
Pathophysiology of Vascular Calcification and Bone Loss: Linked Disorders of Ageing?

 ,
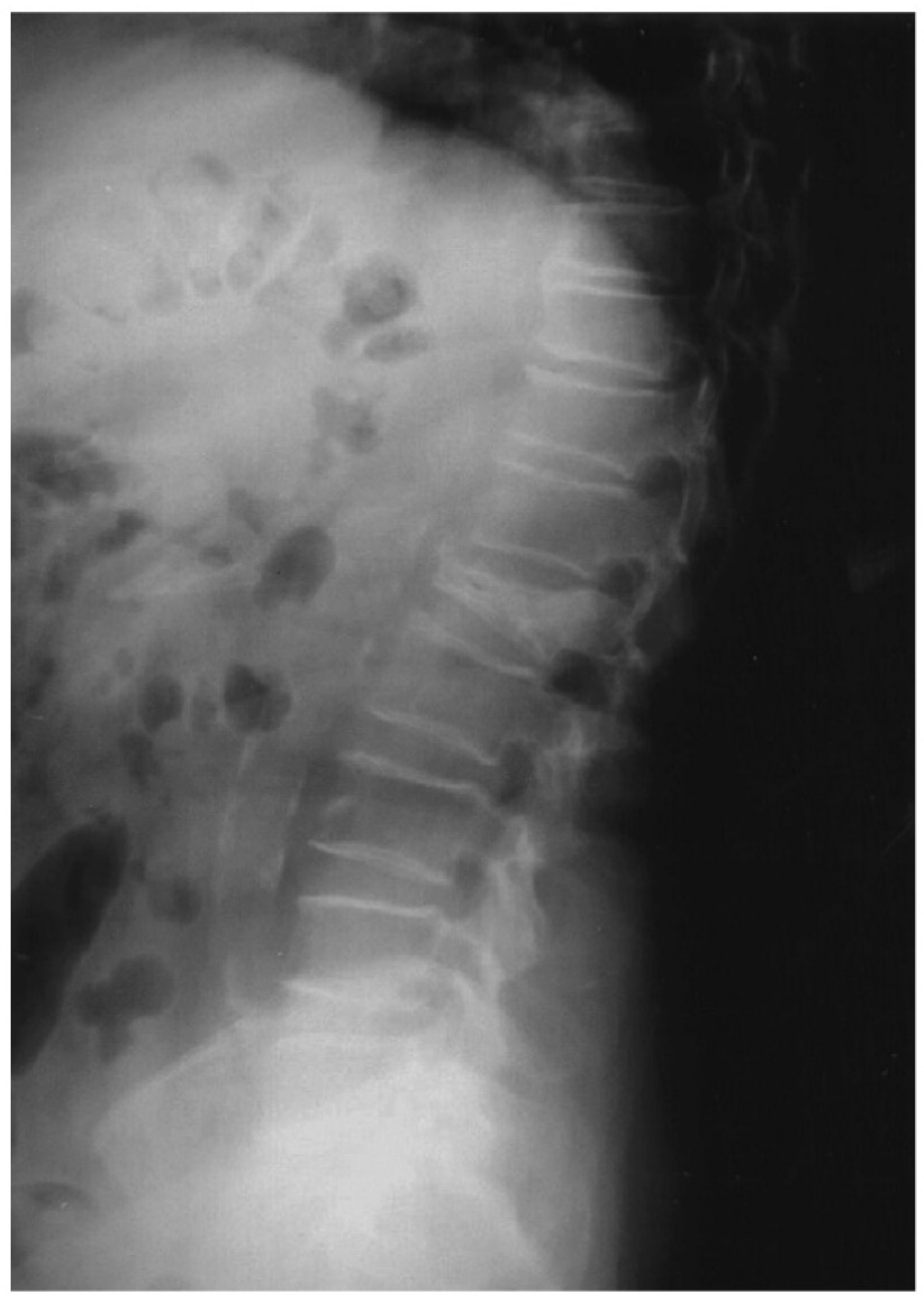
, 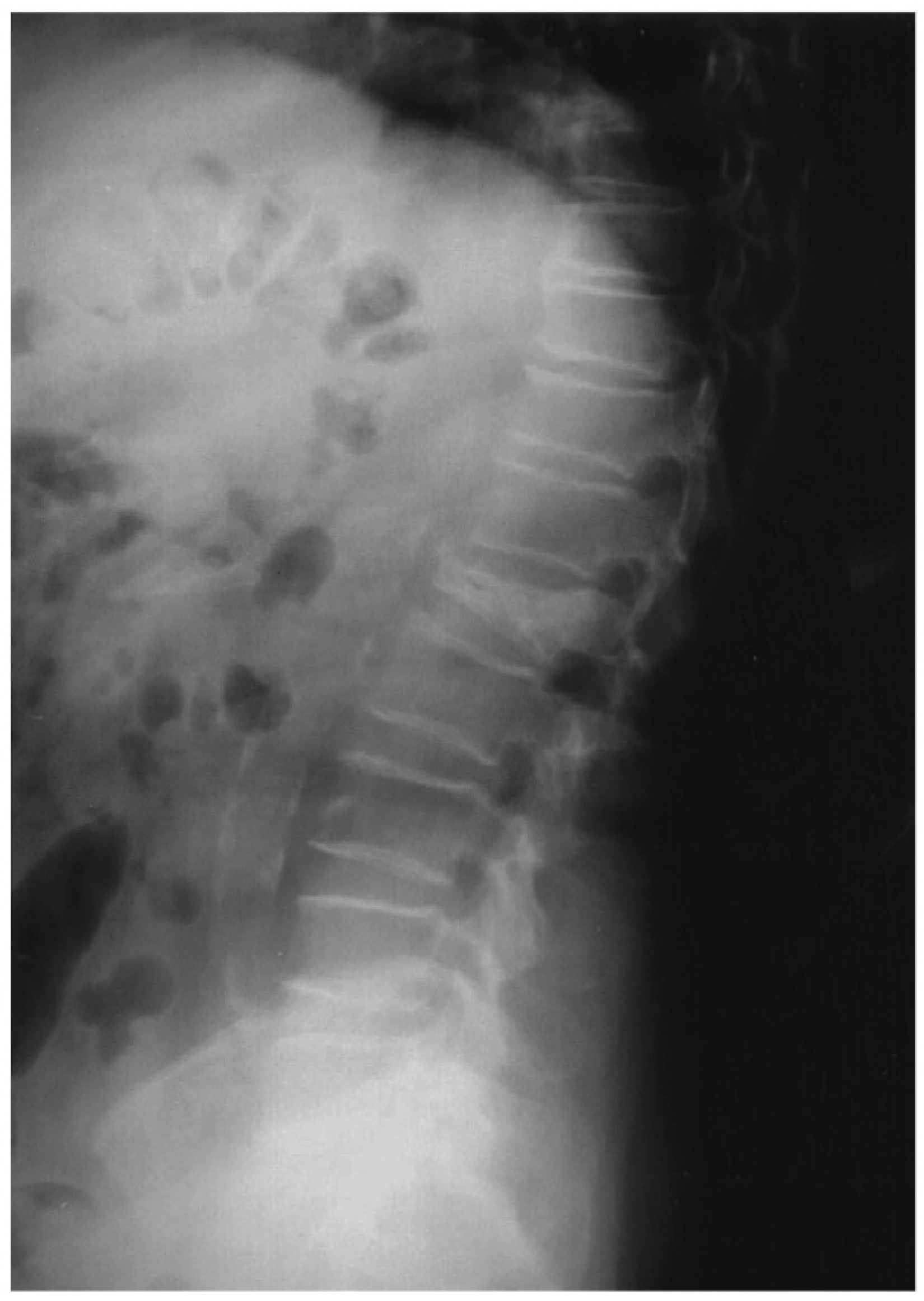{kind=link}
{kind=link}
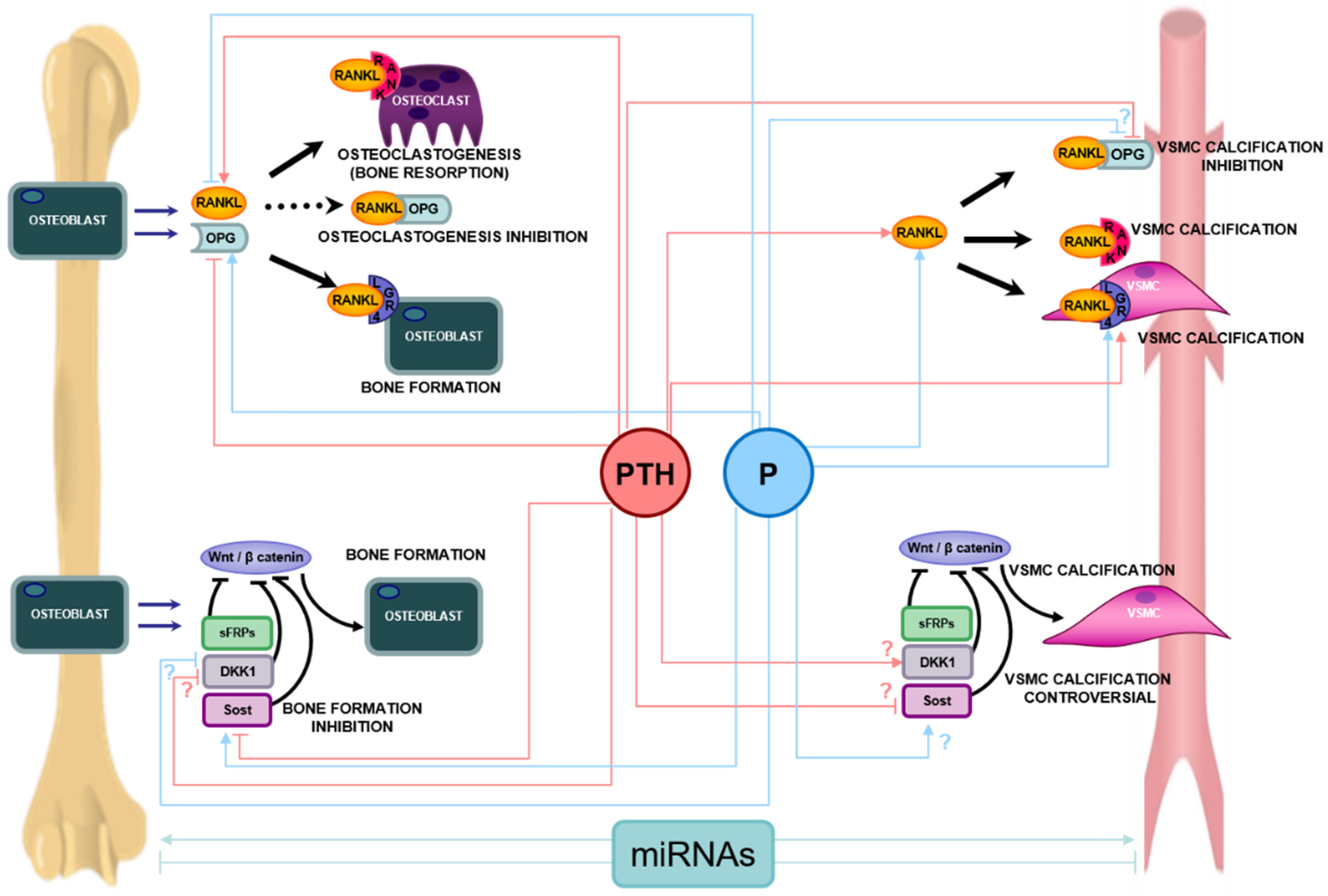{kind=link}
Abstract
:1. Introduction
2. Pathophysiology of Vascular Calcification
3. Pathophysiology of Bone Loss in Osteoporosis
4. Role of Key Regulators of Bone Metabolism on VC
4.1. Parathyroid Hormone and FGF23
4.2. The Role of Phosphorus
4.3. The RANK/RANKL/OPG System
4.4. The Wnt/ß-Catenin Pathway
4.5. The Role of microRNAs in Bone and Vascular Metabolism
4.6. Cellular Senescence
5. Summary and Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Frye, M.A.; Melton, L.J.; Bryant, S.C.; Fitzpatrick, L.A.; Wahner, H.W.; Schwartz, R.S.; Riggs, B.L. Osteoporosis and calcification of the aorta. Bone Miner. 1992, 19, 185–194. [Google Scholar] [CrossRef]
- Kiel, D.; Kauppila, L.I.; Cupples, L.A.; Hannan, M.T.; O’Donnell, C.J.; Wilson, P.W.F. Bone loss and the progression of abdominal aortic calcification over a 25 year period: The Framingham heart study. Calcif. Tissue Int. 2001, 68, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Vogt, M.T.; Valentin, R.S.; Forrest, K.Y.-Z.; Nevitt, M.C.; Cauley, J. Bone Mineral Density and Aortic Calcification: The Study of Osteoporotic Fractures. J. Am. Geriatr. Soc. 1997, 45, 140–145. [Google Scholar] [CrossRef] [PubMed]
- Cannata-Andía, J.B.; Rodríguez-García, M.; Carrillo-Lopez, N.; Naves-Díaz, M.; Díaz-López, B. Vascular Calcifications: Pathogenesis, Management, and Impact on Clinical Outcomes. J. Am. Soc. Nephrol. 2006, 17, S267–S273. [Google Scholar] [CrossRef] [Green Version]
- Garcia, M.R.; Diaz, M.N.; Cannata-Andía, J.B. Bone metabolism, vascular calcifications and mortality: Associations beyond mere coincidence. J. Nephrol. 2005, 18, 458–463. [Google Scholar]
- Schulz, E.; Arfai, K.; Liu, X.; Sayre, J.; Gilsanz, V. Aortic Calcification and the Risk of Osteoporosis and Fractures. J. Clin. Endocrinol. Metab. 2004, 89, 4246–4253. [Google Scholar] [CrossRef]
- Naves, M.; García, M.R.; López, J.B.D.; Alonso, C.G.; Andía, J.B.C. Progression of vascular calcifications is associated with greater bone loss and increased bone fractures. Osteoporos. Int. 2008, 19, 1161–1166. [Google Scholar] [CrossRef]
- Fusaro, M.; Tripepi, G.; Noale, M.; Vajente, N.; Plebani, M.; Zaninotto, M.; Guglielmi, G.; Miotto, D.; Carbonare, L.D.; D’Angelo, A.; et al. High Prevalence of Vertebral Fractures Assessed by Quantitative Morphometry in Hemodialysis Patients, Strongly Associated with Vascular Calcifications. Calcif. Tissue Int. 2013, 93, 39–47. [Google Scholar] [CrossRef]
- Jaminon, A.; Reesink, K.; Kroon, A.; Schurgers, L. The Role of Vascular Smooth Muscle Cells in Arterial Remodeling: Focus on Calcification-Related Processes. Int. J. Mol. Sci. 2019, 20, 5694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amann, K. Media Calcification and Intima Calcification Are Distinct Entities in Chronic Kidney Disease. Clin. J. Am. Soc. Nephrol. 2008, 3, 1599–1605. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-García, M.; Gómez-Alonso, C.; Naves-Díaz, M.; Diaz-Lopez, J.B.; Diaz-Corte, C.; Cannata-Andía, J.B.; the Asturias Study Group. Vascular calcifications, vertebral fractures and mortality in haemodialysis patients. Nephrol. Dial. Transplant. 2008, 24, 239–246. [Google Scholar] [CrossRef] [Green Version]
- Cannata-Andía, J.B.; Roman-Garcia, P.; Hruska, K. The connections between vascular calcification and bone health. Nephrol. Dial. Transplant. 2011, 26, 3429–3436. [Google Scholar] [CrossRef]
