
Therapeutic Potential of Mitophagy-Inducing Microflora Metabolite, Urolithin A for Alzheimer’s Disease

 ,
, 

 ,
,
Abstract
:1. Introduction
2. Alzheimer’s Disease
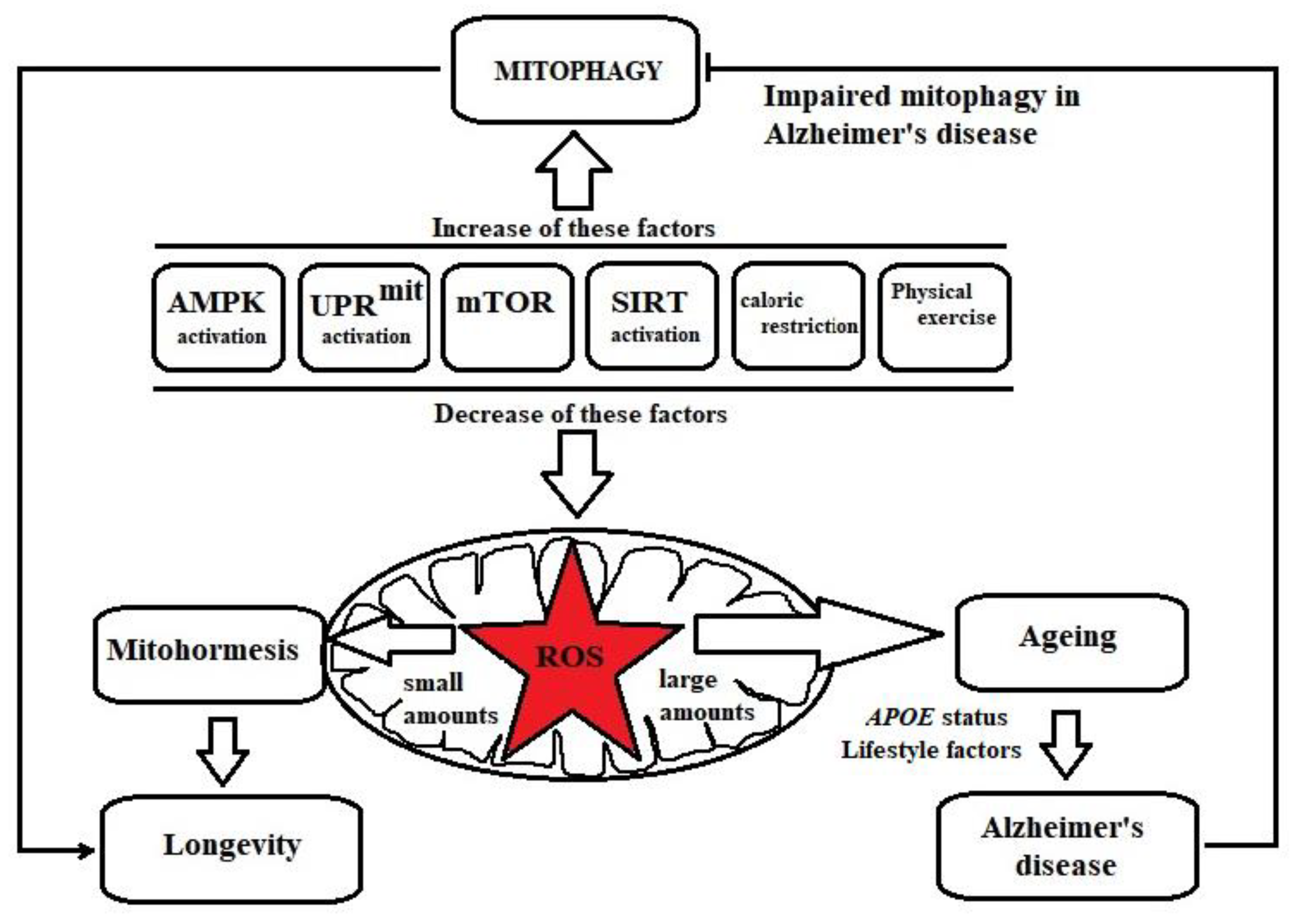
3. Mitophagy Deficits in Alzheimer’s Disease
4. Enhancing Longevity: Molecular Pathways Leading to Mitophagy Activation
5. Polyphenols with Potential to Induce Mitophagy
6. Urolithins
6.1. Biological Effects of Urolithins
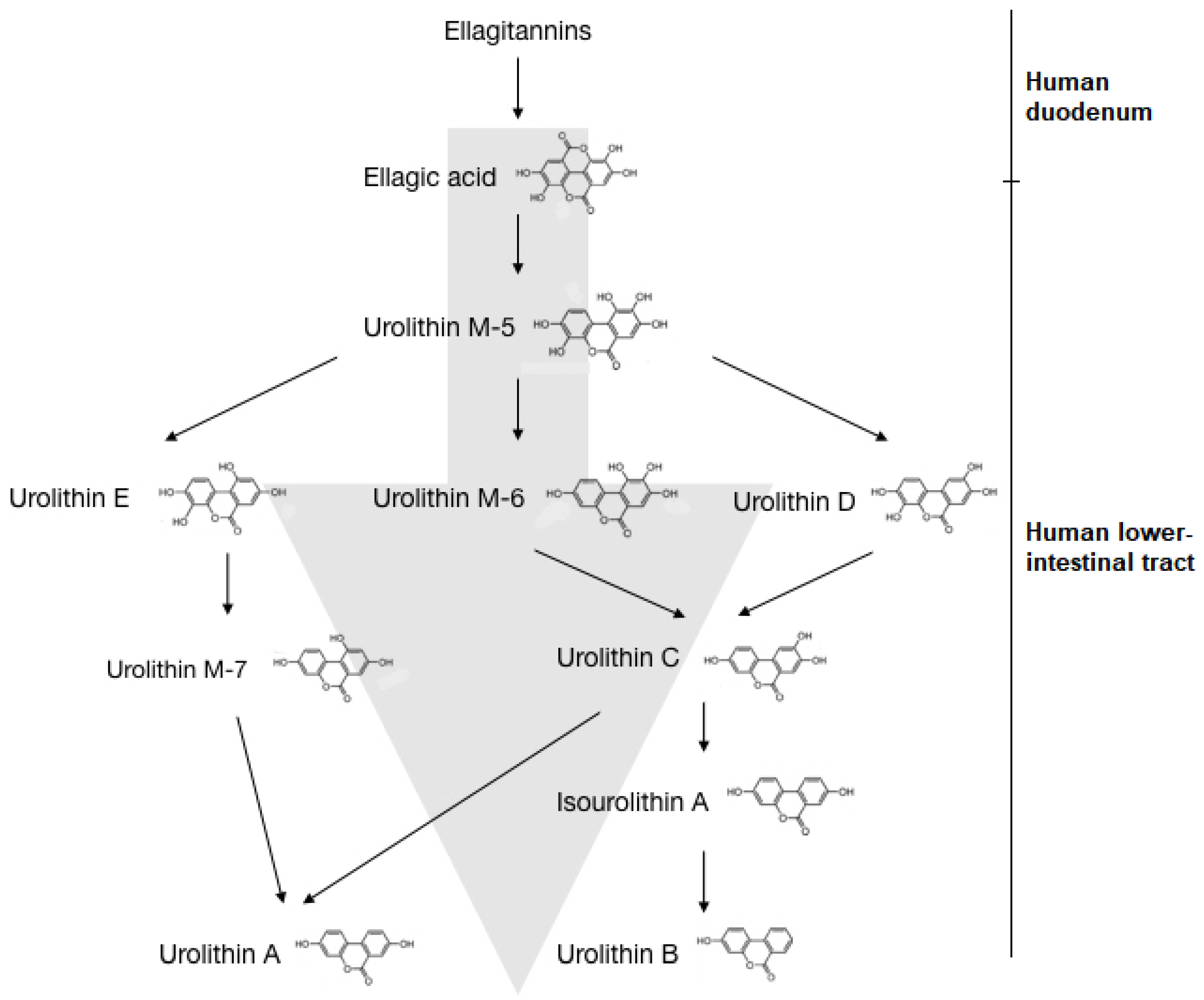
6.1.1. Human Microbiota and Urolithin Metabotypes
6.1.2. Bioavailability and Toxicity of Urolithins
6.1.3. Therapeutic Effects of Urolithins
6.2. Considerations for Urolithin A as a Therapeutic Agent for Alzheimer’s Disease
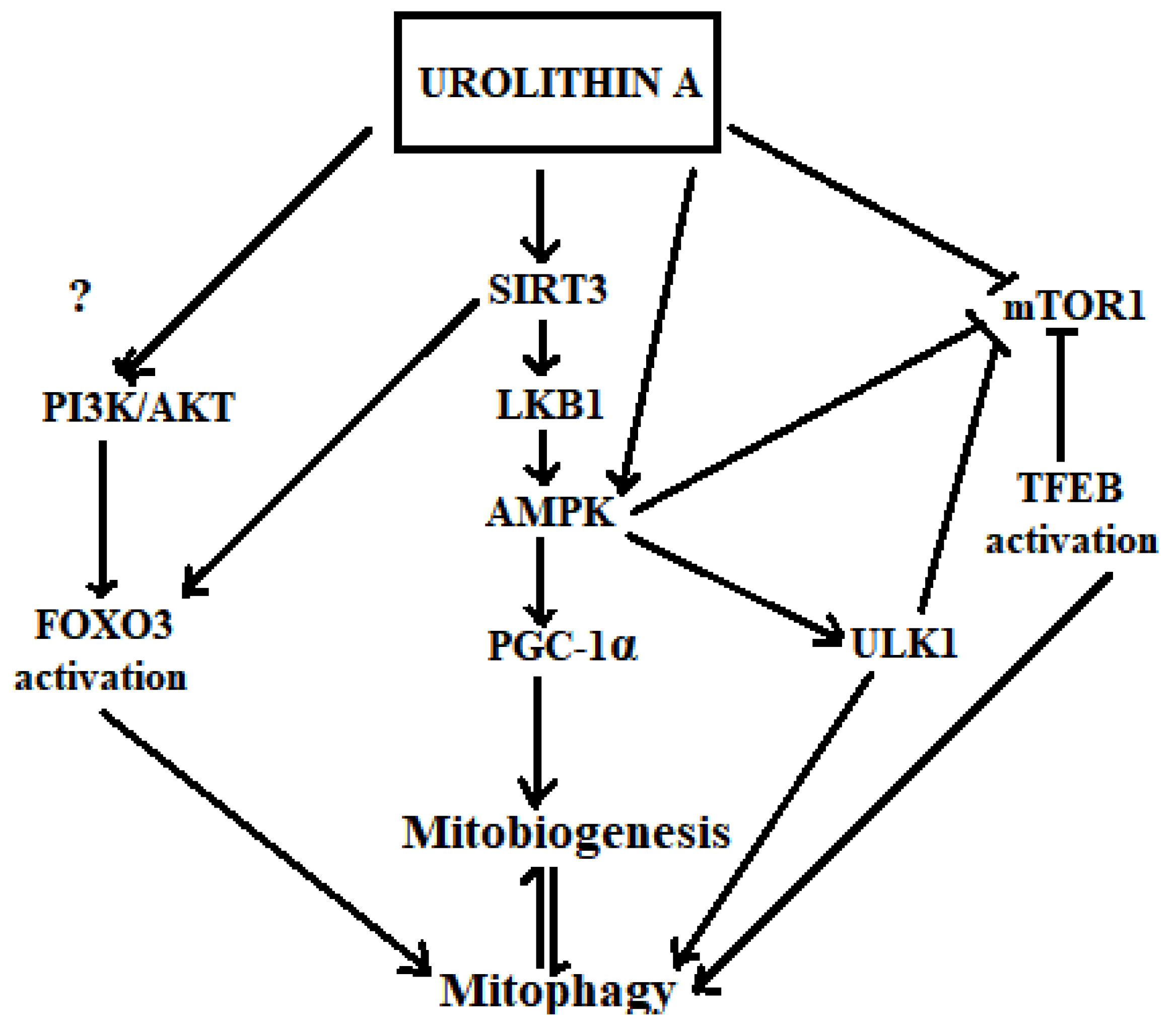
6.3. Insights on How Urolithin A May Trigger Mitophagy
6.3.1. Activation of SIRT1/3, AMPK, PGC1-α and Inhibition of mTOR1
6.3.2. Transcriptional Activation by TFEB and FOXO3
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.-C.; Wei, Y.-H. Role of mitochondria in human aging. J. Biomed. Sci. 1997, 4, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.K. Mitochondria damage checkpoint, aging, and cancer. Ann. N. Y. Acad. Sci. 2006, 1067, 182–190. [Google Scholar] [CrossRef]
- Bratic, A.; Larsson, N.-G. The role of mitochondria in aging. J. Clin. Investig. 2013, 123, 951–957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, N.; Youle, R.J.; Finkel, T. The Mitochondrial Basis of Aging. Mol. Cell 2016, 61, 654–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellerin, L.; Bouzier-Sore, A.K.; Aubert, A.; Serres, S.; Merle, M.; Costalat, R.; Magistretti, P.J. Activity-dependent regulation of energy metabolism by astrocytes: An update. Glia 2007, 55, 1251–1262. [Google Scholar] [CrossRef]
- Riske, L.; Thomas, R.K.; Baker, G.B.; Dursun, S.M. Lactate in the brain: An update on its relevance to brain energy, neurons, glia and panic disorder. Ther. Adv. Psychopharmacol. 2017, 7, 85–89. [Google Scholar] [CrossRef] [Green Version]
- Harman, D. Aging: A theory based on free radical and radiation chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef] [Green Version]
- Harman, D. The biologic clock: The mitochondria? J. Am. Geriatr. Soc. 1972, 20, 145–147. [Google Scholar] [CrossRef]
- Lenaz, G. Role of mitochondria in oxidative stress and ageing. Biochim. Biophys. Acta (BBA) Bioenerg. 1998, 1366, 53–67. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, S. The Mitochondrial Basis of Aging and Age-Related Disorders. Genes 2017, 8, 398. [Google Scholar] [CrossRef] [Green Version]
- Moncada, S.; Bolanos, J.P. Nitric oxide, cell bioenergetics and neurodegeneration. J. Neurochem. 2006, 97, 1676–1689. [Google Scholar] [CrossRef]
- Contestabile, A.; Monti, B.; Polazzi, E. Neuronal-glial Interactions Define the Role of Nitric Oxide in Neural Functional Processes. Curr. Neuropharmacol. 2012, 10, 303–310. [Google Scholar] [CrossRef]
- Halliwell, B. Reactive oxygen species and the central nervous system. J. Neurochem. 1992, 59, 1609–1623. [Google Scholar] [CrossRef]
- Shohami, E.; Beit-Yannai, E.; Horowitz, M.; Kohen, R. Oxidative Stress in Closed-Head Injury: Brain Antioxidant Capacity as an Indicator of Functional Outcome. J. Cereb. Blood Flow Metab. 1997, 17, 1007–1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreira, P.I.; Carvalho, C.; Zhu, X.; Smith, M.A.; Perry, G. Mitochondrial dysfunction is a trigger of Alzheimer’s disease pathophysiology. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2010, 1802, 2–10. [Google Scholar] [CrossRef] [Green Version]
- Alzheimer’s Disease International. Dementia and Risk Reduction: An Analysis of Protective and Modifiable Risk Factors; Alzheimer’s Disease International: London, UK, 2014. [Google Scholar]
- Australian Institute of Health and Welfare. Dementia in Australia. Available online: https://www.aihw.gov.au/reports-data/health-conditions-disability-deaths/dementia/overview (accessed on 21 June 2021).
- Yang, Y.H.; Meguro, K.; Kim, S.Y.; Shim, Y.S.; Yu, X.; Chen, C.L.H.; Wang, H.; Lam, L.; Senanarong, V.; Dominguez, J.; et al. Impact of Alzheimer’s Disease in Nine Asian Countries. Gerontology 2016, 62, 425–433. [Google Scholar] [CrossRef] [PubMed]
- Gorelick, P.B. Risk factors for vascular dementia and Alzheimer disease. Stroke 2004, 35, 2620–2622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riedel, B.C.; Thompson, P.M.; Brinton, R.D. Age, APOE and sex: Triad of risk of Alzheimer’s disease. J. Steroid Biochem. Mol. Biol. 2016, 160, 134–147. [Google Scholar] [CrossRef] [Green Version]
- Edwards Iii, G.A.; Gamez, N.; Escobedo, G., Jr.; Calderon, O.; Moreno-Gonzalez, I. Modifiable Risk Factors for Alzheimer’s Disease. Front. Aging Neurosci. 2019, 11, 146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moller, H.J.; Graeber, M.B. The case described by Alois Alzheimer in 1911. Historical and conceptual perspectives based on the clinical record and neurohistological sections. Euro. Arch. Psychiatry Clin. Neurosci. 1998, 248, 111–122. [Google Scholar] [CrossRef]
- Fox, N.C.; Schott, J.M. Imaging cerebral atrophy: Normal ageing to Alzheimer’s disease. Lancet 2004, 363, 392–394. [Google Scholar] [CrossRef]
- Kern, S.; Zetterberg, H.; Kern, J.; Zettergren, A.; Waern, M.; Höglund, K.; Andreasson, U.; Wetterberg, H.; Börjesson-Hanson, A.; Blennow, K.; et al. Prevalence of preclinical Alzheimer disease. Neurology 2018, 90, e1682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dayan, A.D. Quantitative histological studies on the aged human brain. I. Senile plaques and neurofibrillary tangles in “normal” patients. Acta Neuropathol. 1970, 16, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Villemagne, V.L.; Burnham, S.; Bourgeat, P.; Brown, B.; Ellis, K.A.; Salvado, O.; Szoeke, C.; Macaulay, S.L.; Martins, R.; Maruff, P.; et al. Amyloid beta deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer’s disease: A prospective cohort study. Lancet Neurol. 2013, 12, 357–367. [Google Scholar] [CrossRef]
- Swerdlow, R.H.; Burns, J.M.; Khan, S.M. The Alzheimer’s disease mitochondrial cascade hypothesis: Progress and perspectives. Biochim. Biophys. Acta 2014, 1842, 1219–1231. [Google Scholar] [CrossRef] [Green Version]
- Kerr, J.S.; Adriaanse, B.A.; Greig, N.H.; Mattson, M.P.; Cader, M.Z.; Bohr, V.A.; Fang, E.F. Mitophagy and Alzheimer’s Disease: Cellular and Molecular Mechanisms. Trends Neurosci. 2017. [Google Scholar] [CrossRef] [Green Version]
- Perez Ortiz, J.M.; Swerdlow, R.H. Mitochondrial dysfunction in Alzheimer’s disease: Role in pathogenesis and novel therapeutic opportunities. Br. J. Pharmacol. 2019, 176, 3489–3507. [Google Scholar] [CrossRef]
