
Chlorogenic Acid Protects against Advanced Alcoholic Steatohepatitis in Rats via Modulation of Redox Homeostasis, Inflammation, and Lipogenesis

 , , ,
, , ,  , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Animals & Experimental Design
2.3. Biochemical Analysis
2.4. Isolation of Liver Leukocytes
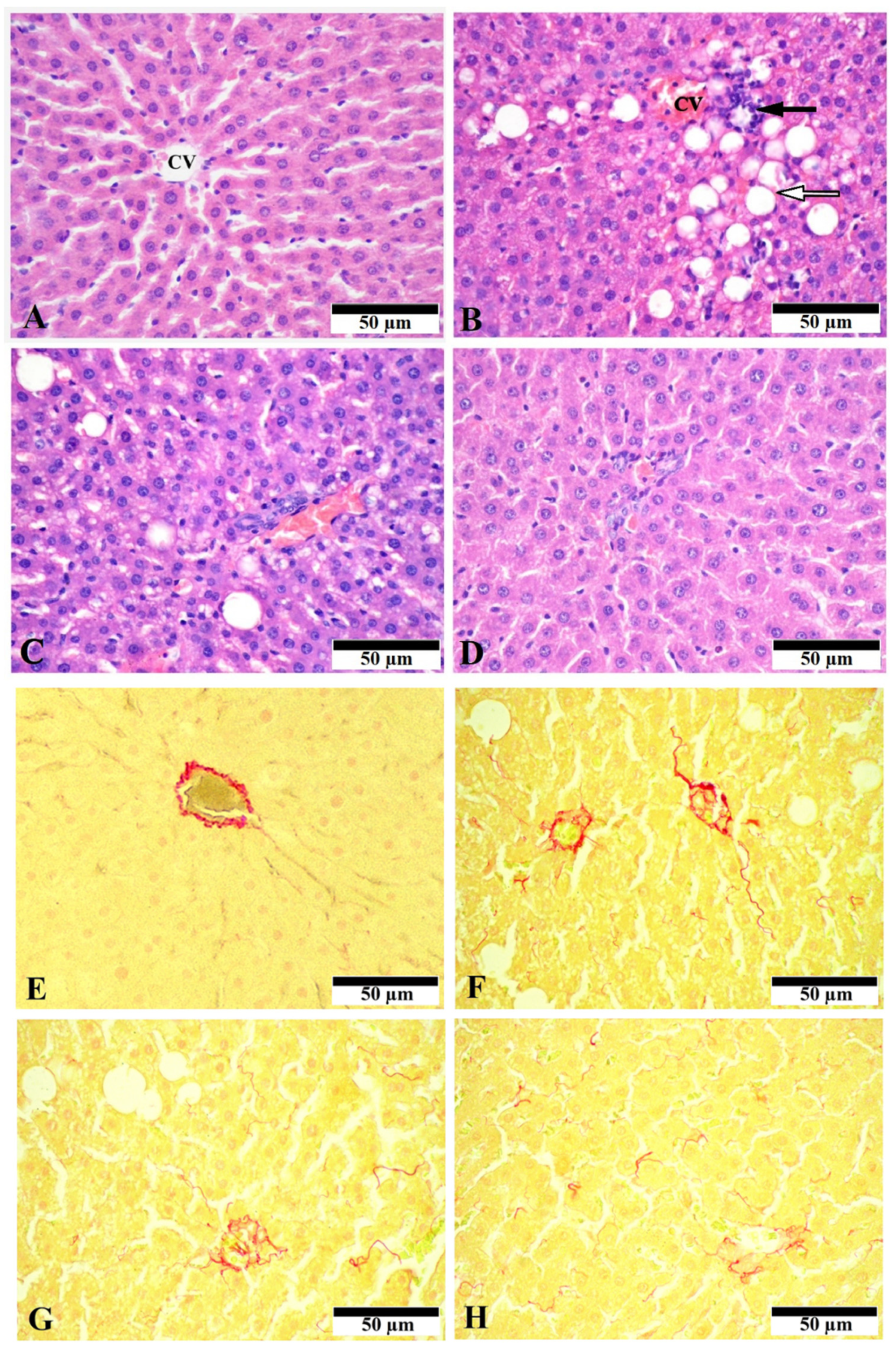
2.5. Histological Evaluation
2.6. Gene Expression Analysis
2.7. Statistical Analysis
3. Results
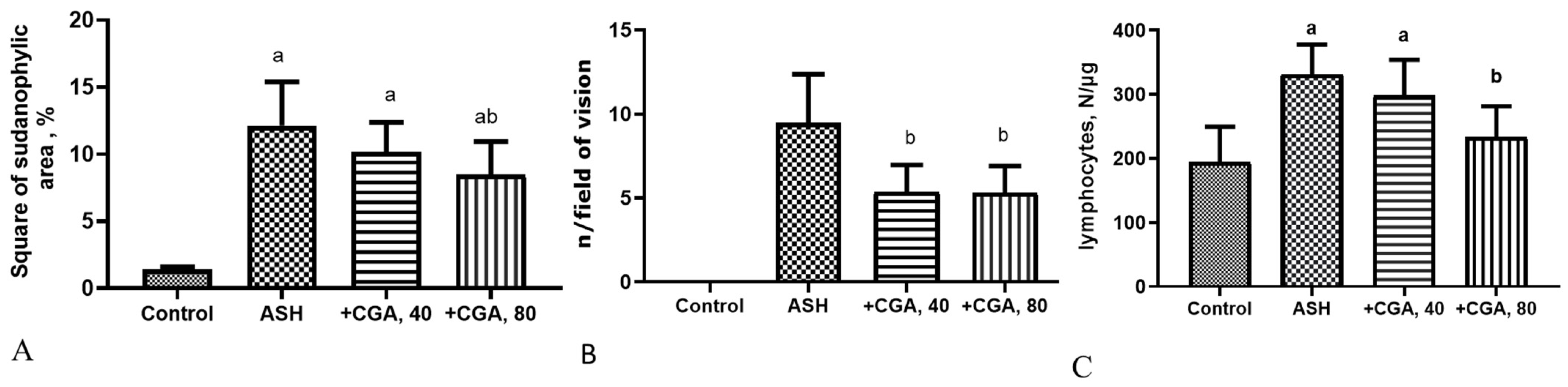
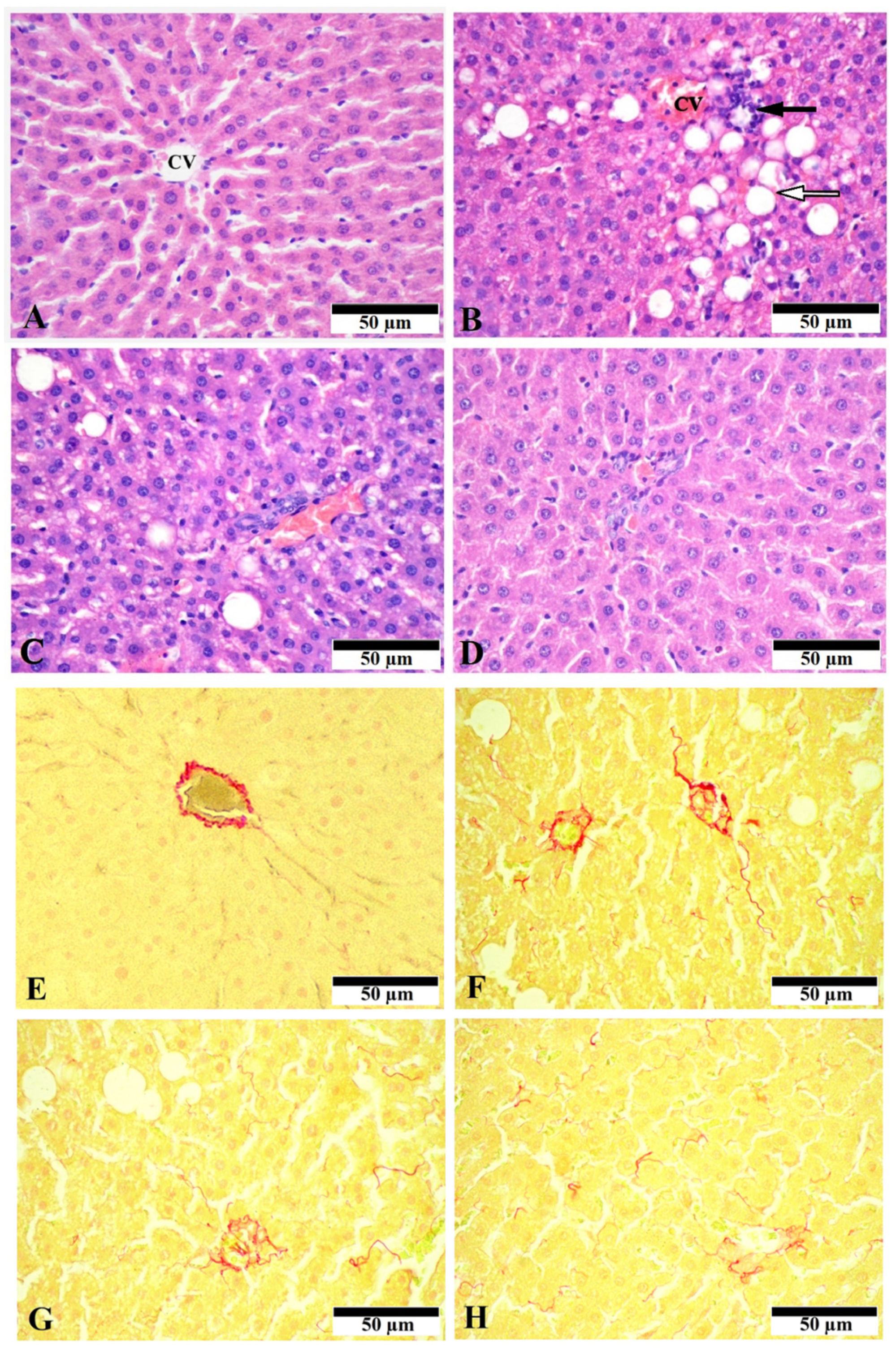
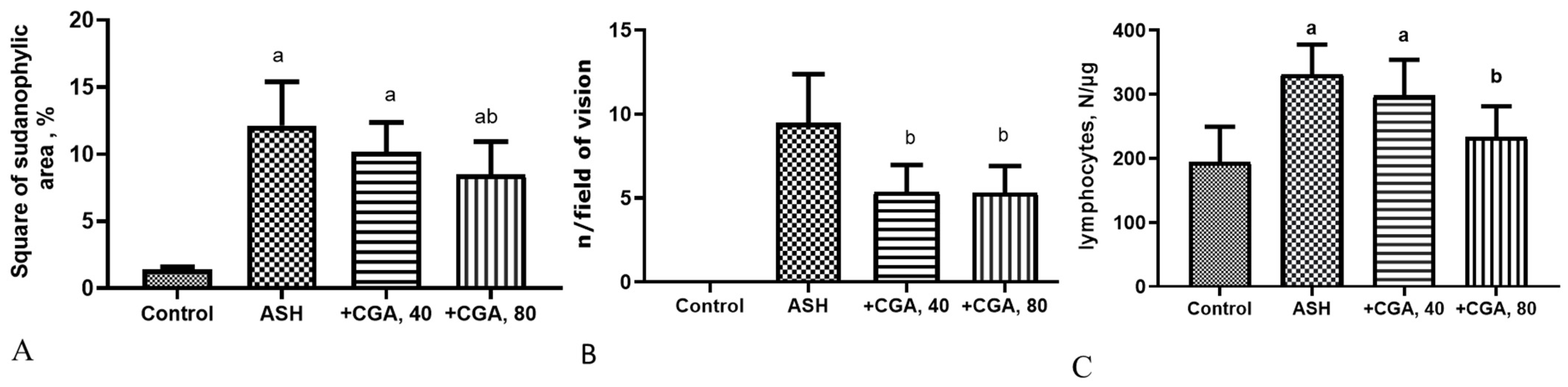
3.1. Effect of CGA on Liver Morphology and Histopathologic Changes
3.2. Effect of CGA on Serum and Liver Biochemical Parameters
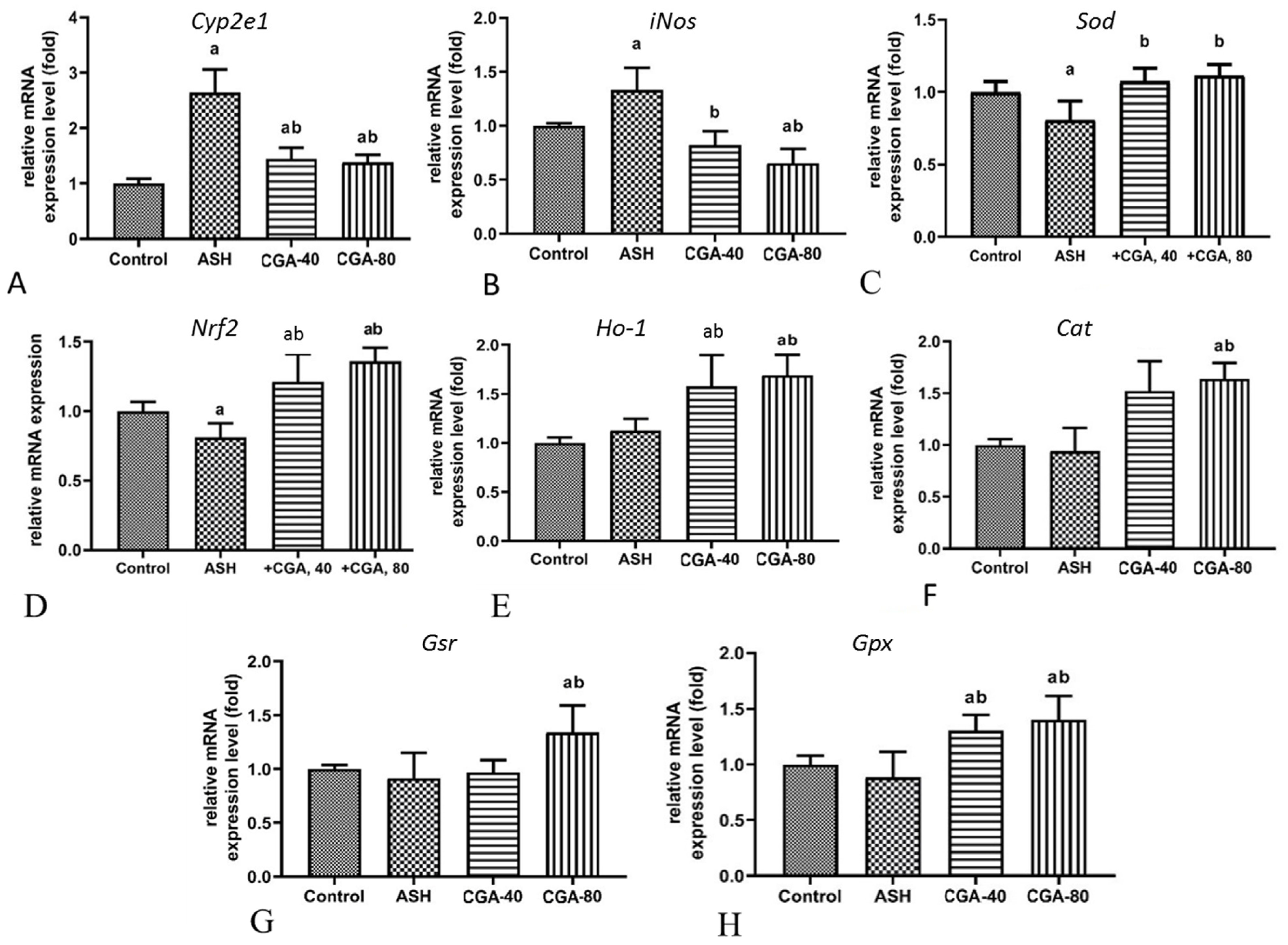
3.3. Effects of CGA on Hepatic Gene Expression of Nrf2, Prooxidant and Antioxidant Enzymes
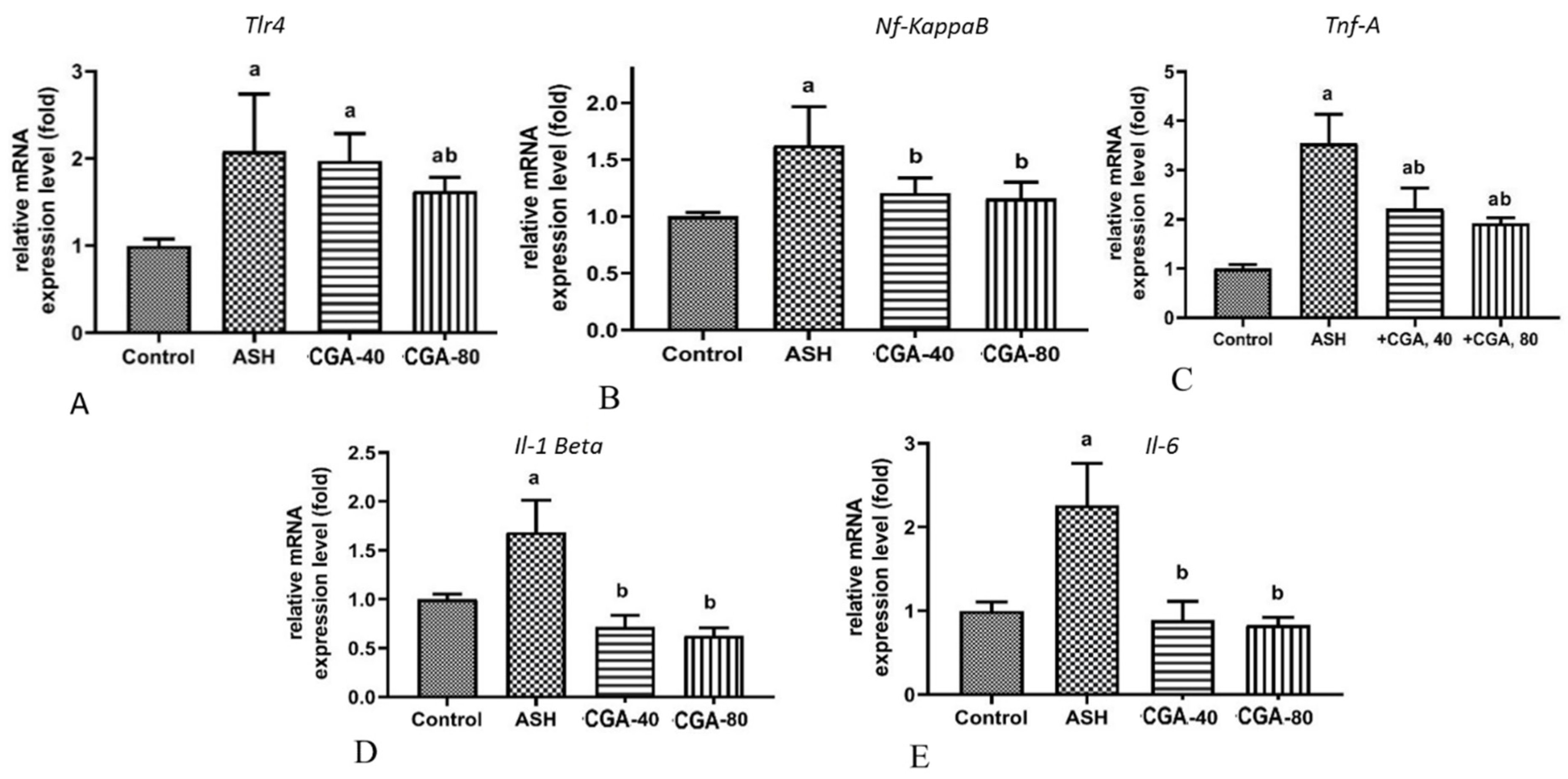
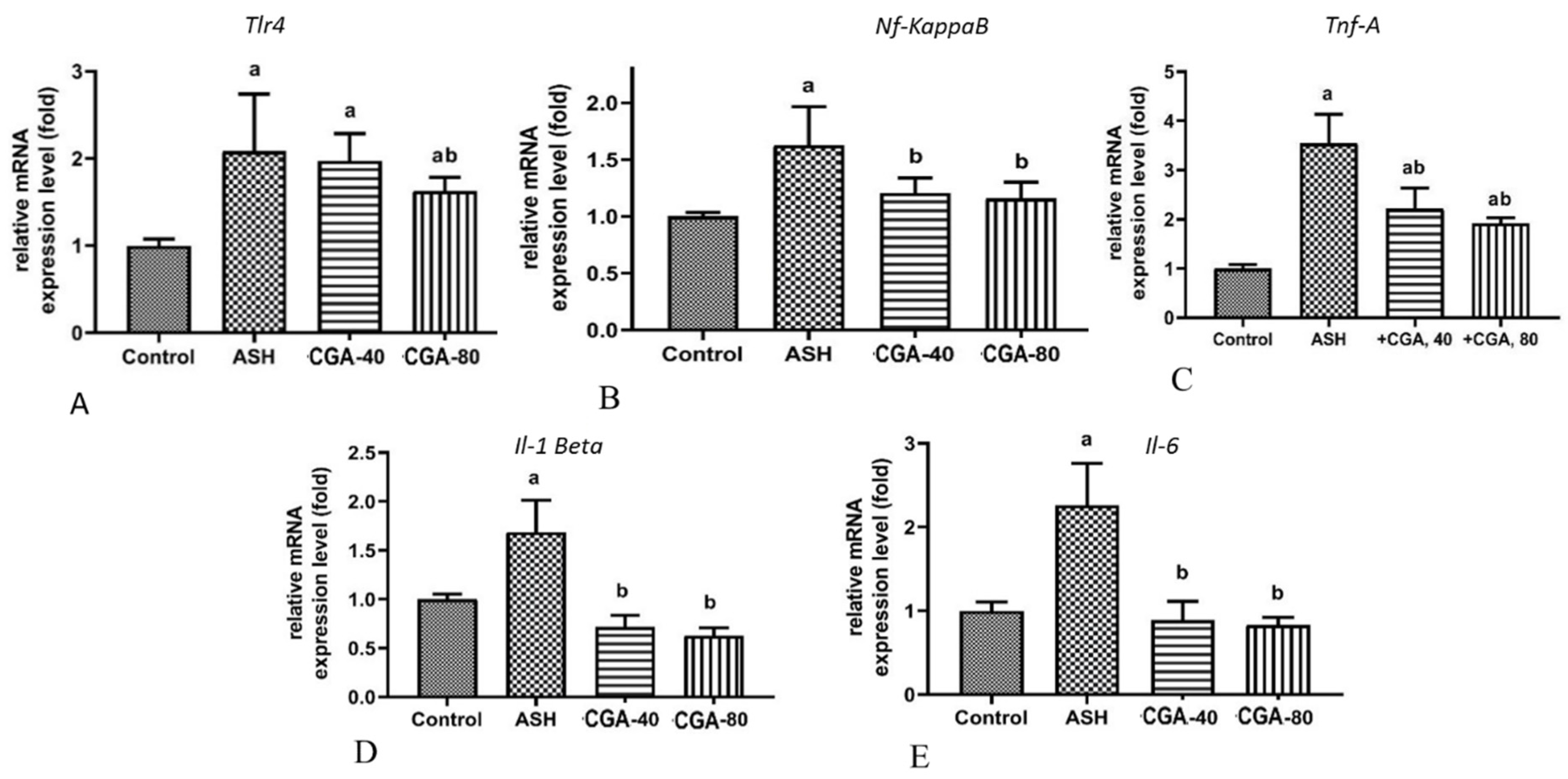
3.4. Effects of CGA on Inflammation-Related Gene Expression
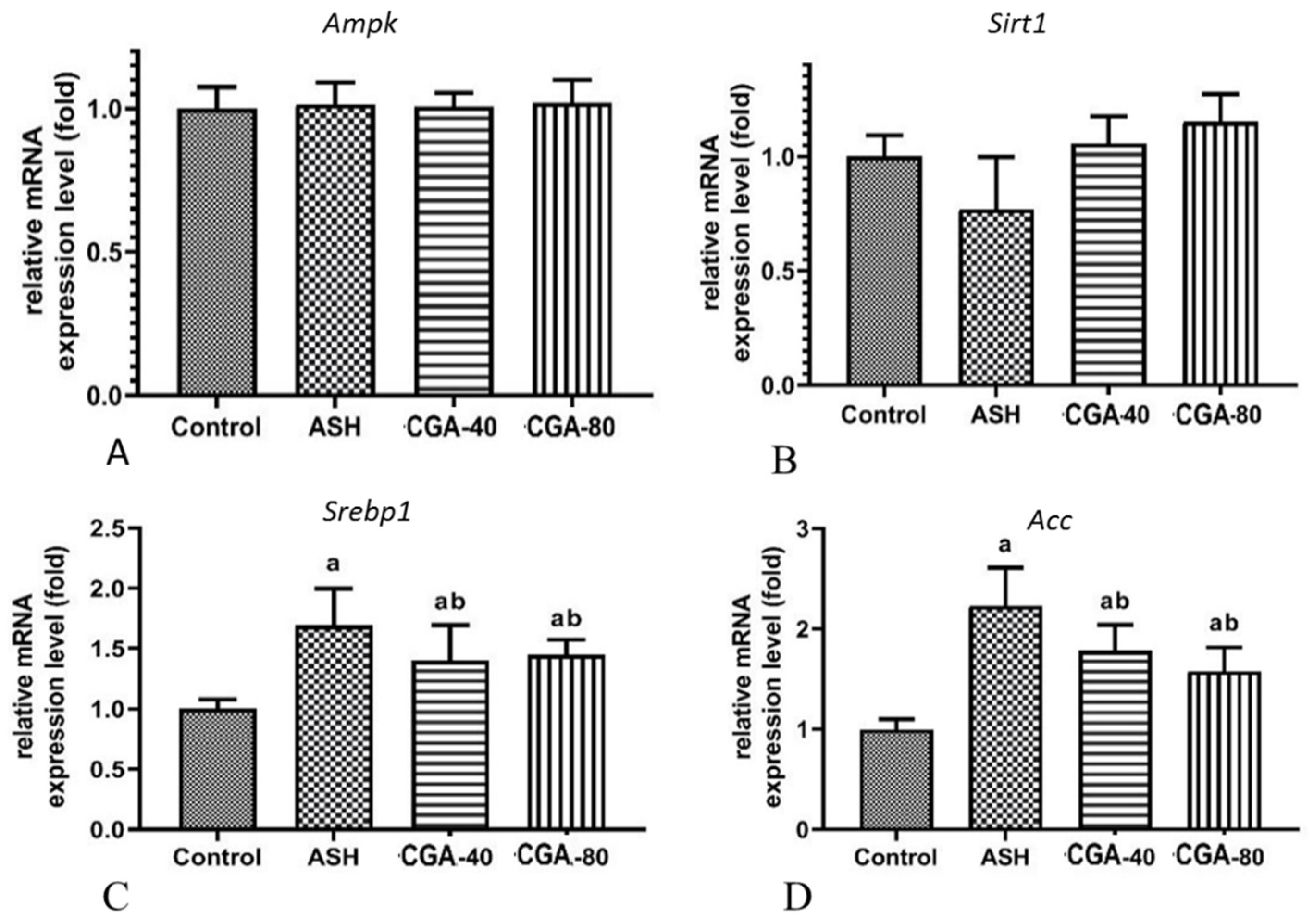
3.5. Effects of CGA on Expression of Genes Related to Lipid Metabolism
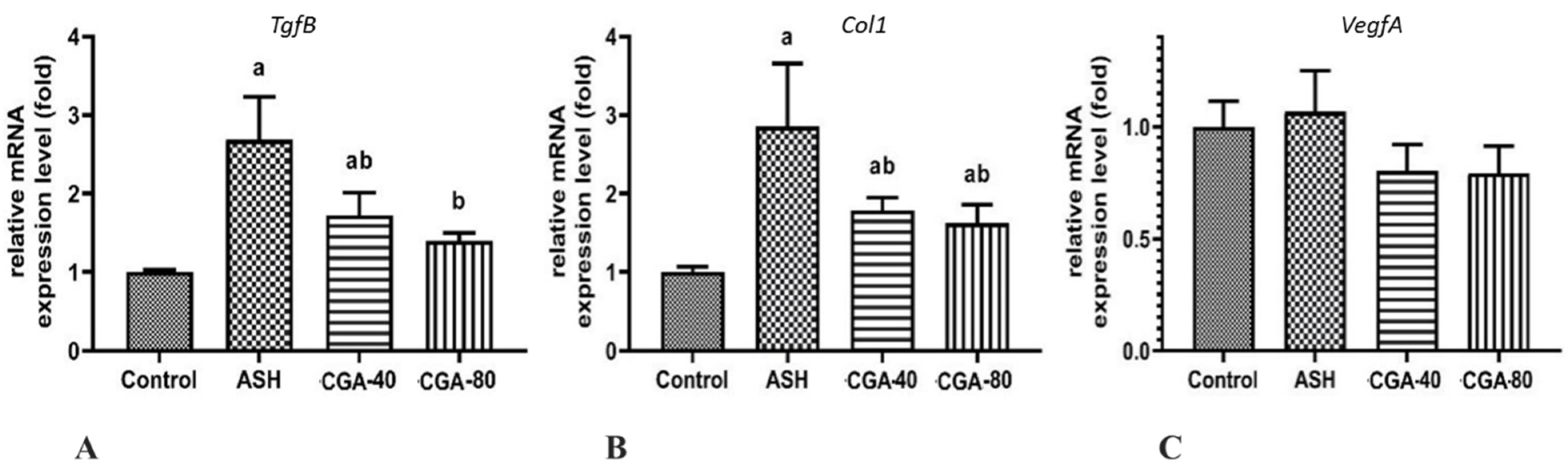
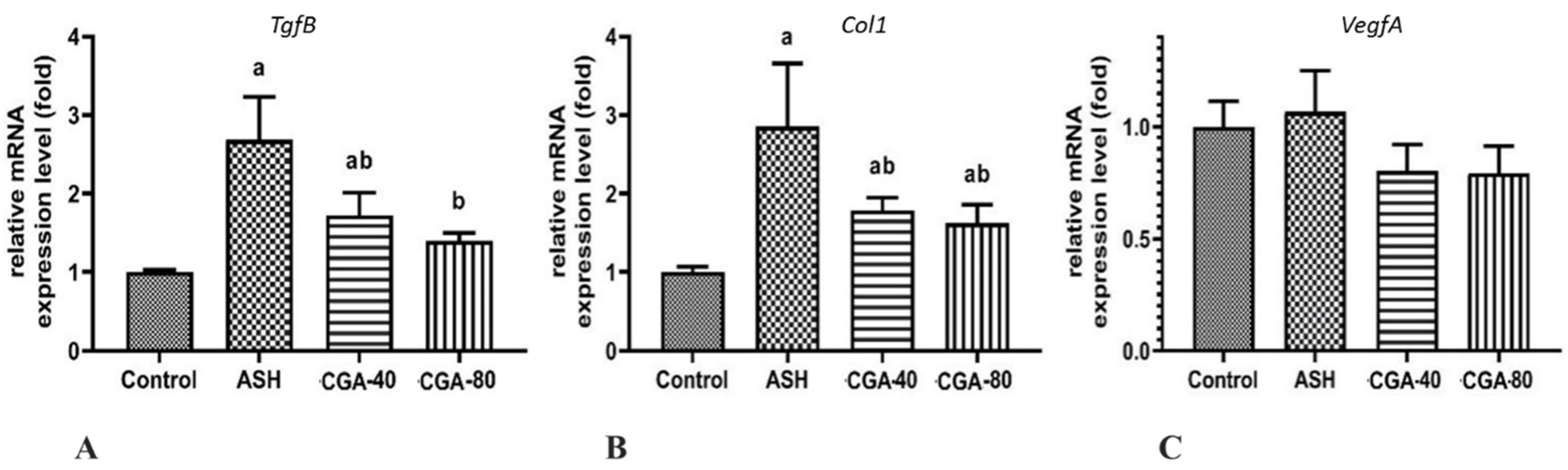
3.6. Effects of CGA on Expression of Genes Related to Liver Fibrosis
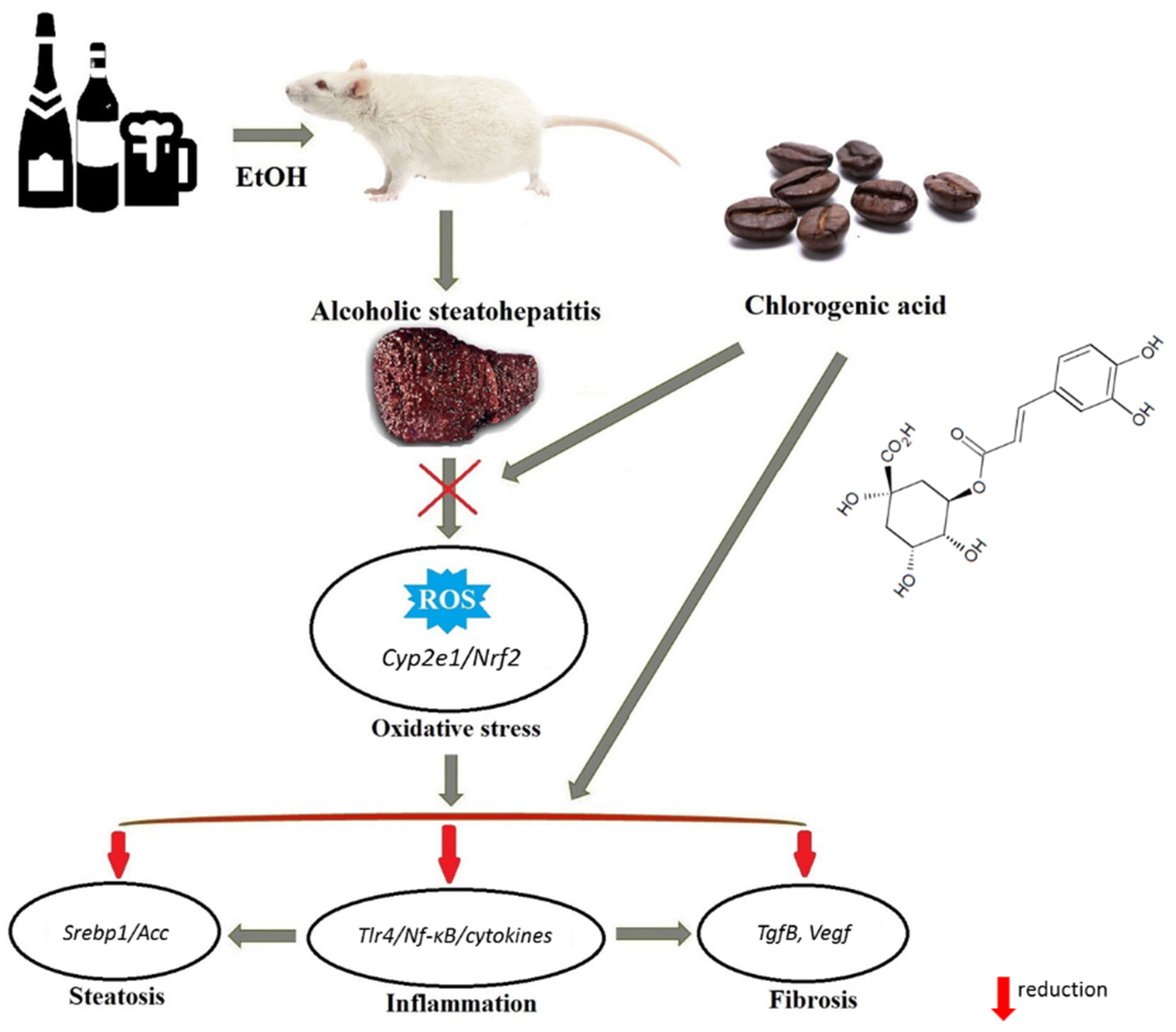
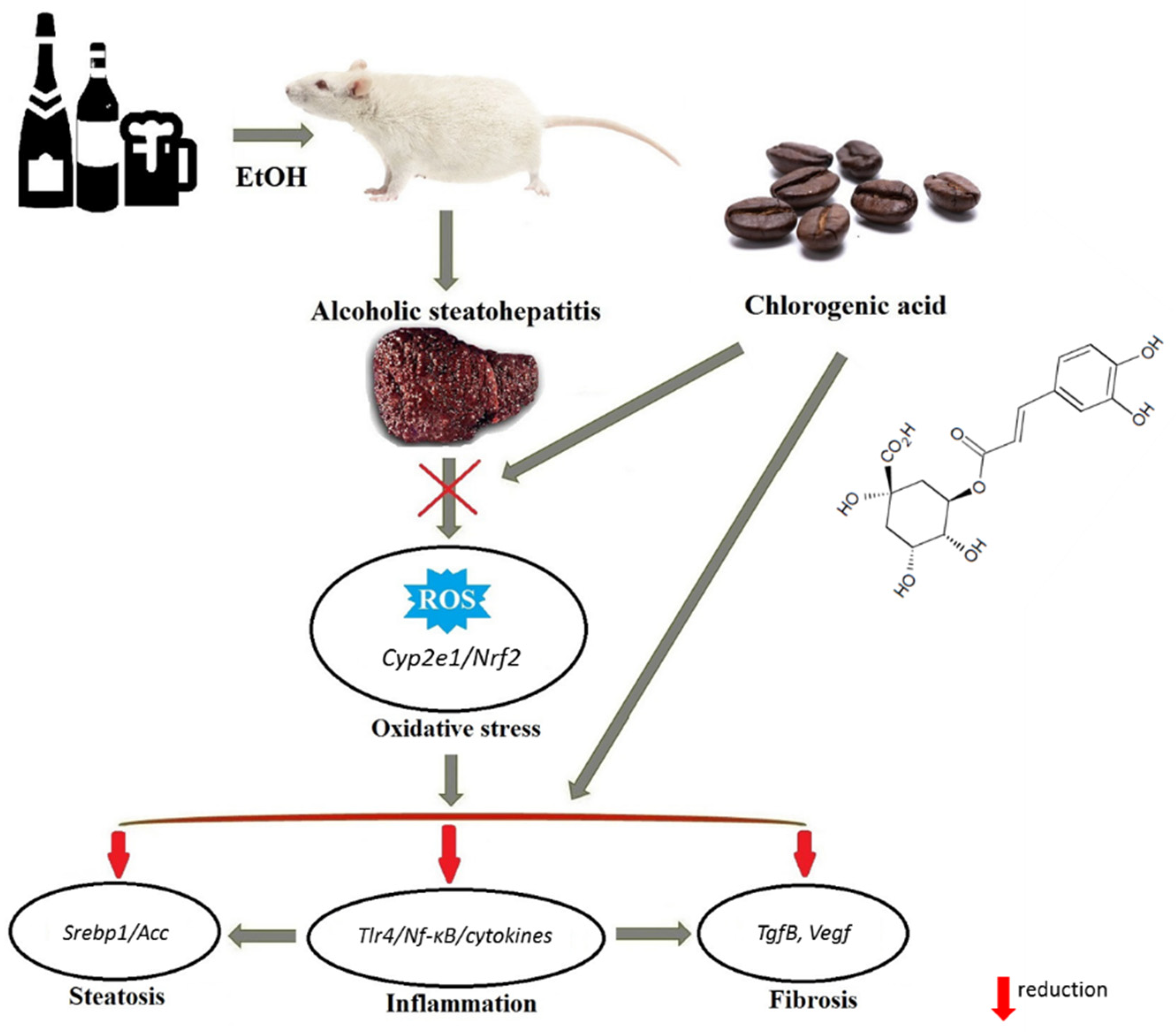
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kim, M.-S.; Ong, M.; Qu, X. Optimal management for alcoholic liver disease: Conventional medications, natural therapy or combination? World J. Gastroenterol. 2016, 22, 8–23. [Google Scholar] [CrossRef]
- Buko, V.; Kuzmitskaya, I.; Kirko, S.; Belonovskaya, E.; Naruta, E.; Lukivskaya, O.; Shlyahtun, A.; Ilyich, T.; Zakreska, A.; Zavodnik, I. Betulin attenuated liver damage by prevention of hepatic mitochondrial dysfunction in rats with alcoholic steatohepatitis. Physiol. Int. 2019, 106, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Tajik, N.; Tajik, M.; Mack, I.; Enk, P. The potential effects of chlorogenic acid, the main phenolic components in coffee, on health: A comprehensive review of the literature. Eur. J. Nutr. 2017, 56, 2215–2244. [Google Scholar] [CrossRef] [PubMed]
- Szkudelski, T.; Szkudelska, K. Potential of resveratrol in mitigating metabolic disturbances induced by ethanol. Biomed. Pharmacother. 2018, 101, 579–584. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Wang, Y.; Liu, J.; Wang, K.; Guo, X.; Ji, B.; Wu, W.; Zhou, F. Protective effects of genistein and puerarin against chronic alcohol-induced liver injury in mice via antioxidant, anti-inflammatory, and anti-apoptotic mechanisms. J. Agric. Food Chem. 2016, 64, 7291–7297. [Google Scholar] [CrossRef]