- Merjanian, R.; Budoff, M.; Adler, S.; Berman, N.; Mehrotra, R. Coronary artery, aortic wall, and valvular calcification in nondialyzed individuals with type 2 diabetes and renal disease. Kidney Int. 2003, 64, 263–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bridgeman, G.; Brookes, M. Blood supply to the human femoral diaphysis in youth and senescence. J. Anat. 1996, 188, 611–621. [Google Scholar]
- Laroche, M.; Pouilles, J.M.; Ribot, C.; Bendayan, P.; Bernard, J.; Boccalon, H.; Mazières, B. Comparison of the bone mineral content of the lower limbs in men with ischaemic atherosclerotic disease. Clin. Rheumatol. 1994, 13, 611–614. [Google Scholar] [CrossRef] [PubMed]
- London, G.M.; Marty, C.; Marchais, S.J.; Guerin, A.P.; Metivier, F.; De Vernejoul, M.-C. Arterial Calcifications and Bone Histomorphometry in End-Stage Renal Disease. J. Am. Soc. Nephrol. 2004, 15, 1943–1951. [Google Scholar] [CrossRef]
- London, G.M.; Marchais, S.J.; Guérin, A.P.; Boutouyrie, P.; Métivier, F.; de Vernejoul, M.-C. Association of Bone Activity, Calcium Load, Aortic Stiffness, and Calcifications in ESRD. J. Am. Soc. Nephrol. 2008, 19, 1827–1835. [Google Scholar] [CrossRef] [Green Version]
- Coen, G.; Ballanti, P.; Mantella, D.; Manni, M.; Lippi, B.; Pierantozzi, A.; Di Giulio, S.; Pellegrino, L.; Romagnoli, A.; Simonetti, G.; et al. Bone Turnover, Osteopenia and Vascular Calcifications in Hemodialysis Patients. Am. J. Nephrol. 2009, 29, 145–152. [Google Scholar] [CrossRef]
- Marcovitz, P.A.; Tran, H.H.; Franklin, B.A.; O’Neill, W.W.; Yerkey, M.; Boura, J.; Kleerekoper, M.; Dickinson, C.Z. Usefulness of Bone Mineral Density to Predict Significant Coronary Artery Disease. Am. J. Cardiol. 2005, 96, 1059–1063. [Google Scholar] [CrossRef]
- Moe, S.M.; Chen, N.X. Mechanisms of Vascular Calcification in Chronic Kidney Disease: Figure 1. J. Am. Soc. Nephrol. 2007, 19, 213–216. [Google Scholar] [CrossRef] [Green Version]
- Román-García, P.; Carrillo-López, N.; Fernández-Martín, J.L.; Naves-Díaz, M.; Ruiz-Torres, M.P.; Cannata-Andía, J.B. High phosphorus diet induces vascular calcification, a related decrease in bone mass and changes in the aortic gene expression. Bone 2010, 46, 121–128. [Google Scholar] [CrossRef]
- Towler, D.A.; Shao, J.; Cheng, S.; Pingsterhaus, J.M.; Loewy, A.P. Osteogenic Regulation of Vascular Calcification. Ann. N. Y. Acad. Sci. 2006, 1068, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Shroff, R.; Shanahan, C. Vascular Calcification in Patients with Kidney Disease: The Vascular Biology of Calcification. Semin. Dial. 2007, 20, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Kapustin, A.N.; Davies, J.D.; Reynolds, J.L.; McNair, R.; Jones, G.T.; Sidibe, A.; Schurgers, L.J.; Skepper, J.N.; Proudfoot, D.; Mayr, M.; et al. Calcium Regulates Key Components of Vascular Smooth Muscle Cell–Derived Matrix Vesicles to Enhance Mineralization. Circ. Res. 2011, 109, e1–e12. [Google Scholar] [CrossRef] [Green Version]
- Giachelli, C.M. Vascular Calcification Mechanisms. J. Am. Soc. Nephrol. 2004, 15, 2959–2964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lomashvili, K.A.; Khawandi, W.; O’Neill, W.C. Reduced Plasma Pyrophosphate Levels in Hemodialysis Patients. J. Am. Soc. Nephrol. 2005, 16, 2495–2500. [Google Scholar] [CrossRef]
- Moe, S.M.; Reslerova, M.; Ketteler, M.; O’Neill, K.; Duan, D.; Koczman, J.; Westenfeld, R.; Jahnen-Dechent, W.; Chen, N.X. Role of calcification inhibitors in the pathogenesis of vascular calcification in chronic kidney disease (CKD). Kidney Int. 2005, 67, 2295–2304. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Zhu, Y.; Jaiswal, S.K.; Liu, N.-F. Mitochondria Homeostasis and Vascular Medial Calcification. Calcif. Tissue Int. 2021, 109, 113–120. [Google Scholar] [CrossRef]
- Ducy, P.; Zhang, R.; Geoffroy, V.; Ridall, A.L.; Karsenty, G. Osf2/Cbfa1: A Transcriptional Activator of Osteoblast Differentiation. Cell 1997, 89, 747–754. [Google Scholar] [CrossRef] [Green Version]