- Leuner, K.; Schütt, T.; Kurz, C.; Eckert, S.H.; Schiller, C.; Occhipinti, A.; Mai, S.; Jendrach, M.; Eckert, G.P.; Kruse, S.E.; et al. Mitochondrion-Derived Reactive Oxygen Species Lead to Enhanced Amyloid Beta Formation. Antioxid. Redox Sgnal. 2012, 16, 1421–1433. [Google Scholar] [CrossRef] [Green Version]
- Lemasters, J.J. Variants of mitochondrial autophagy: Types 1 and 2 mitophagy and micromitophagy (Type 3). Redox Biol. 2014, 2, 749–754. [Google Scholar] [CrossRef] [Green Version]
- Diot, A.; Morten, K.; Poulton, J. Mitophagy plays a central role in mitochondrial ageing. Mamm. Genome 2016, 27, 381–395. [Google Scholar] [CrossRef] [Green Version]
- Du, F.; Yu, Q.; Yan, S.; Hu, G.; Lue, L.F.; Walker, D.G.; Wu, L.; Yan, S.F.; Tieu, K.; Yan, S.S. PINK1 signalling rescues amyloid pathology and mitochondrial dysfunction in Alzheimer’s disease. Brain 2017, 140, 3233–3251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corsetti, V.; Florenzano, F.; Atlante, A.; Bobba, A.; Ciotti, M.T.; Natale, F.; Della Valle, F.; Borreca, A.; Manca, A.; Meli, G.; et al. NH2-truncated human tau induces deregulated mitophagy in neurons by aberrant recruitment of Parkin and UCHL-1: Implications in Alzheimer’s disease. Hum. Mol. Genet. 2015, 24, 3058–3081. [Google Scholar] [CrossRef] [Green Version]
- Hirai, K.; Aliev, G.; Nunomura, A.; Fujioka, H.; Russell, R.L.; Atwood, C.S.; Johnson, A.B.; Kress, Y.; Vinters, H.V.; Tabaton, M.; et al. Mitochondrial abnormalities in Alzheimer’s disease. J. Neurosci. Off. J. Soc. Neurosci. 2001, 21, 3017–3023. [Google Scholar] [CrossRef] [Green Version]
- Moreira, P.I.; Siedlak, S.L.; Wang, X.; Santos, M.S.; Oliveira, C.R.; Tabaton, M.; Nunomura, A.; Szweda, L.I.; Aliev, G.; Smith, M.A.; et al. Increased autophagic degradation of mitochondria in Alzheimer disease. Autophagy 2007, 3, 614–615. [Google Scholar] [CrossRef] [Green Version]
- Baloyannis, S.J. Mitochondrial alterations in Alzheimer’s disease. J. Alzheimer’s Dis. 2006, 9, 119–126. [Google Scholar] [CrossRef]
- Trushina, E.; Nemutlu, E.; Zhang, S.; Christensen, T.; Camp, J.; Mesa, J.; Siddiqui, A.; Tamura, Y.; Sesaki, H.; Wengenack, T.M.; et al. Defects in mitochondrial dynamics and metabolomic signatures of evolving energetic stress in mouse models of familial Alzheimer’s disease. PLoS ONE 2012, 7, e32737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bordi, M.; Berg, M.J.; Mohan, P.S.; Peterhoff, C.M.; Alldred, M.J.; Che, S.; Ginsberg, S.D.; Nixon, R.A. Autophagy flux in CA1 neurons of Alzheimer hippocampus: Increased induction overburdens failing lysosomes to propel neuritic dystrophy. Autophagy 2016, 12, 2467–2483. [Google Scholar] [CrossRef]
- Hu, Y.; Xia-Chun, L.; Zhi-Hao, W.; Yu, L.; Xiangnan, Z.; Xiu-Ping, L.; Qiong Feng, Q.W.; Zhenyu, Y.; Zhong, C.; Keqiang, Y.; et al. Tau accumulation impairs mitophagy via increasing mitochondrial membrane potential and reducing mitochondrial Parkin. Oncotarget 2016, 7, 17356–17368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martín-Maestro, P.; Gargini, R.; Perry, G.; Avila, J.; García-Escudero, V. PARK2 enhancement is able to compensate mitophagy alterations found in sporadic Alzheimer’s disease. Hum. Mol. Genet. 2016, 25, 792–806. [Google Scholar] [CrossRef] [Green Version]
- Jayatunga, D.P.W.; Hone, E.; Bharadwaj, P.; Garg, M.; Verdile, G.; Guillemin, G.J.; Martins, R.N. Targeting Mitophagy in Alzheimer’s Disease. J. Alzheimer’s Dis. 2020, 78, 1273–1297. [Google Scholar] [CrossRef]
- Li, W.; Kui, L.; Demetrios, T.; Gong, X.; Tang, M. A Glimmer of Hope: Maintain Mitochondrial Homeostasis to Mitigate Alzheimer’s Disease. Aging Dis. 2020, 11. [Google Scholar] [CrossRef]
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.M.; Caponio, D.; Dan, X.; et al. Mitophagy inhibits amyloid-beta and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 2019, 22, 401–412. [Google Scholar] [CrossRef]
- Xie, C.; Aman, Y.; Adriaanse, B.A.; Cader, M.Z.; Plun-Favreau, H.; Xiao, J.; Fang, E.F. Culprit or Bystander: Defective Mitophagy in Alzheimer’s Disease. Front. Cell Dev. Biol. 2020, 7, 391. [Google Scholar] [CrossRef]
- Cai, Q.; Jeong, Y.Y. Mitophagy in Alzheimer’s Disease and Other Age-Related Neurodegenerative Diseases. Cells 2020, 9, 150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabiola, S.; Tsitsilonis, O.E.; Athanassios, K.; Ioannis, P.T. Oxidative Stress-mediated Biomolecular Damage and Inflammation in Tumorigenesis. In Vivo 2012, 26, 395–402. [Google Scholar]
- Melov, S.; Ravenscroft, J.; Malik, S.; Gill, M.S.; Walker, D.W.; Clayton, P.E.; Wallace, D.C.; Malfroy, B.; Doctrow, S.R.; Lithgow, G.J. Extension of life-span with superoxide dismutase/catalase mimetics. Science 2000, 289, 1567–1569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruan, H.; Tang, X.D.; Chen, M.L.; Joiner, M.L.; Sun, G.; Brot, N.; Weissbach, H.; Heinemann, S.H.; Iverson, L.; Wu, C.F.; et al. High-quality life extension by the enzyme peptide methionine sulfoxide reductase. Proc. Nat. Acad. Sci. USA 2002, 99, 2748–2753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, D.F.; Santana, L.F.; Vermulst, M.; Tomazela, D.M.; Emond, M.J.; MacCoss, M.J.; Gollahon, K.; Martin, G.M.; Loeb, L.A.; Ladiges, W.C.; et al. Overexpression of catalase targeted to mitochondria attenuates murine cardiac aging. Circulation 2009, 119, 2789–2797. [Google Scholar] [CrossRef]
- Ristow, M.; Schmeisser, S. Extending life span by increasing oxidative stress. Free Radic. Biol. Med. 2011, 51, 327–336. [Google Scholar] [CrossRef] [Green Version]
- Yun, J.; Finkel, T. Mitohormesis. Cell Metab. 2014, 19, 757–766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papaiahgari, S.; Zhang, Q.; Kleeberger, S.R.; Cho, H.Y.; Reddy, S.P. Hyperoxia stimulates an Nrf2-ARE transcriptional response via ROS-EGFR-PI3K-Akt/ERK MAP kinase signaling in pulmonary epithelial cells. Antioxid. Redox Sgnal. 2006, 8, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Ristow, M.; Schmeisser, K. Mitohormesis: Promoting Health and Lifespan by Increased Levels of Reactive Oxygen Species (ROS). Dose Response 2014, 12, 288–341. [Google Scholar] [CrossRef]
- Markaki, M.; Palikaras, K.; Tavernarakis, N. Novel Insights into the Anti-aging Role of Mitophagy. Int. Rev. Cell Mol. Biol. 2018, 340, 169–208. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, B.; Li, T.; Zhu, Y.; Luo, G.; Jiang, Y.; Tang, F.; Jian, Z.; Xiao, Y. AMPK activation serves a critical role in mitochondria quality control via modulating mitophagy in the heart under chronic hypoxia. Int. J. Mol. Med. 2018, 41, 69–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellegrino, M.W.; Haynes, C.M. Mitophagy and the mitochondrial unfolded protein response in neurodegeneration and bacterial infection. BMC Biol. 2015, 13, 22. [Google Scholar] [CrossRef] [Green Version]