- Salomone, F.; Galvano, F.; Li Volti, G. Molecular bases underlying the hepatoprotective effects of coffee. Nutrients. 2017, 9, 85. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.J.; Hsu, J.D.; Lin, W.L.; Kao, S.H.; Wang, C.J. Upregulation of caveolin-1 by mulberry leaf extract and its major components, chlorogenic acid derivatives, attenuates alcoholic steatohepatitis via inhibition of oxidative stress. Food Funct. 2017, 8, 397–405. [Google Scholar] [CrossRef]
- Budryn, G.; Żyżelewicz, D.; Buko, V.; Lukivskaya, O.; Naruta, E.; Belonovskaya, E.; Moroz, V.; Kirko, S.; Grzelczyk, J.; Bojczuk, M.; et al. Evaluation of antifibrotic effects of coffee and cocoa extracts in rats with thioacetamide-induced fibrosis. Eur. Food Res. Technol. 2018, 244, 2107–2115. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Gao, M.; Liu, D. Chlorogenic acid improves high fat diet-induced hepatic steatosis and insulin resistance in mice. Pharm. Res. 2015, 32, 1200–1209. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Gao, Y.Q.; Zhang, Y.; Wang, H.; Liu, G.S.; Lei, J.Y. Chlorogenic acid alleviates autophagy and insulin resistance by suppressing JNK pathway in a rat model of nonalcoholic fatty liver disease. J. Biosci. 2018, 43, 287–294. [Google Scholar] [CrossRef]
- Kim, H.; Pan, J.H.; Kim, S.H.; Lee, J.H.; Park, J.W. Chlorogenic acid ameliorates alcohol-induced liver injuries through species. Biochimie 2018, 150, 131–138. [Google Scholar] [CrossRef]
- Wang, X.Y.; Luo, J.P.; Chen, R.; Zha, X.Q.; Wang, H. The effects of daily supplementation of Dendrobium huoshanense polysaccharide on ethanol-induced subacute liver injury in mice by proteomic analysis. Food Funct. 2014, 9, 2020–2035. [Google Scholar] [CrossRef]
- Stickel, F.; Datz, C.; Hampe, J.; Bataller, R. Pathophysiology and management of alcoholic liver disease: Update 2016. Gut Liver 2017, 11, 173–188. [Google Scholar] [CrossRef] [PubMed]
- Kirpich, I.A.; Warner, D.R.; Feng, W.; Joshi-Barve, S.; McClain, C.J.; Seth, D.; Zhong, W.; Zhou, Z.; Osna, N.A.; Kharbanda, K.K. Mechanisms, biomarkers and targets for therapy in alcohol-associated liver injury: From genetics to nutrition: Summary of the ISBRA 2018 symposium. Alcohol 2020, 83, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Lukivskaya, O.Y.; Naruta, E.; Sadovnichy, V.; Kirko, S.; Buko, V.U. Reversal of experimental ethanol-induced liver steatosis by borage oil. Phytother. Res. 2012, 26, 1626–1631. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Lai, K.Y.; Verlinsky, A.; Lugea, A.; French, S.W.; Cooper, M.P.; Ji, C.; Tsukamoto, H. Synergistic steatohepatitis by moderate obesity and alcohol in mice despite increased adiponectin and p-AMPK. J. Hepatol. 2011, 55, 673–682. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Dong, K.; Ma, Y.; Jin, Q.; Yin, S.; Wang, S. Hepatoprotective effects of chamazulene against alcohol-induced liver damage by alleviation of oxidative stress in rat models. Open Life Sci. 2020, 15, 251–258. [Google Scholar] [CrossRef]
- Lukivskaya, O.; Zavodnik, L.; Knas, M.; Buko, V. Antioxidant mechanism of hepatoprotection by ursodeoxycholic acid in experimental alcoholic steatohepatitis. Adv. Med. Sci. 2006, 51, 54–59. [Google Scholar]
- Dkhil, M.A.; Ab-del Moneim, A.E.; Bauomy, A.A.; Khalil, M.; Al-Shaebi, E.M.; Al-Quraishy, S. Chlorogenic acid prevents hepatotoxicity in arsenic-treated mice: Role of oxidative stress and apoptosis. Mol. Biol. Rep. 2020, 47, 1161–1171. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Bao, C.; Li, L.; Fu, M.; Wang, D.; Xie, J.; Gong, X. Chlorogenic acid protects against cholestatic liver injury in rats. J. Pharmacol. Sci. 2015, 129, 177–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gil-lessen, A.; Schmidt, H.H. Silymarin as Supportive Treatment in Liver Diseases: A Narrative Review. Adv. Ther. 2020, 37, 1279–1301. [Google Scholar] [CrossRef] [Green Version]
- Bhandarkar, N.S.; Brown, L.; Panchal, S.K. Chloro-genic acid attenuates high-carbohydrate, high-fat diet-induced cardiovascular, liver, and metabolic changes in rats. Nutr. Res. 2019, 62, 78–88. [Google Scholar] [CrossRef]
- Buege, J.A.; Aust, S.D. Microsomal lipid peroxidation. Methods Enzymol. 1978, 52, 302–310. [Google Scholar] [PubMed]
- Jouihan, H. Measurement of Liver Triglyceride Content. Bio-Protocol 2012, 2, e223. [Google Scholar] [CrossRef]
- Arai, Y.; Kobayashi, E.; Nazawa, M.; Yamanaka, T. Characteristic changes of hepatic lymphocytes after ischemia-reperfusion. Transplantation 1996, 61, 848–849. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, S.; Vargas, J.; Hoffmann, A. Signaling via the NFκB system. Wiley Interdiscip. Rev. Syst. Biol. Med. 2016, 8, 227–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuo, A.; Wang, S.; Liu, L.; Yao, Y.; Guo, J. Understanding the effect of anthocyanin extracted from Lonicera caerulea L. on alcoholic hepatosteatosis. Biomed. Pharmacother. 2019, 117, 109087. [Google Scholar] [CrossRef]