- Engelse, M.; Neele, J.M.; Bronckers, A.L.; Pannekoek, H.; De Vries, C.J. Vascular calcification: Expression patterns of the osteoblast-specific gene core binding factor α-1 and the protective factor matrix gla protein in human atherogenesis. Cardiovasc. Res. 2001, 52, 281–289. [Google Scholar] [CrossRef] [Green Version]
- O’Neill, W.C. Pyrophosphate, Alkaline Phosphatase, and Vascular Calcification. Circ. Res. 2006, 99, e2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oury, F.; Khrimian, L.; Denny, C.A.; Gardin, A.; Chamouni, A.; Goeden, N.; Huang, Y.-Y.; Lee, H.; Srinivas, P.; Gao, X.-B.; et al. Maternal and Offspring Pools of Osteocalcin Influence Brain Development and Functions. Cell 2013, 155, 228–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, N.K.; Sowa, H.; Hinoi, E.; Ferron, M.; Ahn, J.D.; Confavreux, C.; Dacquin, R.; Mee, P.J.; McKee, M.D.; Jung, D.Y.; et al. Endocrine Regulation of Energy Metabolism by the Skeleton. Cell 2007, 130, 456–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ducy, P.; Desbois, C.; Boyce, B.; Pinero, G.; Story, B.; Dunstan, C.; Smith, E.; Bonadio, J.; Goldstein, S.; Gundberg, C.; et al. Increased bone formation in osteocalcin-deficient mice. Nat. Cell Biol. 1996, 382, 448–452. [Google Scholar] [CrossRef] [Green Version]
- Idelevich, A.; Rais, Y.; Monsonego-Ornan, E. Bone Gla Protein Increases HIF-1α–Dependent Glucose Metabolism and Induces Cartilage and Vascular Calcification. Arter. Thromb. Vasc. Biol. 2011, 31, e55–e71. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Dang, K.; Huai, Y.; Qian, A. Osteoimmunology: The Regulatory Roles of T Lymphocytes in Osteoporosis. Front. Endocrinol. 2020, 11, 465. [Google Scholar] [CrossRef]
- Stenvinkel, P.; Ketteler, M.; Johnson, R.; Lindholm, B.; Pecoits-Filho, R.; Riella, M.; Heimbürger, O.; Cederholm, T.; Girndt, M. IL-10, IL-6, and TNF-α: Central factors in the altered cytokine network of uremia—The good, the bad, and the ugly. Kidney Int. 2005, 67, 1216–1233. [Google Scholar] [CrossRef] [Green Version]
- Tintut, Y.; Patel, J.; Parhami, F.; Demer, L.L. Tumor Necrosis Factor-α Promotes In Vitro Calcification of Vascular Cells via the cAMP Pathway. Circulation 2000, 102, 2636–2642. [Google Scholar] [CrossRef] [Green Version]
- Kearns, A.E.; Khosla, S.; Kostenuik, P.J. Receptor activator of nuclear factor kappaB ligand and osteoprotegerin regulation of bone remodeling in health and disease. Endocr. Rev. 2008, 29, 155–192. [Google Scholar] [CrossRef]
- Eghbali-Fatourechi, G.; Khosla, S.; Sanyal, A.; Boyle, W.J.; Lacey, D.L.; Riggs, B.L. Role of RANK ligand in mediating increased bone resorption in early postmenopausal women. J. Clin. Investig. 2003, 111, 1221–1230. [Google Scholar] [CrossRef]
- Cummings, S.R.; Martin, J.S.; McClung, M.R.; Siris, E.S.; Eastell, R.; Reid, I.R.; Delmas, P.; Zoog, H.B.; Austin, M.; Wang, A.; et al. Denosumab for Prevention of Fractures in Postmenopausal Women with Osteoporosis. N. Engl. J. Med. 2009, 361, 756–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sousa, S.; Clézardin, P. Bone-Targeted Therapies in Cancer-Induced Bone Disease. Calcif. Tissue Int. 2017, 102, 227–250. [Google Scholar] [CrossRef] [PubMed]
- Ke, H.Z.; Richards, W.G.; Li, X.; Ominsky, M.S. Sclerostin and Dickkopf-1 as Therapeutic Targets in Bone Diseases. Endocr. Rev. 2012, 33, 747–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kranenburg, G.; Bartstra, J.W.; De Jong, P.; Cosman, F.; Crittenden, D.B.; Grauer, A. Romosozumab Treatment in Postmenopausal Osteoporosis. N. Engl. J. Med. 2017, 376, 395–397. [Google Scholar] [CrossRef]
- Baron, R.; Rawadi, G. Targeting the Wnt/beta-catenin pathway to regulate bone formation in the adult skeleton. Endocrinology 2007, 148, 2635–2643. [Google Scholar] [CrossRef] [Green Version]