- Jang, S.Y.; Kang, H.T.; Hwang, E.S. Nicotinamide-induced mitophagy: Event mediated by high NAD+/NADH ratio and SIRT1 protein activation. J. Biol. Chem. 2012, 287, 19304–19314. [Google Scholar] [CrossRef] [Green Version]
- Gilkerson, R.W.; De Vries, R.L.; Lebot, P.; Wikstrom, J.D.; Torgyekes, E.; Shirihai, O.S.; Przedborski, S.; Schon, E.A. Mitochondrial autophagy in cells with mtDNA mutations results from synergistic loss of transmembrane potential and mTORC1 inhibition. Hum. Mol. Genet. 2012, 21, 978–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardie, D.G.; Scott, J.W.; Pan, D.A.; Hudson, E.R. Management of cellular energy by the AMP-activated protein kinase system. FEBS Lett. 2003, 546, 113–120. [Google Scholar] [CrossRef]
- Woods, A.; Johnstone, S.R.; Dickerson, K.; Leiper, F.C.; Fryer, L.G.D.; Neumann, D.; Schlattner, U.; Wallimann, T.; Carlson, M.; Carling, D. LKB1 Is the Upstream Kinase in the AMP-Activated Protein Kinase Cascade. Curr. Biol. 2003, 13, 2004–2008. [Google Scholar] [CrossRef] [Green Version]
- Zong, H.; Ren, J.M.; Young, L.H.; Pypaert, M.; Mu, J.; Birnbaum, M.J.; Shulman, G.I. AMP kinase is required for mitochondrial biogenesis in skeletal muscle in response to chronic energy deprivation. Proc. Nat. Acad. Sci. USA 2002, 99, 15983–15987. [Google Scholar] [CrossRef] [Green Version]
- Song, S.B.; Hwang, E.S. A Rise in ATP, ROS, and Mitochondrial Content upon Glucose Withdrawal Correlates with a Dysregulated Mitochondria Turnover Mediated by the Activation of the Protein Deacetylase SIRT1. Cells 2018, 8, 11. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.S.; Lin, S.C. AMPK Promotes Autophagy by Facilitating Mitochondrial Fission. Cell Metab. 2016, 23, 399–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laker, R.C.; Drake, J.C.; Wilson, R.J.; Lira, V.A.; Lewellen, B.M.; Ryall, K.A.; Fisher, C.C.; Zhang, M.; Saucerman, J.J.; Goodyear, L.J.; et al. Ampk phosphorylation of Ulk1 is required for targeting of mitochondria to lysosomes in exercise-induced mitophagy. Nat. Commun. 2017, 8, 548. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Liang, B.; Shirwany, N.A.; Zou, M.-H. 2-Deoxy-D-Glucose Treatment of Endothelial Cells Induces Autophagy by Reactive Oxygen Species-Mediated Activation of the AMP-Activated Protein Kinase. PLoS ONE 2011, 6, e17234. [Google Scholar] [CrossRef] [Green Version]
- Jovaisaite, V.; Mouchiroud, L.; Auwerx, J. The mitochondrial unfolded protein response, a conserved stress response pathway with implications in health and disease. J. Exp. Biol. 2014, 217, 137–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mouchiroud, L.; Houtkooper, R.H.; Moullan, N.; Katsyuba, E.; Ryu, D.; Cantó, C.; Mottis, A.; Jo, Y.-S.; Viswanathan, M.; Schoonjans, K.; et al. The NAD(+)/sirtuin pathway modulates longevity through activation of mitochondrial UPR and FOXO signaling. Cell 2013, 154, 430–441. [Google Scholar] [CrossRef] [Green Version]
- Imaizumi, K.; Miyoshi, K.; Katayama, T.; Yoneda, T.; Taniguchi, M.; Kudo, T.; Tohyama, M. The unfolded protein response and Alzheimer’s disease. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2001, 1536, 85–96. [Google Scholar] [CrossRef] [Green Version]
- Scheper, W.; Hoozemans, J.J. The unfolded protein response in neurodegenerative diseases: A neuropathological perspective. Acta Neuropathol. 2015, 130, 315–331. [Google Scholar] [CrossRef] [Green Version]
- Hetz, C.; Papa, F.R. The Unfolded Protein Response and Cell Fate Control. Mol. Cell 2017, 69, 169–181. [Google Scholar] [CrossRef] [Green Version]
- Rose, G.; Dato, S.; Altomare, K.; Bellizzi, D.; Garasto, S.; Greco, V.; Passarino, G.; Feraco, E.; Mari, V.; Barbi, C.; et al. Variability of the SIRT3 gene, human silent information regulator Sir2 homologue, and survivorship in the elderly. Exp. Gerontol. 2003, 38, 1065–1070. [Google Scholar] [CrossRef]
- Bellizzi, D.; Rose, G.; Cavalcante, P.; Covello, G.; Dato, S.; De Rango, F.; Greco, V.; Maggiolini, M.; Feraco, E.; Mari, V.; et al. A novel VNTR enhancer within the SIRT3 gene, a human homologue of SIR2, is associated with survival at oldest ages. Genomics 2005, 85, 258–263. [Google Scholar] [CrossRef] [PubMed]
- Webster, B.R.; Lu, Z.; Sack, M.N.; Scott, I. The role of sirtuins in modulating redox stressors. Free Radic. Biol. Med. 2012, 52, 281–290. [Google Scholar] [CrossRef] [Green Version]
- Merksamer, P.I.; Liu, Y.; He, W.; Hirschey, M.D.; Chen, D.; Verdin, E. The sirtuins, oxidative stress and aging: An emerging link. Aging 2013, 5, 144–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmeisser, K.; Mansfeld, J.; Kuhlow, D.; Weimer, S.; Priebe, S.; Heiland, I.; Birringer, M.; Groth, M.; Segref, A.; Kanfi, Y.; et al. Role of sirtuins in lifespan regulation is linked to methylation of nicotinamide. Nat. Chem. Biol. 2013, 9, 693–700. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Song, M.Y.; Song, E.K.; Kim, E.K.; Moon, W.S.; Han, M.K.; Park, J.W.; Kwon, K.B.; Park, B.H. Overexpression of SIRT1 protects pancreatic beta-cells against cytokine toxicity by suppressing the nuclear factor-kappaB signaling pathway. Diabetes 2009, 58, 344–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodgers, J.T.; Lerin, C.; Haas, W.; Gygi, S.P.; Spiegelman, B.M.; Puigserver, P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature 2005, 434, 113–118. [Google Scholar] [CrossRef]
- Kim, H.S.; Patel, K.; Muldoon-Jacobs, K.; Bisht, K.S.; Aykin-Burns, N.; Pennington, J.D.; van der Meer, R.; Nguyen, P.; Savage, J.; Owens, K.M.; et al. SIRT3 is a mitochondria-localized tumor suppressor required for maintenance of mitochondrial integrity and metabolism during stress. Cancer Cell 2010, 17, 41–52. [Google Scholar] [CrossRef] [Green Version]
- Sundaresan, N.R.; Gupta, M.; Kim, G.; Rajamohan, S.B.; Isbatan, A.; Gupta, M.P. Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a-dependent antioxidant defense mechanisms in mice. J. Clin. Investig. 2009, 119, 2758–2771. [Google Scholar] [CrossRef] [Green Version]