- Fabregat, I.; Caballero-Díaz, D. Transforming growth factor-β-induced cell plasticity in liver fibrosis and hepatocarcinogenesis. Front. Oncol. 2018, 8, 357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.H.; Batey, R.G.; George, J. Role of ethanol in the regulation of hepatic stellate cell function. World J. Gastroenterol. 2006, 12, 6926–6932. [Google Scholar] [CrossRef]
- Zadorozhna, M.; Di Gioia, S.; Conese, M.; Mangieri, D. Neovascularization is a key feature of liver fibrosis progression: Anti-angiogenesis as an innovative way of liver fibrosis treatment. Mol. Biol. Rep. 2020, 47, 2279–2288. [Google Scholar] [CrossRef]
- Gao, B.; Bataller, R. Alcoholic liver disease: Pathogenesis and new therapeutic targets. Gastroenterology 2011, 141, 1572–1585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Z.F.; Yu, W.L.; Zhao, Y.P. Study on the scavenging of ROS and anti-lipid peroxidation by chlorogenic acid. Food Sci. 2006, 2, 128–130. [Google Scholar]
- Ji, L.; Jiang, P.; Lu, B.; Sheng, Y.; Wang, X.; Wang, Z. Chlorogenic acid, a dietary polyphenol, protects acetaminophen-induced liver injury and its mechanism. J. Nutr. Biochem. 2013, 24, 1911–1919. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Shi, A.; Dong, L.; Lu, X.; Wang, Y.; Zhao, J.; Dai, F.; Guo, X. Chlorogenic acid protects against liver fibrosis in vivo and in vitro through inhibition of oxidative stress. Clin. Nutr. 2016, 35, 1366–1373. [Google Scholar] [CrossRef] [PubMed]
- Yun, N.; Kang, J.W.; Lee, S.M. Protective effects of chlorogenic acid against ischemia/reperfusion injury in rat liver: Molecular evidence of its antioxidant and anti-inflammatory properties. J. Nutr. Biochem. 2012, 23, 1249–1255. [Google Scholar] [CrossRef] [PubMed]
- Nordmann, R.; Ribière, C.; Rouach, H. Implication of free radical mechanisms in ethanol-induced cellular injury. Free Radic. Biol. 1992, 12, 219–240. [Google Scholar] [CrossRef]
- Lu, Y.; Cederbaum, A.I. CYP2E1 and oxidative liver injury by alcohol. Free Radic. Biol. Med. 2008, 44, 723–738. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.E.; Koh, H.; Joo, D.J.; Nedumaran, B.; Jeon, H.J.; Park, C.S.; Harris, R.A.; Kim, Y.D. Induction of SIRT1 by melatonin improves alcohol-mediated oxidative liver injury by disrupting the CRBN-YY1-CYP2E1 signaling pathway. J. Pineal. Res. 2020, 68, e12638. [Google Scholar] [CrossRef]
- Li, Z.; Lian, Y.; Wei, R.; Jin, L.; Cao, H.; Zhao, T.; Ma, X.; Zhong, M.; Gao, Y.; Zhang, K. Effects of taraxasterol against ethanol and high-fat diet-induced liver injury by regulating TLR4/MyD88/NF-κB and Nrf2/HO-1 signaling pathways. Life Sci. 2020, 262, 118546. [Google Scholar] [CrossRef]
