyer, H.; Tsuji, M.; Hashimoto, Y.; Shudo, K.; et al. Retinoid x receptor-antagonistic diazepinylbenzoic acids. Chem. Pharm. Bull. 1999, 47, 1778–1786. [Google Scholar] [CrossRef] [Green Version]
- Mullan, M.; Crawford, F.; Axelman, K.; Houlden, H.; Lilius, L.; Winblad, B.; Lannfelt, L. A pathogenic mutation for probable Alzheimer’s disease in the app gene at the n–terminus of β–amyloid. Nat. Genet. 1992, 1, 345–347. [Google Scholar] [CrossRef] [PubMed]
- Bloom, G.S. Amyloid-β and tau: The trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 2014, 71, 505–508. [Google Scholar] [CrossRef] [Green Version]
- Ketteler, M.; Block, G.A.; Evenepoel, P.; Fukagawa, M.; Herzog, C.A.; McCann, L.; Moe, S.M.; Shroff, R.; Tonelli, M.A.; Toussaint, N.D.; et al. Executive summary of the 2017 kdigo chronic kidney disease-mineral and bone disorder (ckd-mbd) guideline update: What’s changed and why it matters. Kidney Int. 2017, 92, 26–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Fu, Y.; Yasvoina, M.; Shao, P.; Hitt, B.; O’Connor, T.; Logan, S.; Maus, E.; Citron, M.; Berry, R.; et al. Β-site amyloid precursor protein cleaving enzyme 1 levels become elevated in neurons around amyloid plaques: Implications for Alzheimer’s disease pathogenesis. J. Neurosci. 2007, 27, 3639–3649. [Google Scholar] [CrossRef]
- Kandalepas, P.C.; Sadleir, K.R.; Eimer, W.A.; Zhao, J.; Nicholson, D.A.; Vassar, R. The Alzheimer’s β-secretase bace1 localizes to normal presynaptic terminals and to dystrophic presynaptic terminals surrounding amyloid plaques. Acta Neuropathol. 2013, 126, 329–352. [Google Scholar] [CrossRef] [Green Version]
- Dotzenrath, C.M.E.; Kaetsch, A.K.; Pfingsten, H.; Cupisti, K.; Weyerbrock, N.; Vossough, A.; Verde, P.E.; Ohmann, C. Neuropsychiatric and cognitive changes after surgery for primary hyperparathyroidism. World J. Surg. 2006, 30, 680–685. [Google Scholar] [CrossRef]
- Roman, S.A.; Sosa, J.A.; Mayes, L.; Desmond, E.; Boudourakis, L.; Lin, R.; Snyder, P.J.; Holt, E.; Udelsman, R. Parathyroidectomy improves neurocognitive deficits in patients with primary hyperparathyroidism. Surgery 2005, 138, 1121–1129. [Google Scholar] [CrossRef]
- Sneddon, W.B.; Barry, E.L.R.; Coutermarsh, B.A.; Gesek, F.A.; Liu, F.; Friedman, P.A. Regulation of renal parathyroid hormone receptor expression by 1,25-dihydroxyvitamin d3 and retinoic acid. Cell. Physiol. Biochem. 1998, 8, 261–277. [Google Scholar] [CrossRef] [PubMed]
- Cramer, P.E.; Cirrito, J.R.; Wesson, D.W.; Lee, C.Y.D.; Karlo, J.C.; Zinn, A.E.; Casali, B.T.; Restivo, J.L.; Goebel, W.D.; James, M.J.; et al. Apoe-directed therapeutics rapidly clear β-amyloid and reverse deficits in ad mouse models. Science 2012, 335, 1503–1506. [Google Scholar] [CrossRef] [Green Version]
- Vidal, V.; Puente, A.; García-Cerro, S.; García Unzueta, M.T.; Rueda, N.; Riancho, J.; Martínez-Cué, C. Bexarotene impairs cognition and produces hypothyroidism in a mouse model of down syndrome and Alzheimer’s disease. Front. Pharmacol. 2021, 12, 613211. [Google Scholar] [CrossRef] [PubMed]
- Casali, B.T.; Reed-Geaghan, E.G.; Landreth, G.E. Nuclear receptor agonist-driven modification of inflammation and amyloid pathology enhances and sustains cognitive improvements in a mouse model of Alzheimer’s disease. J. Neuroinflamm. 2018, 15, 43. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, H.-J.; Jung, Y.-S.; Jung, Y.-J.; Kim, O.-H.; Oh, B.-C. High-Phytate Diets Increase Amyloid β Deposition and Apoptotic Neuronal Cell Death in a Rat Model. Nutrients 2021, 13, 4370. https://doi.org/10.3390/nu13124370
Kim H-J, Jung Y-S, Jung Y-J, Kim O-H, Oh B-C. High-Phytate Diets Increase Amyloid β Deposition and Apoptotic Neuronal Cell Death in a Rat Model. Nutrients. 2021; 13(12):4370. https://doi.org/10.3390/nu13124370
Chicago/Turabian StyleKim, Hyo-Jung, Yun-Shin Jung, Yun-Jae Jung, Ok-Hee Kim, and Byung-Chul Oh. 2021. "High-Phytate Diets Increase Amyloid β Deposition and Apoptotic Neuronal Cell Death in a Rat Model" Nutrients 13, no. 12: 4370. https://doi.org/10.3390/nu13124370
APA StyleKim, H.-J., Jung, Y.-S., Jung, Y.-J., Kim, O.-H., & Oh, B.-C. (2021). High-Phytate Diets Increase Amyloid β Deposition and Apoptotic Neuronal Cell Death in a Rat Model. Nutrients, 13(12), 4370. https://doi.org/10.3390/nu13124370