- Samant, S.A.; Zhang, H.J.; Hong, Z.; Pillai, V.B.; Sundaresan, N.R.; Wolfgeher, D.; Archer, S.L.; Chan, D.C.; Gupta, M.P. SIRT3 Deacetylates and Activates OPA1 To Regulate Mitochondrial Dynamics during Stress. Mol. Cell. Biol. 2014, 34, 807–819. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Nguyen, M.; Qin, F.X.; Tong, Q. SIRT2 deacetylates FOXO3a in response to oxidative stress and caloric restriction. Aging Cell 2007, 6, 505–514. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.C.; Rabinovitch, P.S.; Kaeberlein, M. mTOR is a key modulator of ageing and age-related disease. Nature 2013, 493, 338–345. [Google Scholar] [CrossRef] [Green Version]
- Kaeberlein, M. Targeting mTOR signaling to promote healthy longevity. FASEB J. 2017, 31, 256.4. [Google Scholar] [CrossRef]
- Weichhart, T. mTOR as Regulator of Lifespan, Aging, and Cellular Senescence: A Mini-Review. Gerontology 2018, 64, 127–134. [Google Scholar] [CrossRef]
- Lamming, D.W.; Ye, L.; Katajisto, P.; Goncalves, M.D.; Saitoh, M.; Stevens, D.M.; Davis, J.G.; Salmon, A.B.; Richardson, A.; Ahima, R.S.; et al. Rapamycin-induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity. Science 2012, 335, 1638–1643. [Google Scholar] [CrossRef] [Green Version]
- Polak, P.; Cybulski, N.; Feige, J.N.; Auwerx, J.; Ruegg, M.A.; Hall, M.N. Adipose-specific knockout of raptor results in lean mice with enhanced mitochondrial respiration. Cell Metab. 2008, 8, 399–410. [Google Scholar] [CrossRef] [PubMed]
- Bartolomé, A.; García-Aguilar, A.; Asahara, S.-I.; Kido, Y.; Guillén, C.; Pajvani, U.B.; Benito, M. MTORC1 Regulates both General Autophagy and Mitophagy Induction after Oxidative Phosphorylation Uncoupling. Mol. Cell. Biol. 2017, 37, e00441-17. [Google Scholar] [CrossRef] [Green Version]
- Fontana, L.; Partridge, L.; Longo, V.D. Extending healthy life span—From yeast to humans. Science 2010, 328, 321–326. [Google Scholar] [CrossRef] [Green Version]
- Vendelbo, M.H.; Nair, K.S. Mitochondrial longevity pathways. Biochim. Biophys. Acta 2011, 1813, 634–644. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, S.; Sharma, S.; Agrawal, V.; Roy, N. Caloric restriction augments ROS defense in S. cerevisiae, by a Sir2p independent mechanism. Free Rad. Res. 2005, 39, 55–62. [Google Scholar] [CrossRef]
- Schulz, T.J.; Zarse, K.; Voigt, A.; Urban, N.; Birringer, M.; Ristow, M. Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab. 2007, 6, 280–293. [Google Scholar] [CrossRef] [Green Version]
- Sharma, P.K.; Agrawal, V.; Roy, N. Mitochondria-mediated hormetic response in life span extension of calorie-restricted Saccharomyces cerevisiae. Age 2011, 33, 143–154. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Dong, W.; Wang, R.; Li, Y.; Xu, B.; Zhang, J.; Zhao, Z.; Wang, Y. Effect of caloric restriction on the SIRT1/mTOR signaling pathways in senile mice. Brain Res. Bull. 2015, 116, 67–72. [Google Scholar] [CrossRef]
- Palacios, O.M.; Carmona, J.J.; Michan, S.; Chen, K.Y.; Manabe, Y.; Ward, J.L., 3rd; Goodyear, L.J.; Tong, Q. Diet and exercise signals regulate SIRT3 and activate AMPK and PGC-1alpha in skeletal muscle. Aging 2009, 1, 771–783. [Google Scholar] [CrossRef]
- Lanza, I.R.; Zabielski, P.; Klaus, K.A.; Morse, D.M.; Heppelmann, C.J.; Bergen, H.R.; Dasari, S.; Walrand, S.; Short, K.R.; Johnson, M.L.; et al. Chronic Caloric Restriction Preserves Mitochondrial Function in Senescence Without Increasing Mitochondrial Biogenesis. Cell Metab. 2012, 16, 777–788. [Google Scholar] [CrossRef] [Green Version]
- Ferreira-Marques, M.; Aveleira, C.A.; Carmo-Silva, S.; Botelho, M.; Pereira de Almeida, L.; Cavadas, C. Caloric restriction stimulates autophagy in rat cortical neurons through neuropeptide Y and ghrelin receptors activation. Aging 2016, 8, 1470–1484. [Google Scholar] [CrossRef] [Green Version]
- Cui, J.; Shi, S.; Sun, X.; Cai, G.; Cui, S.; Hong, Q.; Chen, X.; Bai, X.-Y. Mitochondrial Autophagy Involving Renal Injury and Aging Is Modulated by Caloric Intake in Aged Rat Kidneys. PLoS ONE 2013, 8, e69720. [Google Scholar] [CrossRef] [Green Version]
- Arumugam, T.V.; Gleichmann, M.; Tang, S.C.; Mattson, M.P. Hormesis/preconditioning mechanisms, the nervous system and aging. Ageing Res. Rev. 2006, 5, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D. Physical activity is medicine for older adults. Postgrad. Med. J. 2014, 90, 26–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLeod, M.; Breen, L.; Hamilton, D.L.; Philp, A. Live strong and prosper: The importance of skeletal muscle strength for healthy ageing. Biogerontology 2016, 17, 497–510. [Google Scholar] [CrossRef] [Green Version]
- McPhee, J.S.; French, D.P.; Jackson, D.; Nazroo, J.; Pendleton, N.; Degens, H. Physical activity in older age: Perspectives for healthy ageing and frailty. Biogerontology 2016, 17, 567–580. [Google Scholar] [CrossRef]
- Cheng, A.; Yang, Y.; Zhou, Y.; Maharana, C.; Lu, D.; Peng, W.; Liu, Y.; Wan, R.; Marosi, K.; Misiak, M.; et al. Mitochondrial SIRT3 Mediates Adaptive Responses of Neurons to Exercise, and Metabolic and Excitatory Challenges. Cell Metab. 2016, 23, 128–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baar, K. Involvement of PPAR gamma co-activator-1, nuclear respiratory factors 1 and 2, and PPAR alpha in the adaptive response to endurance exercise. Proc. Nutr. Soc. 2004, 63, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Akimoto, T.; Pohnert, S.C.; Li, P.; Zhang, M.; Gumbs, C.; Rosenberg, P.B.; Williams, R.S.; Yan, Z. Exercise stimulates Pgc-1alpha transcription in skeletal muscle through activation of the p38 MAPK pathway. J. Biol. Chem. 2005, 280, 19587–19593. [Google Scholar] [CrossRef] [Green Version]
- Philp, A.; Chen, A.; Lan, D.; Meyer, G.A.; Murphy, A.N.; Knapp, A.E.; Olfert, I.M.; McCurdy, C.E.; Marcotte, G.R.; Hogan, M.C.; et al. Sirtuin 1 (SIRT1) deacetylase activity is not required for mitochondrial biogenesis or peroxisome proliferator-activated receptor-gamma coactivator-1alpha (PGC-1alpha) deacetylation following endurance exercise. J. Biol. Chem. 2011, 286, 30561–30570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atherton, P.J.; Babraj, J.; Smith, K.; Singh, J.; Rennie, M.J.; Wackerhage, H. Selective activation of AMPK-PGC-1alpha or PKB-TSC2-mTOR signaling can explain specific adaptive responses to endurance or resistance training-like electrical muscle stimulation. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2005, 19, 786–788. [Google Scholar] [CrossRef]
- Arbogast, S.; Reid, M.B. Oxidant activity in skeletal muscle fibers is influenced by temperature, CO2 level, and muscle-derived nitric oxide. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004, 287, R698–R705. [Google Scholar] [CrossRef]