- Mantena, S.K.; King, A.L.; Andringa, K.K.; Landar, A.; Darley-Usmar, V.; Bailey, S.M. Novel interactions of mitochondria and reactive oxygen/nitrogen species in alcohol mediated liver disease. World J. Gastroenterol. 2007, 13, 4967–4973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikeda, H.; Kimura, Y.; Masaki, M.; Iwahashi, H. Caffeic acid inhibits the formation of 1-hydroxyethyl radical in the reaction mixture of rat liver microsomes with ethanol partly through its metal chelating activity. J. Clin. Biochem. Nutr. 2011, 48, 187–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albano, E.; Clot, P.; Morimoto, M.; Tomasi, A.; Ingelman-Sundberg, M.; French, S.W. Role of cytochrome P4502E1-dependent formation of hydroxyethyl free radical in the development of liver damage in rats intragastrically fed with ethanol. Hepatology 1996, 23, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Domitrović, R.; Cvijanović, O.; Šušnić, V.; Katalinić, N. Renoprotective mechanisms of chlorogenic acid in cisplatin-induced kidney injury. Toxicology 2014, 324, 98–107. [Google Scholar] [CrossRef]
- Diesinger, T.; Buko, V.; Lautwein, A.; Dvorsky, R.; Belonovskaya, E.; Lukivskaya, O.; Naruta, E.; Kirko, S.; Andreev, V.; Buckert, D.; et al. Drug targeting CYP2E1 for the treatment of early-stage alcoholic steatohepatitis. PLoS ONE 2020, 15, e0235990. [Google Scholar] [CrossRef] [PubMed]
- Jeong, W.S.; Jun, M.; Kong, A.N. Nrf2: A potential molecular target for cancer chemoprevention by natural compounds. Antioxid. Redox Signal. 2006, 8, 99–106. [Google Scholar] [CrossRef]
- Li, J.P.; Gao, Y.; Chu, S.F.; Zhang, Z.; Xia, C.Y.; Mou, Z.; Song, X.Y.; He, W.B.; Guo, X.F.; Chen, N.H. Nrf2 pathway activation contributes to anti-fibrosis effects of ginsenoside Rg1 in a rat model of alcohol- and CCl4-induced hepatic fibrosis. Acta Pharmacol. Sin. 2014, 35, 1031–1044. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Tian, L.; Chai, G.; Wen, B.; Wang, B. Targeting heme oxygenase-1 by quercetin ameliorates alcohol-induced acute liver injury via inhibiting NLRP3 inflammasome activation. Food Funct. 2018, 9, 4184–4193. [Google Scholar] [CrossRef]
- Wang, X.; Chang, X.; Zhan, H.; Zhang, Q.; Li, C.; Gao, Q.; Yang, M.; Luo, Z.; Li, S.; Sun, Y. Curcumin and Baicalin ameliorate ethanol-induced liver oxidative damage via the Nrf2/HO-1 pathway. J. Food Biochem. 2020, 8, e13425. [Google Scholar] [CrossRef] [PubMed]
- Shi, A.; Shi, H.; Wang, Y.; Liu, X.; Cheng, Y.; Li, H.; Zhao, H.; Wang, S.; Dong, L. Activation of Nrf2 pathway and inhibition of NLRP3 inflammasome activation contribute to the protective effect of chlorogenic acid on acute liver injury. Int. Immunopharmacol. 2018, 54, 125–130. [Google Scholar] [CrossRef]