- Carrillo-Lopez, N.; Panizo, S.; Montes, C.A.; Roman-Garcia, P.; Rodríguez, I.; Martínez-Salgado, C.; Dusso, A.S.; Naves, M.; Cannata-Andía, J.B. Direct inhibition of osteoblastic Wnt pathway by fibroblast growth factor 23 contributes to bone loss in chronic kidney disease. Kidney Int. 2016, 90, 77–89. [Google Scholar] [CrossRef] [Green Version]
- Cannata-Andía, J.B.; Carrillo-López, N.; Rodriguez-García, M.; Torregrosa, J.-V. Mineral and Bone Disorders in Chronic Kidney Disease. In Management of Chronic Kidney Disease; Springer: Berlin/Heidelberg, Germany, 2014; pp. 223–239. [Google Scholar]
- Shao, J.-S.; Cheng, S.-L.; Charlton-Kachigian, N.; Loewy, A.P.; Towler, D. Teriparatide (Human Parathyroid Hormone (1–34)) Inhibits Osteogenic Vascular Calcification in Diabetic Low Density Lipoprotein Receptor-deficient Mice. J. Biol. Chem. 2003, 278, 50195–50202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vattikuti, R.; Towler, D.A. Osteogenic regulation of vascular calcification: An early perspective. Am. J. Physiol. Metab. 2004, 286, E686–E696. [Google Scholar] [CrossRef]
- Graciolli, F.G.; Neves, K.R.; Dos Reis, L.; Noronha, I.L.; Moyses, R.; Jorgetti, V. Phosphorus overload and PTH induce aortic expression of Runx2 in experimental uraemia. Nephrol. Dial. Transplant. 2009, 24, 1416–1421. [Google Scholar] [CrossRef] [PubMed]
- Carrillo-Lopez, N.; Panizo, S.; Alonso-Montes, C.; Martínez-Arias, L.; Avello, N.; Sosa, P.; Dusso, A.S.; Cannata-Andía, J.B.; Naves-Díaz, M. High-serum phosphate and parathyroid hormone distinctly regulate bone loss and vascular calcification in experimental chronic kidney disease. Nephrol. Dial. Transplant. 2019, 34, 934–941. [Google Scholar] [CrossRef]
- Carrillo-López, N.; Martínez-Arias, L.; Alonso-Montes, C.; Martín-Carro, B.; Martín-Vírgala, J.; Ruiz-Ortega, M.; Fernández-Martín, J.L.; Dusso, A.S.; Rodriguez-García, M.; Naves-Díaz, M.; et al. The receptor activator of nuclear factor κΒ ligand receptor leucine-rich repeat-containing G-protein-coupled receptor 4 contributes to parathyroid hormone-induced vascular calcification. Nephrol. Dial. Transplant. 2021, 36, 618–631. [Google Scholar] [CrossRef]
- Cannata-Andia, J.B.; Roman-Garcia, P.; Carrillo-Lopez, N.; Dusso, A. Clinical and Preclinical Evidence of the Skeletal and Vascular Adverse Health Effects of High Dietary Phosphorus; Taylor and Francis: London, UK, 2016. [Google Scholar]
- Yuan, Q.; Sato, T.; Densmore, M.; Saito, H.; Schüler, C.; Erben, R.G.; Lanske, B. FGF-23/Klotho signaling is not essential for the phosphaturic and anabolic functions of PTH. J. Bone Miner. Res. 2011, 26, 2026–2035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hruska, K.A.; Mathew, S.; Lund, R.; Qiu, P.; Pratt, R. Hyperphosphatemia of chronic kidney disease. Kidney Int. 2008, 74, 148–157. [Google Scholar] [CrossRef] [Green Version]
- Cannata-Andía, J.B.; Naves-Díaz, M. Phosphorus and Survival: Key Questions That Need Answers. J. Am. Soc. Nephrol. 2009, 20, 234–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Román-García, P.; Carrillo-López, N.; Cannata-Andía, J.B. Pathogenesis of Bone and Mineral Related Disorders in Chronic Kidney Disease: Key Role of Hyperphosphatemia. J. Ren. Care 2009, 35, 34–38. [Google Scholar] [CrossRef]
- Campos-Obando, N.; Koek, W.N.H.; Hooker, E.R.; van der Eerden, B.; Pols, H.; Hofman, A.; Van Leeuwen, J.P.; Uitterlinden, A.G.; Nielson, C.M.; Zillikens, M.C. Serum Phosphate Is Associated With Fracture Risk: The Rotterdam Study and MrOS. J. Bone Miner. Res. 2017, 32, 1182–1193. [Google Scholar] [CrossRef] [PubMed]