- St-Pierre, J.; Drori, S.; Uldry, M.; Silvaggi, J.M.; Rhee, J.; Jager, S.; Handschin, C.; Zheng, K.; Lin, J.; Yang, W.; et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell 2006, 127, 397–408. [Google Scholar] [CrossRef] [Green Version]
- Tam, B.T.; Siu, P.M. Autophagic cellular responses to physical exercise in skeletal muscle. Sports Med. 2014, 44, 625–640. [Google Scholar] [CrossRef]
- Saleem, A.; Carter, H.N.; Hood, D.A. p53 is necessary for the adaptive changes in cellular milieu subsequent to an acute bout of endurance exercise. American journal of physiology. Cell Physiol. 2014, 306, C241–C249. [Google Scholar] [CrossRef] [Green Version]
- Drake, J.C.; Wilson, R.J.; Yan, Z. Molecular mechanisms for mitochondrial adaptation to exercise training in skeletal muscle. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2016, 30, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Schwalm, C.; Deldicque, L.; Francaux, M. Lack of Activation of Mitophagy during Endurance Exercise in Human. Med. Sci. Sports Exerc. 2017, 49, 1552–1561. [Google Scholar] [CrossRef] [PubMed]
- Ogborn, D.I.; McKay, B.R.; Crane, J.D.; Safdar, A.; Akhtar, M.; Parise, G.; Tarnopolsky, M.A. Effects of age and unaccustomed resistance exercise on mitochondrial transcript and protein abundance in skeletal muscle of men. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2015, 308, R734–R741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- May, J.M. Vitamin C transport and its role in the central nervous system. Sub Cell. Biochem. 2012, 56, 85–103. [Google Scholar] [CrossRef] [Green Version]
- Nualart, F.; Mack, L.; García, A.; Cisternas, P.; Bongarzone, E.R.; Heitzer, M.; Jara, N.; Martínez, F.; Ferrada, L.; Espinoza, F.; et al. Vitamin C Transporters, Recycling and the Bystander Effect in the Nervous System: SVCT2 versus Gluts. J. Stem Cell Res. Ther. 2014, 4, 209. [Google Scholar] [CrossRef] [Green Version]
- Bolanos, J.P.; Almeida, A. The pentose-phosphate pathway in neuronal survival against nitrosative stress. IUBMB Life 2010, 62, 14–18. [Google Scholar] [CrossRef]
- Shefa, U.; Jeong, N.Y.; Song, I.O.; Chung, H.-J.; Kim, D.; Jung, J.; Huh, Y. Mitophagy links oxidative stress conditions and neurodegenerative diseases. Neural. Regen. Res. 2019, 14, 749–756. [Google Scholar] [CrossRef]
- Fan, P.; Xie, X.-H.; Chen, C.-H.; Peng, X.; Zhang, P.; Yang, C.; Wang, Y.-T. Molecular Regulation Mechanisms and Interactions Between Reactive Oxygen Species and Mitophagy. DNA Cell Biol. 2018, 38, 10–22. [Google Scholar] [CrossRef]
- Apetz, N.; Munch, G.; Govindaraghavan, S.; Gyengesi, E. Natural compounds and plant extracts as therapeutics against chronic inflammation in Alzheimer’s disease—A translational perspective. CNS Neurol. Dis. Drug Targets 2014, 13, 1175–1191. [Google Scholar] [CrossRef]
- Reddy, V.P.; Aryal, P.; Robinson, S.; Rafiu, R.; Obrenovich, M.; Perry, G. Polyphenols in Alzheimer’s Disease and in the Gut-Brain Axis. Microorganisms 2020, 8, 199. [Google Scholar] [CrossRef] [Green Version]
- Pandareesh, M.D.; Mythri, R.B.; Srinivas Bharath, M.M. Bioavailability of dietary polyphenols: Factors contributing to their clinical application in CNS diseases. Neurochem. Int. 2015, 89, 198–208. [Google Scholar] [CrossRef]
- Figueira, I.; Garcia, G.; Pimpão, R.C.; Terrasso, A.P.; Costa, I.; Almeida, A.F.; Tavares, L.; Pais, T.F.; Pinto, P.; Ventura, M.R.; et al. Polyphenols journey through blood-brain barrier towards neuronal protection. Sci. Rep. 2017, 7, 11456. [Google Scholar] [CrossRef]
- Garcia-Alloza, M.; Borrelli, L.A.; Rozkalne, A.; Hyman, B.T.; Bacskai, B.J. Curcumin labels amyloid pathology in vivo, disrupts existing plaques, and partially restores distorted neurites in an Alzheimer mouse model. J. Neurochem. 2007, 102, 1095–1104. [Google Scholar] [CrossRef] [PubMed]
- Maiti, P.; Dunbar, G.L. Comparative Neuroprotective Effects of Dietary Curcumin and Solid Lipid Curcumin Particles in Cultured Mouse Neuroblastoma Cells after Exposure to Abeta42. Int. J. Alzheimer’s Dis. 2017, 2017, 4164872. [Google Scholar] [CrossRef] [Green Version]
- Draczynska-Lusiak, B.; Doung, A.; Sun, A.Y. Oxidized lipoproteins may play a role in neuronal cell death in Alzheimer disease. Mol. Chem. Neuropathol. 1998, 33, 139–148. [Google Scholar] [CrossRef]
- Ma, T.; Tan, M.-S.; Yu, J.-T.; Tan, L. Resveratrol as a Therapeutic Agent for Alzheimer’s Disease. BioMed. Res. Int. 2014, 2014, 13. [Google Scholar] [CrossRef]
- Sawda, C.; Moussa, C.; Turner, R.S. Resveratrol for Alzheimer’s disease. Ann. N. Y. Acad. Sci. 2017, 1403, 142–149. [Google Scholar] [CrossRef]
- Jung, S.H.; Murphy, E.A.; McClellan, J.L.; Carmichael, M.D.; Davis, J.M. The dietary flavonoid quercetin decreases neuroinflammation in a mouse model of Alzheimer’s disease. FASEB J. 2010, 24, 604.17. [Google Scholar] [CrossRef]
- Maria, S.-G.A.; Ignacio, M.-M.J.; Ramírez-Pineda Jose, R.; Marisol, L.-R.; Edison, O.; Patricia, C.-G.G. The flavonoid quercetin ameliorates Alzheimer’s disease pathology and protects cognitive and emotional function in aged triple transgenic Alzheimer’s disease model mice. Neuropharmacology 2015, 93, 134–145. [Google Scholar] [CrossRef] [Green Version]
- Singh, N.A.; Mandal, A.K.; Khan, Z.A. Potential neuroprotective properties of epigallocatechin-3-gallate (EGCG). Nutr. J. 2016, 15, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Currais, A.; Prior, M.; Dargusch, R.; Armando, A.; Ehren, J.; Schubert, D.; Maher, P. Modulation of p25 and inflammatory pathways by fisetin maintains cognitive function in Alzheimer’s disease transgenic mice. Aging Cell 2014, 13, 379–390. [Google Scholar] [CrossRef]
- Sandoval-Acuna, C.; Ferreira, J.; Speisky, H. Polyphenols and mitochondria: An update on their increasingly emerging ROS-scavenging independent actions. Arch. Biochem. Biophys. 2014, 559, 75–90. [Google Scholar] [CrossRef]
- Ryu, D.; Mouchiroud, L.; Andreux, P.A.; Katsyuba, E.; Moullan, N.; Nicolet-Dit-Félix, A.A.; Williams, E.G.; Jha, P.; Lo Sasso, G.; Huzard, D.; et al. Urolithin A induces mitophagy and prolongs lifespan in C. elegans and increases muscle function in rodents. Nat. Med. 2016, 22. [Google Scholar] [CrossRef] [PubMed]
- Amakura, Y.; Okada, M.; Tsuji, S.; Tonogai, Y. Determination of phenolic acids in fruit juices by isocratic column liquid chromatography. J. Chromatogr. A 2000, 891, 183–188. [Google Scholar] [CrossRef]
- Fischer, U.A.; Carle, R.; Kammerer, D.R. Identification and quantification of phenolic compounds from pomegranate (Punica granatum L.) peel, mesocarp, aril and differently produced juices by HPLC-DAD–ESI/MSn. Food Chem. 2011, 127, 807–821. [Google Scholar] [CrossRef] [PubMed]