- Lawrence, T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb. Perspect. Biol. 2009, 1, a001651. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, S.M.; Luo, L.; Namani, A.; Wang, X.J.; Tang, X. Nrf2 signaling pathway: Pivotal roles in inflammation. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 63, 585–597. [Google Scholar] [CrossRef]
- Mandrekar, P.; Szabo, G. Signalling pathways in alcohol-induced liver inflammation. J. Hepatol. 2009, 50, 1258–1266. [Google Scholar] [CrossRef] [Green Version]
- Kagan, J.C.; Medzhitov, R. Phosphoinositide-mediated adaptor recruitment controls Toll-like receptor signaling. Cell 2006, 125, 943–955. [Google Scholar] [CrossRef] [Green Version]
- You, M.; Crabb, D.W. Recent advances in alcoholic liver disease II. Minireview: Molecular mechanisms of alcoholic fatty liver. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 287, G1–G6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- You, M.; Rogers, C.Q. Adiponectin: A key adipokine in alcoholic fatty liver. Exp. Biol. Med. (Maywood) 2009, 234, 850–859. [Google Scholar] [CrossRef]
- You, M.; Liang, X.; Ajmo, J.M.; Ness, G.C. Involvement of mammalian sirtuin 1 in the action of ethanol in the liver. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G892–G898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, V.; Ubaid, S. Role of Silent Information Regulator 1 (SIRT1) in regulating oxidative stress and inflammation. Inflammation 2020, 43, 1589–1598. [Google Scholar] [CrossRef]
- Pietrzyk, N.; Zakłos-Szyda, M.; Koziołkiewicz, M.; Podsędek, A. Viburnum opulus L. fruit phenolic compounds protect against FFA-induced steatosis of HepG2 cells via AMPK pathway. J. Funct. Foods 2021, 80, 104437. [Google Scholar] [CrossRef]
- Liu, B.; Deng, X.; Jiang, Q.; Li, G.; Zhang, J.; Zhang, N.; Xin, S.; Xu, K. Scoparone alleviates inflammation, apoptosis and fibrosis of non-alcoholic steatohepatitis by suppressing the TLR4/NF-κB signaling pathway in mice. Int. Immunopharmacol. 2019, 105797. [Google Scholar] [CrossRef]
- Zhang, K.; Xu, Q.; Gao, Y.; Cao, H.; Lian, Y.; Li, Z.; Xu, J.; Zhong, M.; Li, J.; Wei, R.; et al. Polysaccharides from Diclipterachinensis ameliorate liver disturbance by regulating TLR-4/NF-κB and AMPK/Nrf2 signaling pathways. J. Cell Mol. Med. 2020, 24, 6397–6409. [Google Scholar] [CrossRef] [Green Version]