- Huttunen, M.M.; Tillman, I.; Viljakainen, H.T.; Tuukkanen, J.; Peng, Z.; Pekkinen, M.; Lamberg-Allardt, C.J. High Dietary Phosphate Intake Reduces Bone Strength in the Growing Rat Skeleton. J. Bone Miner. Res. 2006, 22, 83–92. [Google Scholar] [CrossRef]
- Conrads, K.A.; Yi, M.; Simpson, K.A.; Lucas, D.A.; Camalier, C.E.; Yu, L.-R.; Veenstra, T.D.; Stephens, R.M.; Conrads, T.P.; Beck, G.R. A Combined Proteome and Microarray Investigation of Inorganic Phosphate-induced Pre-osteoblast Cells. Mol. Cell. Proteom. 2005, 4, 1284–1296. [Google Scholar] [CrossRef] [Green Version]
- Camalier, C.E.; Yi, M.; Yu, L.-R.; Hood, B.L.; Conrads, K.A.; Lee, Y.J.; Lin, Y.; Garneys, L.M.; Bouloux, G.F.; Young, M.R.; et al. An integrated understanding of the physiological response to elevated extracellular phosphate. J. Cell. Physiol. 2013, 228, 1536–1550. [Google Scholar] [CrossRef] [Green Version]
- Dallas, S.L.; Prideaux, M.; Bonewald, L.F. The Osteocyte: An Endocrine Cell … and More. Endocr. Rev. 2013, 34, 658–690. [Google Scholar] [CrossRef] [Green Version]
- Capulli, M.; Paone, R.; Rucci, N. Osteoblast and osteocyte: Games without frontiers. Arch. Biochem. Biophys. 2014, 561, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Kanatani, M.; Sugimoto, T.; Kano, J.; Kanzawa, M.; Chihara, K. Effect of high phosphate concentration on osteoclast differentiation as well as bone-resorbing activity. J. Cell. Physiol. 2003, 196, 180–189. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yang, H.-Y.; Giachelli, C.M. Role of the Sodium-Dependent Phosphate Cotransporter, Pit-1, in Vascular Smooth Muscle Cell Calcification. Circ. Res. 2006, 98, 905–912. [Google Scholar] [CrossRef] [Green Version]
- Panizo, S.; Naves-Díaz, M.; Carrillo-López, N.; Martínez-Arias, L.; Fernández-Martín, J.L.; Ruiz-Torres, M.P.; Cannata-Andía, J.B.; Rodríguez, I. MicroRNAs 29b, 133b, and 211 Regulate Vascular Smooth Muscle Calcification Mediated by High Phosphorus. J. Am. Soc. Nephrol. 2016, 27, 824–834. [Google Scholar] [CrossRef] [Green Version]
- Boyce, B.F.; Xing, L. Functions of RANKL/RANK/OPG in bone modeling and remodeling. Arch. Biochem. Biophys. 2008, 473, 139–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panizo, S.; Cardus, A.; Encinas, M.; Parisi, E.; Valcheva, P.; López-Ongil, S.; Coll, B.; Fernandez, E.; Valdivielso, J.M. RANKL Increases Vascular Smooth Muscle Cell Calcification Through a RANK-BMP4–Dependent Pathway. Circ. Res. 2009, 104, 1041–1048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhore, C.R.; Cleutjens, J.P.M.; Lutgens, E.; Cleutjens, K.B.J.M.; Geusens, P.P.M.; Kitslaar, P.J.E.H.M.; Tordoir, J.H.M.; Spronk, H.M.H.; Vermeer, C.; Daemen, M.J.A.P. Differential Expression of Bone Matrix Regulatory Proteins in Human Atherosclerotic Plaques. Arter. Thromb. Vasc. Biol. 2001, 21, 1998–2003. [Google Scholar] [CrossRef] [Green Version]
- Simonet, W.; Lacey, D.; Dunstan, C.; Kelley, M.; Chang, M.-S.; Lüthy, R.; Nguyen, H.; Wooden, S.; Bennett, L.; Boone, T.; et al. Osteoprotegerin: A Novel Secreted Protein Involved in the Regulation of Bone Density. Cell 1997, 89, 309–319. [Google Scholar] [CrossRef] [Green Version]
- Malyankar, U.M.; Scatena, M.; Suchland, K.L.; Yun, T.J.; Clark, E.A.; Giachelli, C.M. Osteoprotegerin is an alpha vbeta 3-induced, NF-kappa B-dependent survival factor for endothelial cells. J. Biol. Chem. 2000, 275, 20959–20962. [Google Scholar] [CrossRef] [Green Version]
- Hofbauer, L.C.; Shui, C.; Riggs, B.L.; Dunstan, C.R.; Spelsberg, T.C.; O’Brien, T.; Khosla, S. Effects of immunosuppressants on receptor activator of NF-kappaB ligand and osteoprotegerin pro-duction by human osteoblastic and coronary artery smooth muscle cells. Biochem. Biophys. Res. Commun. 2001, 280, 334–339. [Google Scholar] [CrossRef]