- Gil, M.I.; Tomas-Barberan, F.A.; Hess-Pierce, B.; Holcroft, D.M.; Kader, A.A. Antioxidant activity of pomegranate juice and its relationship with phenolic composition and processing. J. Agric. Food Chem. 2000, 48, 4581–4589. [Google Scholar] [CrossRef]
- Gil-Izquierdo, A.; Zafrilla, P.; Tomás-Barberán, F.A. An in vitro method to simulate phenolic compound release from the food matrix in the gastrointestinal tract. Euro. Food Res. Technol. 2002, 214, 155–159. [Google Scholar] [CrossRef]
- Larrosa, M.; Tomas-Barberan, F.A.; Espin, J.C. The dietary hydrolysable tannin punicalagin releases ellagic acid that induces apoptosis in human colon adenocarcinoma Caco-2 cells by using the mitochondrial pathway. J. Nutr. Biochem. 2006, 17, 611–625. [Google Scholar] [CrossRef]
- Espin, J.C.; Gonzalez-Barrio, R.; Cerda, B.; Lopez-Bote, C.; Rey, A.I.; Tomas-Barberan, F.A. Iberian pig as a model to clarify obscure points in the bioavailability and metabolism of ellagitannins in humans. J. Agric. Food Chem. 2007, 55, 10476–10485. [Google Scholar] [CrossRef]
- Walker, A.W.; Duncan, S.H.; Louis, P.; Flint, H.J. Phylogeny, culturing, and metagenomics of the human gut microbiota. Trends Microbiol. 2014, 22, 267–274. [Google Scholar] [CrossRef]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [Green Version]
- D’Argenio, V.; Salvatore, F. The role of the gut microbiome in the healthy adult status. Clin. Chim. Acta 2015, 451, 97–102. [Google Scholar] [CrossRef] [Green Version]
- Onoue, M.; Kado, S.; Sakaitani, Y.; Uchida, K.; Morotomi, M. Specific species of intestinal bacteria influence the induction of aberrant crypt foci by 1,2-dimethylhydrazine in rats. Cancer Lett. 1997, 113, 179–186. [Google Scholar] [CrossRef]
- Vogt, N.M.; Kerby, R.L.; Dill-McFarland, K.A.; Harding, S.J.; Merluzzi, A.P.; Johnson, S.C.; Carlsson, C.M.; Asthana, S.; Zetterberg, H.; Blennow, K.; et al. Gut microbiome alterations in Alzheimer’s disease. Sci. Rep. 2017, 7, 13537. [Google Scholar] [CrossRef]
- Hu, X.; Wang, T.; Jin, F. Alzheimer’s disease and gut microbiota. Sci. China Life Sci. 2016, 59, 1006–1023. [Google Scholar] [CrossRef] [Green Version]
- Solfrizzi, V.; Panza, F.; Frisardi, V.; Seripa, D.; Logroscino, G.; Imbimbo, B.P.; Pilotto, A. Diet and Alzheimer’s disease risk factors or prevention: The current evidence. Exp. Rev. Neurother. 2011, 11, 677–708. [Google Scholar] [CrossRef]
- Hu, N.; Yu, J.-T.; Tan, L.; Wang, Y.-L.; Sun, L.; Tan, L. Nutrition and the Risk of Alzheimer’s Disease. BioMed Res. Int. 2013, 2013, 524820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.C.; Jenner, A.M.; Low, C.S.; Lee, Y.K. Effect of tea phenolics and their aromatic fecal bacterial metabolites on intestinal microbiota. Res. Microbiol. 2006, 157, 876–884. [Google Scholar] [CrossRef] [PubMed]
- De Vadder, F.; Kovatcheva-Datchary, P.; Goncalves, D.; Vinera, J.; Zitoun, C.; Duchampt, A.; Bäckhed, F.; Mithieux, G. Microbiota-Generated Metabolites Promote Metabolic Benefits via Gut-Brain Neural Circuits. Cell 2014, 156, 84–96. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Villalba, R.; Beltran, D.; Espin, J.C.; Selma, M.V.; Tomas-Barberan, F.A. Time course production of urolithins from ellagic acid by human gut microbiota. J. Agric. Food Chem. 2013, 61, 8797–8806. [Google Scholar] [CrossRef] [PubMed]
- Selma, M.V.; Beltran, D.; Garcia-Villalba, R.; Espin, J.C.; Tomas-Barberan, F.A. Description of urolithin production capacity from ellagic acid of two human intestinal Gordonibacter species. Food Funct. 2014, 5, 1779–1784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bialonska, D.; Ramnani, P.; Kasimsetty, S.G.; Muntha, K.R.; Gibson, G.R.; Ferreira, D. The influence of pomegranate by-product and punicalagins on selected groups of human intestinal microbiota. Int. J. Food Microbiol. 2010, 140, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Ito, H.; Iguchi, A.; Hatano, T. Identification of urinary and intestinal bacterial metabolites of ellagitannin geraniin in rats. J. Agric. Food Chem. 2008, 56, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-F.; Li, X.-L.; Zhang, Z.-L.; Qiu, L.; Ding, S.-X.; Xue, J.-X.; Zhao, G.-P.; Li, J. Antiaging Effects of Urolithin A on Replicative Senescent Human Skin Fibroblasts. Rejuvenation Res. 2018, 22, 191–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, J.; Pierre, N.; Naslain, D.; Bontemps, F.; Ferreira, D.; Priem, F.; Deldicque, L.; Francaux, M. Urolithin B, a newly identified regulator of skeletal muscle mass. J. Cachexia Sarcopenia Muscle 2017. [Google Scholar] [CrossRef] [PubMed]
- Kang, I.; Tomás-Barberán, F.; Carlos Espín, J.; Chung, S. Urolithin C, a Gut Microbiota Metabolite Derived from Ellagic Acid, Attenuates Triglyceride Accumulation in Human Adipocytes and Hepatoma Huh7 Cells. FASEB J. 2015, 29, 130.1. [Google Scholar] [CrossRef]
- Piwowarski, J.P.; Granica, S.; Stefanska, J.; Kiss, A.K. Differences in Metabolism of Ellagitannins by Human Gut Microbiota ex Vivo Cultures. J. Nat. Prod. 2016, 79, 3022–3030. [Google Scholar] [CrossRef]
- Li, Z.; Henning, S.M.; Lee, R.P.; Lu, Q.Y.; Summanen, P.H.; Thames, G.; Corbett, K.; Downes, J.; Tseng, C.H.; Finegold, S.M.; et al. Pomegranate extract induces ellagitannin metabolite formation and changes stool microbiota in healthy volunteers. Food Funct. 2015, 6, 2487–2495. [Google Scholar] [CrossRef]
- Tomas-Barberan, F.A.; Garcia-Villalba, R.; Gonzalez-Sarrias, A.; Selma, M.V.; Espin, J.C. Ellagic acid metabolism by human gut microbiota: Consistent observation of three urolithin phenotypes in intervention trials, independent of food source, age, and health status. J. Agric. Food Chem. 2014, 62, 6535–6538. [Google Scholar] [CrossRef]
- Romo-Vaquero, M.; Cortés-Martín, A.; Loria-Kohen, V.; Ramírez-de-Molina, A.; García-Mantrana, I.; Collado, M.C.; Espín, J.C.; Selma, M.V. Deciphering the Human Gut Microbiome of Urolithin Metabotypes: Association with Enterotypes and Potential Cardiometabolic Health Implications. Mol. Nutr. Food Res. 2019, 63, 1800958. [Google Scholar] [CrossRef]