- Roderburg, C.; Urban, G.W.; Bettermann, K.; Vucur, M.; Zimmermann, H.; Schmidt, S.; Janssen, J.; Koppe, C.; Knolle, P.; Castoldi, M.; et al. Micro-RNA profiling reveals a role for miR-29 in human and murine liver fibrosis. Hepatology 2011, 53, 209–218. [Google Scholar] [CrossRef]
- Blazka, M.E.; Germolec, D.R.; Simeonova, P.; Bruccoleri, A.; Pennypacker, K.R.; Luster, M.I. Acetaminophen-induced hepatotoxicity is associated with early changes in NF-kB and NF-IL6 DNA binding activity. J. Inflamm. 1995, 47, 138–150. [Google Scholar]
- Ding, H.; Huang, J.A.; Tong, J.; Yu, X.; Yu, J.P. Influence of Kupffer cells on hepatic signal transduction as demonstrated by second messengers and nuclear transcription factors. World J. Gastroenterol. 2003, 9, 2519–2522. [Google Scholar] [CrossRef]
- Adachi, Y.; Bradford, B.U.; Gao, W.; Bojes, H.K.; Thurman, R.G. Inactivation of Kupffer cells prevents early alcohol-induced liver injury. Hepatology 1994, 20, 453–460. [Google Scholar] [CrossRef]
- Endo, M.; Masaki, T.; Seike, M.; Yoshimatsu, H. TNF-alpha induces hepatic steatosis in mice by enhancing gene expression of sterol regulatory element binding protein-1c (SREBP-1c). Exp. Biol. Med. 2007, 232, 614–621. [Google Scholar] [CrossRef]
- Yang, F.; Luo, L.; Zhu, Z.D.; Zhou, X.; Wang, Y.; Xue, J.; Zhang, J.; Cai, X.; Chen, Z.L.; Ma, Q.; et al. Chlorogenic acid inhibits liver fibrosis by blocking the mir-21-regulated TGF-β1/Smad7 signaling pathway in vitro and in vivo. Front. Pharmacol. 2017, 8, 929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Yang, F.; Xue, J.; Zhou, X.; Luo, L.; Ma, Q.; Chen, Y.F.; Zhang, J.; Zhang, S.L.; Zhao, L. Antischistosomiasis liver fibrosis effects of chlorogenic acid through IL-13/miR-1/Smad7 signaling interactions in vivo and in vitro. Antimicrob. Agents Chemother. 2017, 61, e01347-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luedde, T.; Schwabe, R.F. NF-κB in the liver–linking injury, fibrosis and hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 108–118. [Google Scholar] [CrossRef] [Green Version]
- Shi, H.; Dong, L.; Jiang, J.; Zhao, J.; Zhao, G.; Dang, X.; Lu, X.; Jia, M. Chlorogenic acid reduces liver inflammation and fibrosis through inhibition of toll-like receptor 4 signaling pathway. Toxicology 2013, 303, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Morbidelli, L.; Donnini, S.; Ziche, M. Role of nitric oxide in the modulation of angiogenesis. Curr. Pharm. Des. 2003, 9, 521–530. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Diet | ||
|---|---|---|
| Control Group Diet | EtOH-Treated Groups Diet | |
| Sunflower oil | 13 | 13 |
| Lard | 13 | 13 |
| Cellulose | 6.5 | 12 |
| Corn starch | 41.5 | 35.5 |