- Yasuda, H.; Shima, N.; Nakagawa, N.; Yamaguchi, K.; Kinosaki, M.; Mochizuki, S.-I.; Tomoyasu, A.; Yano, K.; Goto, M.; Murakami, A.; et al. Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc. Natl. Acad. Sci. USA 1998, 95, 3597–3602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bucay, N.; Sarosi, I.; Dunstan, C.; Morony, S.; Tarpley, J.; Capparelli, C.; Scully, S.; Tan, H.L.; Xu, W.; Lacey, D.L.; et al. osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes Dev. 1998, 12, 1260–1268. [Google Scholar] [CrossRef]
- Min, H.; Morony, S.; Sarosi, I.; Dunstan, C.; Capparelli, C.; Scully, S.; Van, G.; Kaufman, S.; Kostenuik, P.J.; Lacey, D.L.; et al. Osteoprotegerin Reverses Osteoporosis by Inhibiting Endosteal Osteoclasts and Prevents Vascular Calcification by Blocking a Process Resembling Osteoclastogenesis. J. Exp. Med. 2000, 192, 463–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rinotas, V.; Niti, A.; Dacquin, R.; Bonnet, N.; Stolina, M.; Han, C.-Y.; Kostenuik, P.; Jurdic, P.; Ferrari, S.; Douni, E. Novel Genetic Models of Osteoporosis by Overexpression of Human RANKL in Transgenic Mice. J. Bone Miner. Res. 2014, 29, 1158–1169. [Google Scholar] [CrossRef]
- Luo, J.; Yang, Z.; Ma, Y.; Yue, Z.; Lin, H.; Qu, G.; Huang, J.; Dai, W.; Li, C.; Zheng, C.; et al. LGR4 is a receptor for RANKL and negatively regulates osteoclast differentiation and bone resorption. Nat. Med. 2016, 22, 539–546. [Google Scholar] [CrossRef] [PubMed]
- de Lau, W.; Barker, N.; Low, T.Y.; Koo, B.K.; Li, V.S.; Teunissen, H.; Kujala, P.; Haegebarth, A.; Peters, P.J.; van de Wetering, M.; et al. Lgr5 homologues associate with Wnt receptors and mediate R-spondin signalling. Nature 2011, 476, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Zhou, W.; Zhou, X.; Li, D.; Weng, J.; Yi, Z.; Cho, S.-G.; Li, C.; Yi, T.; Wu, X.; et al. Regulation of bone formation and remodeling by G-protein-coupled receptor 48. Development 2009, 136, 2747–2756. [Google Scholar] [CrossRef] [Green Version]
- Collin-Osdoby, P. Regulation of Vascular Calcification by Osteoclast Regulatory Factors RANKL and Osteoprotegerin. Circ. Res. 2004, 95, 1046–1057. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, R.K.; Dar, H.Y.; Mishra, P.K. Immunoporosis: Immunology of Osteoporosis—Role of T Cells. Front. Immunol. 2018, 9, 657. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Suematsu, A.; Okamoto, K.; Yamaguchi, A.; Morishita, Y.; Kadono, Y.; Tanaka, S.; Kodama, T.; Akira, S.; Iwakura, Y.; et al. Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J. Exp. Med. 2006, 203, 2673–2682. [Google Scholar] [CrossRef] [Green Version]
- Hofbauer, L.C.; Schoppet, M. Clinical implications of the osteoprotegerin/RANKL/RANK system for bone and vascular diseases. JAMA 2004, 292, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Nitta, K.; Akiba, T.; Uchida, K.; Otsubo, S.; Takei, T.; Yumura, W.; Kabaya, T.; Nihei, H. Serum osteoprotegerin levels and the extent of vascular calcification in haemodialysis patients. Nephrol. Dial. Transplant. 2004, 19, 1886–1889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.H.; Liu, X.; Wang, J.; Chen, X.; Zhang, H.; Kim, S.H.; Cui, J.; Li, R.; Zhang, W.; Kong, Y.; et al. Wnt signaling in bone formation and its therapeutic potential for bone diseases. Ther. Adv. Musculoskelet. Dis. 2013, 5, 13–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rashdan, N.; Sim, A.M.; Cui, L.; Phadwal, K.; Roberts, F.; Carter, R.; Ozdemir, D.D.; Hohenstein, P.; Hung, J.; Kaczynski, J.; et al. Osteocalcin Regulates Arterial Calcification Via Altered Wnt Signaling and Glucose Metabolism. J. Bone Miner. Res. 2020, 35, 357–367. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, V.; Bryant, H.U.; MacDougald, O. Regulation of bone mass by Wnt signaling. J. Clin. Investig. 2006, 116, 1202–1209. [Google Scholar] [CrossRef]