- Selma, M.V.; Gonzalez-Sarrias, A.; Salas-Salvado, J.; Andres-Lacueva, C.; Alasalvar, C.; Orem, A.; Tomas-Barberan, F.A.; Espin, J.C. The gut microbiota metabolism of pomegranate or walnut ellagitannins yields two urolithin-metabotypes that correlate with cardiometabolic risk biomarkers: Comparison between normoweight, overweight-obesity and metabolic syndrome. Clin. Nutr. 2017. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Villalba, R.; Selma, M.V.; Espin, J.C.; Tomas-Barberan, F.A. Identification of Novel Urolithin Metabolites in Human Feces and Urine after the Intake of a Pomegranate Extract. J. Agric. Food Chem. 2019, 67, 11099–11107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Espín, J.C.; Larrosa, M.; García-Conesa, M.T.; Tomás-Barberán, F. Biological significance of urolithins, the gut microbial ellagic acid-derived metabolites: The evidence so far. Evid.-Based Complement. Altern. Med. 2013, 2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerda, B.; Llorach, R.; Ceron, J.J.; Espin, J.C.; Tomas-Barberan, F.A. Evaluation of the bioavailability and metabolism in the rat of punicalagin, an antioxidant polyphenol from pomegranate juice. Eur. J. Nutr. 2003, 42, 18–28. [Google Scholar] [CrossRef]
- Larrosa, M.; Gonzalez-Sarrias, A.; Garcia-Conesa, M.T.; Tomas-Barberan, F.A.; Espin, J.C. Urolithins, ellagic acid-derived metabolites produced by human colonic microflora, exhibit estrogenic and antiestrogenic activities. J. Agric. Food Chem. 2006, 54, 1611–1620. [Google Scholar] [CrossRef]
- Heilman, J.; Andreux, P.; Tran, N.; Rinsch, C.; Blanco-Bose, W. Safety assessment of Urolithin A, a metabolite produced by the human gut microbiota upon dietary intake of plant derived ellagitannins and ellagic acid. Food Chem. Toxicol. Biol. Res. Assoc. 2017, 108, 289–297. [Google Scholar] [CrossRef]
- Bobowska, A.; Granica, S.; Filipek, A.; Melzig, M.F.; Moeslinger, T.; Zentek, J.; Kruk, A.; Piwowarski, J.P. Comparative studies of urolithins and their phase II metabolites on macrophage and neutrophil functions. Eur. J. Nutr. 2021, 60, 1957–1972. [Google Scholar] [CrossRef]
- Bayle, M.; Roques, C.; Marion, B.; Audran, M.; Oiry, C.; Bressolle-Gomeni, F.M.; Cros, G. Development and validation of a liquid chromatography-electrospray ionization-tandem mass spectrometry method for the determination of urolithin C in rat plasma and its application to a pharmacokinetic study. J. Pharm. Biomed. Anal. 2016, 131, 33–39. [Google Scholar] [CrossRef]
- Yuan, T.; Ma, H.; Liu, W.; Niesen, D.B.; Shah, N.; Crews, R.; Rose, K.N.; Vattem, D.A.; Seeram, N.P. Pomegranate’s Neuroprotective Effects against Alzheimer’s Disease Are Mediated by Urolithins, Its Ellagitannin-Gut Microbial Derived Metabolites. ACS Chem. Neurosci. 2016, 7, 26–33. [Google Scholar] [CrossRef]
- Gasperotti, M.; Passamonti, S.; Tramer, F.; Masuero, D.; Guella, G.; Mattivi, F.; Vrhovsek, U. Fate of microbial metabolites of dietary polyphenols in rats: Is the brain their target destination? ACS Chem. Neurosci. 2015, 6, 1341–1352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kujawska, M.; Jourdes, M.; Kurpik, M.; Szulc, M.; Szaefer, H.; Chmielarz, P.; Kreiner, G.; Krajka-Kuźniak, V.; Mikołajczak, P.; Teissedre, P.L.; et al. Neuroprotective Effects of Pomegranate Juice against Parkinson’s Disease and Presence of Ellagitannins-Derived Metabolite-Urolithin A-In the Brain. Int. J. Mol. Sci. 2020, 21, 202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreux, P.A.; Blanco-Bose, W.; Ryu, D.; Burdet, F.; Ibberson, M.; Aebischer, P.; Auwerx, J.; Singh, A.; Rinsch, C. The mitophagy activator urolithin A is safe and induces a molecular signature of improved mitochondrial and cellular health in humans. Nat. Metab. 2019, 1, 595–603. [Google Scholar] [CrossRef] [PubMed]
- Patel, C.; Dadhaniya, P.; Hingorani, L.; Soni, M.G. Safety assessment of pomegranate fruit extract: Acute and subchronic toxicity studies. Food Chem. Toxicol. 2008, 46, 2728–2735. [Google Scholar] [CrossRef] [PubMed]
- Johanningsmeier, S.D.; Harris, G.K. Pomegranate as a functional food and nutraceutical source. Annu. Rev. Food Sci. Technol. 2011, 2, 181–201. [Google Scholar] [CrossRef] [PubMed]
- Gramec Skledar, D.; Tomasic, T.; Sollner Dolenc, M.; Peterlin Masic, L.; Zega, A. Evaluation of endocrine activities of ellagic acid and urolithins using reporter gene assays. Chemosphere 2019, 220, 706–713. [Google Scholar] [CrossRef] [PubMed]
- Savi, M.; Bocchi, L.; Mena, P.; Dall’Asta, M.; Crozier, A.; Brighenti, F.; Stilli, D.; Del Rio, D. In vivo administration of urolithin A and B prevents the occurrence of cardiac dysfunction in streptozotocin-induced diabetic rats. Cardiovasc. Diabetol. 2017, 16, 80. [Google Scholar] [CrossRef] [PubMed]
- Kasimsetty, S.G.; Bialonska, D.; Reddy, K.K.; Ma, G.; Khan, S.I.; Ferreira, D. Colon Cancer Chemopreventive Activities of Pomegranate Ellagitannins and Urolithins. J. Agric. Food Chem. 2010, 58, 2180–2187. [Google Scholar] [CrossRef]
- Qiu, Z.; Zhou, B.; Jin, L.; Yu, H.; Liu, L.; Liu, Y.; Zhu, F. In vitro antioxidant and antiproliferative effects of ellagic acid and its colonic metabolite, urolithins, on human bladder cancer T24 cells. Food Chem. Toxicol. 2013, 59, 428–437. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Shi, F.; Guo, Z.; Zhao, J.; Song, X.; Yang, H. Metabolite of ellagitannins, urolithin A induces autophagy and inhibits metastasis in human sw620 colorectal cancer cells. Mol. Carcinog. 2018, 57, 193–200. [Google Scholar] [CrossRef] [Green Version]
- Yin, P.; Zhang, J.; Yan, L.; Yang, L.; Sun, L.; Shi, L.; Ma, C.; Liu, Y. Urolithin C, a gut metabolite of ellagic acid, induces apoptosis in PC12 cells through a mitochondria-mediated pathway. RSC Adv. 2017, 7, 17254–17263. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Sarrias, A.; Gimenez-Bastida, J.A.; Nunez-Sanchez, M.A.; Larrosa, M.; Garcia-Conesa, M.T.; Tomas-Barberan, F.A.; Espin, J.C. Phase-II metabolism limits the antiproliferative activity of urolithins in human colon cancer cells. Eur. J. Nutr. 2014, 53, 853–864. [Google Scholar] [CrossRef]
- Colombo, E.; Sangiovanni, E.; Dell’agli, M. A review on the anti-inflammatory activity of pomegranate in the gastrointestinal tract. Evid. Based Complement. Altern. Med. 2013, 2013, 247145. [Google Scholar] [CrossRef] [Green Version]
- Larrosa, M.; Gonzalez-Sarrias, A.; Yanez-Gascon, M.J.; Selma, M.V.; Azorin-Ortuno, M.; Toti, S.; Tomas-Barberan, F.; Dolara, P.; Espin, J.C. Anti-inflammatory properties of a pomegranate extract and its metabolite urolithin-A in a colitis rat model and the effect of colon inflammation on phenolic metabolism. J. Nutr. Biochem. 2010, 21, 717–725. [Google Scholar] [CrossRef] [PubMed]
- Neyrinck, A.M.; Van Hee, V.F.; Bindels, L.B.; De Backer, F.; Cani, P.D.; Delzenne, N.M. Polyphenol-rich extract of pomegranate peel alleviates tissue inflammation and hypercholesterolaemia in high-fat diet-induced obese mice: Potential implication of the gut microbiota. Br. J. Nutr. 2013, 109, 802–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, A.; Andreux, P.; Blanco, W.; Auwerx, J.; Rinsch, C. Urolithin A, a Gut Microbiome Derived Metabolite Improves Mitochondrial and Cellular Health: Results from a Randomized, Placebo-controlled, Double-blind Clinical Trial (FS09-06-19). Curr. Dev. Nutr. 2019, 3, FS09-06-19. [Google Scholar] [CrossRef]