| Mineral mix 1 | 3.5 | 3.5 |
| Vitamin mix 2 | 1.0 | 1.0 |
| DL-methionine | 0.3 | 0.3 |
| Choline chloride | 0.2 | 0.2 |
| Control | EtOH | EtOH + CGA, 40 mg/kg | EtOH + CGA, 80 mg/kg | |
|---|---|---|---|---|
| Liver/body mass × 100 | 2.69 ± 0.07 | 3.15 ± 0.06 a | 2.96 ± 0.05 ab | 2.88 ± 0.06 b |
| AST, IU/L | 92.2 ± 8.71 | 135.9 ± 19.63 a | 113.8 ± 14.12 b | 112.9 ± 17.14 b |
| ALT, IU/L | 50.7 ± 7.23 | 80.2 ± 20.15 a | 59.8 ± 9.32 b | 60.1 ± 10.77 b |
| Alkaline phosphatase, IU/L | 102.2 ± 14.2 | 218.9± 69.4 a | 166.0 ± 28.7 ab | 144.2 ± 31.1 ab |
| Serum triglycerides, mmol/L | 2.89 ± 0.149 | 4.12 ± 0.305 a | 3.40 ± 0.376 ab | 3.24 ± 0.263 ab |
| Liver triglycerides, mg/g tissue | 1.41 ± 0.30 | 4.83 ± 0.77 a | 3.34 ± 0.43 ab | 3.04 ± 0.58 ab |
| Serum TNFα, pg/mL | 90.2 ± 2.95 | 179.8 ± 9.82 a | 164.0 ± 8.07 a | 144.5 ± 12.44 ab |
| Serum Il-6, pg/mL | 88.9 ± 2.06 | 181.6 ± 8.61 a | 145.2 ± 16.22 a | 102.0 ± 5.05 ab |
| Liver TBARS, nmol/g tissue | 39.9 ± 6.06 | 104.6 ± 15.02 a | 59.3 ± 6.53 ab | 52.3 ± 4.87 ab |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buko, V.; Zavodnik, I.; Budryn, G.; Zakłos-Szyda, M.; Belonovskaya, E.; Kirko, S.; Żyżelewicz, D.; Zakrzeska, A.; Bakunovich, A.; Rusin, V.; et al. Chlorogenic Acid Protects against Advanced Alcoholic Steatohepatitis in Rats via Modulation of Redox Homeostasis, Inflammation, and Lipogenesis. Nutrients 2021, 13, 4155. https://doi.org/10.3390/nu13114155
Buko V, Zavodnik I, Budryn G, Zakłos-Szyda M, Belonovskaya E, Kirko S, Żyżelewicz D, Zakrzeska A, Bakunovich A, Rusin V, et al. Chlorogenic Acid Protects against Advanced Alcoholic Steatohepatitis in Rats via Modulation of Redox Homeostasis, Inflammation, and Lipogenesis. Nutrients. 2021; 13(11):4155. https://doi.org/10.3390/nu13114155
Chicago/Turabian StyleBuko, Vyacheslav, Ilya Zavodnik, Grażyna Budryn, Małgorzata Zakłos-Szyda, Elena Belonovskaya, Siarhei Kirko, Dorota Żyżelewicz, Agnieszka Zakrzeska, Aliaksei Bakunovich, Viktor Rusin, and et al. 2021. "Chlorogenic Acid Protects against Advanced Alcoholic Steatohepatitis in Rats via Modulation of Redox Homeostasis, Inflammation, and Lipogenesis" Nutrients 13, no. 11: 4155. https://doi.org/10.3390/nu13114155
APA StyleBuko, V., Zavodnik, I., Budryn, G., Zakłos-Szyda, M., Belonovskaya, E., Kirko, S., Żyżelewicz, D., Zakrzeska, A., Bakunovich, A., Rusin, V., & Moroz, V. (2021). Chlorogenic Acid Protects against Advanced Alcoholic Steatohepatitis in Rats via Modulation of Redox Homeostasis, Inflammation, and Lipogenesis. Nutrients, 13(11), 4155. https://doi.org/10.3390/nu13114155