- Fang, Y.; Ginsberg, C.; Seifert, M.; Agapova, O.; Sugatani, T.; Register, T.C.; Freedman, B.I.; Monier-Faugere, M.-C.; Malluche, H.; Hruska, K.A. CKD-Induced Wingless/Integration1 Inhibitors and Phosphorus Cause the CKD–Mineral and Bone Disorder. J. Am. Soc. Nephrol. 2014, 25, 1760–1773. [Google Scholar] [CrossRef] [Green Version]
- Shalhoub, V.; Shatzen, E.; Henley, C.; Boedigheimer, M.; McNinch, J.; Manoukian, R.; Damore, M.; Fitzpatrick, D.; Haas, K.; Twomey, B.; et al. Calcification Inhibitors and Wnt Signaling Proteins Are Implicated in Bovine Artery Smooth Muscle Cell Calcification in the Presence of Phosphate and Vitamin D Sterols. Calcif. Tissue Int. 2006, 79, 431–442. [Google Scholar] [CrossRef]
- Woldt, E.; Terrand, J.; Mlih, M.; Matz, R.L.; Bruban, V.; Coudane, F.; Foppolo, S.; El Asmar, Z.; Chollet, M.E.; Ninio, E.; et al. The nuclear hormone receptor PPARgamma counteracts vascular calcification by inhibiting Wnt5a signal-ling in vascular smooth muscle cells. Nat. Commun. 2012, 3, 1077. [Google Scholar]
- Deng, D.; Diao, Z.; Han, X.; Liu, W. Secreted Frizzled-Related Protein 5 Attenuates High Phosphate-Induced Calcification in Vascular Smooth Muscle Cells by Inhibiting the Wnt/ß-Catenin Pathway. Calcif. Tissue Int. 2016, 99, 66–75. [Google Scholar] [CrossRef]
- Goettsch, C.; Hutcheson, J.D.; Aikawa, E. MicroRNA in cardiovascular calcification: Focus on targets and extracellular vesicle delivery mechanisms. Circ. Res. 2013, 112, 1073–10784. [Google Scholar] [CrossRef] [Green Version]
- Goettsch, C.; Rauner, M.; Pacyna, N.; Hempel, U.; Bornstein, S.R.; Hofbauer, L.C. miR-125b Regulates Calcification of Vascular Smooth Muscle Cells. Am. J. Pathol. 2011, 179, 1594–1600. [Google Scholar] [CrossRef] [PubMed]
- Carè, A.; Catalucci, D.; Felicetti, F.; Bonci, D.; Addario, A.; Gallo, P.; Bang, M.-L.; Segnalini, P.; Gu, Y.; Dalton, N.D.; et al. MicroRNA-133 controls cardiac hypertrophy. Nat. Med. 2007, 13, 613–618. [Google Scholar] [CrossRef]
- Zhou, S.; Liu, Y.; Prater, K.; Zheng, Y.; Cai, L. Roles of microRNAs in pressure overload- and ischemia-related myocardial remodeling. Life Sci. 2013, 93, 855–862. [Google Scholar] [CrossRef] [PubMed]
- Panizo, S.; Carrillo-Lopez, N.; Naves-Díaz, M.; Berrocal, G.S.; Arias, L.M.; Díez, R.R.; Fernández-Vázquez, A.; Martínez-Salgado, C.; Ruiz-Ortega, M.; Dusso, A.; et al. Regulation of miR-29b and miR-30c by vitamin D receptor activators contributes to attenuate uraemia-induced cardiac fibrosis. Nephrol. Dial. Transplant. 2017, 32, 1831–1840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papaioannou, G. miRNAs in Bone Development. Curr. Genom. 2015, 16, 427–434. [Google Scholar] [CrossRef] [Green Version]
- Papaioannou, G.; Mirzamohammadi, F.; Kobayashi, T. MicroRNAs involved in bone formation. Cell. Mol. Life Sci. 2014, 71, 4747–4761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandourah, A.Y.; Ranganath, L.; Barraclough, R.; Vinjamuri, S.; Hof, R.V.; Hamill, S.; Czanner, G.; Dera, A.A.; Wang, D.; Barraclough, D. Circulating microRNAs as potential diagnostic biomarkers for osteoporosis. Sci. Rep. 2018, 8, 1–10. [Google Scholar] [CrossRef]
- Kelch, S.; Balmayor, E.R.; Seeliger, C.; Vester, H.; Kirschke, J.S.; Van Griensven, M. miRNAs in bone tissue correlate to bone mineral density and circulating miRNAs are gender independent in osteoporotic patients. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Gu, H.; Wu, L.; Chen, H.; Huang, Z.; Xu, J.; Zhou, K.; Zhang, Y.; Chen, J.; Xia, J.; Yin, X. Identification of differentially ex-pressed microRNAs in the bone marrow of osteoporosis patients. Am. J. Transl. Res. 2019, 11, 2940–2954. [Google Scholar]
- Lian, W.-S.; Ko, J.-Y.; Chen, Y.-S.; Ke, H.-J.; Hsieh, C.-K.; Kuo, C.-W.; Wang, S.-Y.; Huang, B.-W.; Tseng, J.-G.; Wang, F.-S. MicroRNA-29a represses osteoclast formation and protects against osteoporosis by regulating PCAF-mediated RANKL and CXCL12. Cell Death Dis. 2019, 10, 1–14. [Google Scholar] [CrossRef]
- Badi, I.; Mancinelli, L.; Polizzotto, A.; Ferri, D.; Zeni, F.; Burba, I.; Milano, G.; Brambilla, F.; Saccu, C.; Bianchi, M.E.; et al. miR-34a Promotes Vascular Smooth Muscle Cell Calcification by Downregulating SIRT1 (Sirtuin 1) and Axl (AXL Receptor Tyrosine Kinase). Arter. Thromb. Vasc. Biol. 2018, 38, 2079–2090. [Google Scholar] [CrossRef]
- Lin, X.; Li, F.; Xu, F.; Cui, R.-R.; Xiong, D.; Zhong, J.-Y.; Zhu, T.; Shan, S.-K.; Wu, F.; Xie, X.-B.; et al. Aberration methylation of miR-34b was involved in regulating vascular calcification by targeting Notch1. Aging 2019, 11, 3182–3197. [Google Scholar] [CrossRef] [PubMed]
- Rangrez, A.Y.; M’Baya-Moutoula, E.; Meuth, V.M.-L.; Hénaut, L.; Djelouat, M.S.E.I.; Benchitrit, J.; Massy, Z.A.; Metzinger, L. Inorganic Phosphate Accelerates the Migration of Vascular Smooth Muscle Cells: Evidence for the Involvement of miR-223. PLoS ONE 2012, 7, e47807. [Google Scholar] [CrossRef] [PubMed]
- Ai, J.; Zhang, R.; Li, Y.; Pu, J.; Lu, Y.; Jiao, J.; Li, K.; Yu, B.; Li, Z.; Wang, R.; et al. Circulating microRNA-1 as a potential novel biomarker for acute myocardial infarction. Biochem. Biophys. Res. Commun. 2010, 391, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Burton, D.G.; Giles, P.J.; Sheerin, A.N.; Smith, S.K.; Lawton, J.J.; Ostler, E.L.; Rhys-Williams, W.; Kipling, D.; Faragher, R.G. Microarray analysis of senescent vascular smooth muscle cells: A link to atherosclerosis and vascular calcification. Exp. Gerontol. 2009, 44, 659–665. [Google Scholar] [CrossRef] [Green Version]
- Burton, D.; Matsubara, H.; Ikeda, K. Pathophysiology of vascular calcification: Pivotal role of cellular senescence in vascular smooth muscle cells. Exp. Gerontol. 2010, 45, 819–824. [Google Scholar] [CrossRef]
- Durosier, C.; van Lierop, A.; Ferrari, S.; Chevalley, T.; Papapoulos, S.; Rizzoli, R. Association of Circulating Sclerostin With Bone Mineral Mass, Microstructure, and Turnover Biochemical Markers in Healthy Elderly Men and Women. J. Clin. Endocrinol. Metab. 2013, 98, 3873–3883. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cannata-Andía, J.B.; Carrillo-López, N.; Messina, O.D.; Hamdy, N.A.T.; Panizo, S.; Ferrari, S.L.; on behalf of the International Osteoporosis Foundation (IOF) Working Group on Bone and Cardiovascular Diseases. Pathophysiology of Vascular Calcification and Bone Loss: Linked Disorders of Ageing? Nutrients 2021, 13, 3835. https://doi.org/10.3390/nu13113835
Cannata-Andía JB, Carrillo-López N, Messina OD, Hamdy NAT, Panizo S, Ferrari SL, on behalf of the International Osteoporosis Foundation (IOF) Working Group on Bone and Cardiovascular Diseases. Pathophysiology of Vascular Calcification and Bone Loss: Linked Disorders of Ageing? Nutrients. 2021; 13(11):3835. https://doi.org/10.3390/nu13113835
Chicago/Turabian StyleCannata-Andía, Jorge B., Natalia Carrillo-López, Osvaldo D. Messina, Neveen A. T. Hamdy, Sara Panizo, Serge L. Ferrari, and on behalf of the International Osteoporosis Foundation (IOF) Working Group on Bone and Cardiovascular Diseases. 2021. "Pathophysiology of Vascular Calcification and Bone Loss: Linked Disorders of Ageing?" Nutrients 13, no. 11: 3835. https://doi.org/10.3390/nu13113835
APA StyleCannata-Andía, J. B., Carrillo-López, N., Messina, O. D., Hamdy, N. A. T., Panizo, S., Ferrari, S. L., & on behalf of the International Osteoporosis Foundation (IOF) Working Group on Bone and Cardiovascular Diseases. (2021). Pathophysiology of Vascular Calcification and Bone Loss: Linked Disorders of Ageing? Nutrients, 13(11), 3835. https://doi.org/10.3390/nu13113835