- Kwak, H.M.; Jeon, S.Y.; Sohng, B.H.; Kim, J.G.; Lee, J.M.; Lee, K.B.; Jeong, H.H.; Hur, J.M.; Kang, Y.H.; Song, K.S. J3-Secretase (BACE1) Inhibitors from Pomegranate (Punica granatum) Husk. Arch. Pharm. Res. 2005, 28, 1328–1332. [Google Scholar] [CrossRef] [PubMed]
- Youn, K.; Jun, M. In vitro BACE1 inhibitory activity of geraniin and corilagin from Geranium thunbergii. Planta Med. 2013, 79, 1038–1042. [Google Scholar] [CrossRef] [Green Version]
- Mele, L.; Mena, P.; Piemontese, A.; Marino, V.; Lopez-Gutierrez, N.; Bernini, F.; Brighenti, F.; Zanotti, I.; Del Rio, D. Antiatherogenic effects of ellagic acid and urolithins in vitro. Arch. Biochem. Biophys. 2016, 599, 42–50. [Google Scholar] [CrossRef]
- Olajide, O.A.; Kumar, A.; Velagapudi, R.; Okorji, U.P.; Fiebich, B.L. Punicalagin inhibits neuroinflammation in LPS-activated rat primary microglia. Mol. Nutr. Food Res. 2014, 58, 1843–1851. [Google Scholar] [CrossRef]
- Velagapudi, R.; Baco, G.; Khela, S.; Okorji, U.; Olajide, O. Pomegranate inhibits neuroinflammation and amyloidogenesis in IL-1beta-stimulated SK-N-SH cells. Eur. J. Nutr. 2016, 55, 1653–1660. [Google Scholar] [CrossRef] [Green Version]
- Forouzanfar, F.; Afkhami Goli, A.; Asadpour, E.; Ghorbani, A.; Sadeghnia, H.R. Protective Effect of Punica granatum L. against Serum/Glucose Deprivation-Induced PC12 Cells Injury. Evid. Based Complement. Altern. Med. 2013, 2013, 716730. [Google Scholar] [CrossRef] [PubMed]
- Morzelle, M.C.; Salgado, J.M.; Telles, M.; Mourelle, D.; Bachiega, P.; Buck, H.S.; Viel, T.A. Neuroprotective Effects of Pomegranate Peel Extract after Chronic Infusion with Amyloid-beta Peptide in Mice. PLoS ONE 2016, 11, e0166123. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.J.; Lee, J.H.; Heo, H.J.; Cho, H.Y.; Kim, H.K.; Kim, C.J.; Kim, M.O.; Suh, S.H.; Shin, D.H. Punica granatum protects against oxidative stress in PC12 cells and oxidative stress-induced Alzheimer’s symptoms in mice. J. Med. Food 2011, 14, 695–701. [Google Scholar] [CrossRef]
- Subash, S.; Essa, M.M.; Al-Asmi, A.; Al-Adawi, S.; Vaishnav, R.; Braidy, N.; Manivasagam, T.; Guillemin, G.J. Pomegranate from Oman Alleviates the Brain Oxidative Damage in Transgenic Mouse Model of Alzheimer’s disease. J. Trad. Complement. Med. 2014, 4, 232–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verzelloni, E.; Pellacani, C.; Tagliazucchi, D.; Tagliaferri, S.; Calani, L.; Costa, L.G.; Brighenti, F.; Borges, G.; Crozier, A.; Conte, A.; et al. Antiglycative and neuroprotective activity of colon-derived polyphenol catabolites. Mol. Nutr. Food Res. 2011, 55, S35–S43. [Google Scholar] [CrossRef]
- DaSilva, N.A.; Nahar, P.P.; Ma, H.; Eid, A.; Wei, Z.; Meschwitz, S.; Zawia, N.H.; Slitt, A.L.; Seeram, N.P. Pomegranate ellagitannin-gut microbial-derived metabolites, urolithins, inhibit neuroinflammation in vitro. Nutr. Neurosci. 2017, 1–11. [Google Scholar] [CrossRef]
- Xu, J.; Yuan, C.; Wang, G.; Luo, J.; Ma, H.; Xu, L.; Mu, Y.; Li, Y.; Seeram, N.P.; Huang, X.; et al. Urolithins Attenuate LPS-Induced Neuroinflammation in BV2Microglia via MAPK, Akt, and NF-κB Signaling Pathways. J. Agric. Food Chem. 2018, 66, 571–580. [Google Scholar] [CrossRef]
- Bialonska, D.; Kasimsetty, S.G.; Khan, S.I.; Ferreira, D. Urolithins, intestinal microbial metabolites of Pomegranate ellagitannins, exhibit potent antioxidant activity in a cell-based assay. J. Agric. Food Chem. 2009, 57, 10181–10186. [Google Scholar] [CrossRef]
- Xu, L.; He, S.; Yin, P.; Li, D.; Mei, C.; Yu, X.; Shi, Y.; Jiang, L.; Liu, F. Punicalagin induces Nrf2 translocation and HO-1 expression via PI3K/Akt, protecting rat intestinal epithelial cells from oxidative stress. Int. J. Hyperth. 2016, 32, 465–473. [Google Scholar] [CrossRef]
- Akiyama, H.; Barger, S.; Barnum, S.; Bradt, B.; Bauer, J.; Cole, G.M.; Cooper, N.R.; Eikelenboom, P.; Emmerling, M.; Fiebich, B.L.; et al. Inflammation and Alzheimer’s disease. Neurobiol. Aging 2000, 21, 383–421. [Google Scholar] [CrossRef]
- Wyss-Coray, T.; Rogers, J. Inflammation in Alzheimer Disease—A Brief Review of the Basic Science and Clinical Literature. Cold Spring Harb. Perspect. Med. 2012, 2, a006346. [Google Scholar] [CrossRef] [PubMed]
- Hamid Reza Rahimia, M.A.a.S.N.O. A Comprehensive Review of Punica granatum (Pomegranate) Properties in Toxicological, Pharmacological, Cellular and Molecular Biology Researches. Iran. J. Pharm. Res. 2012, 11, 385–400. [Google Scholar]
- Green, P.S.; Simpkins, J.W. Neuroprotective effects of estrogens: Potential mechanisms of action. Int. J. Dev. Neurosci. Off. J. Int. Soc. Develop. Neurosci. 2000, 18, 347–358. [Google Scholar] [CrossRef]
- Brann, D.W.; Dhandapani, K.; Wakade, C.; Mahesh, V.B.; Khan, M.M. Neurotrophic and Neuroprotective Actions of Estrogen: Basic Mechanisms and Clinical Implications. Steroids 2007, 72, 381–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papoutsi, Z.; Kassi, E.; Tsiapara, A.; Fokialakis, N.; Chrousos, G.P.; Moutsatsou, P. Evaluation of estrogenic/antiestrogenic activity of ellagic acid via the estrogen receptor subtypes ERalpha and ERbeta. J. Agric. Food Chem. 2005, 53, 7715–7720. [Google Scholar] [CrossRef]
- Zhang, W.; Chen, J.H.; Aguilera-Barrantes, I.; Shiau, C.W.; Sheng, X.; Wang, L.S.; Stoner, G.D.; Huang, Y.W. Urolithin A suppresses the proliferation of endometrial cancer cells by mediating estrogen receptor-alpha-dependent gene expression. Mol. Nutr. Food Res. 2016, 60, 2387–2395. [Google Scholar] [CrossRef] [PubMed]
- Rojo, L.; Sjöberg, M.K.; Hernández, P.; Zambrano, C.; Maccioni, R.B. Roles of Cholesterol and Lipids in the Etiopathogenesis of Alzheimer’s Disease. J. Biomed. Biotechnol. 2006, 2006, 73976. [Google Scholar] [CrossRef] [Green Version]
- Ghribi, O. Potential Mechanisms Linking Cholesterol to Alzheimer’s Disease-like Pathology in Rabbit Brain, Hippocampal Organotypic Slices, and Skeletal Muscle. J. Alzheimer’s Dis. JAD 2008, 15, 673–684. [Google Scholar] [CrossRef]
- Hong, C.; Tontonoz, P. Liver X receptors in lipid metabolism: Opportunities for drug discovery. Nat. Rev. Drug Discov. 2014, 13, 433–444. [Google Scholar] [CrossRef]
- Park, S.H.; Kim, J.L.; Lee, E.S.; Han, S.Y.; Gong, J.H.; Kang, M.K.; Kang, Y.H. Dietary ellagic acid attenuates oxidized LDL uptake and stimulates cholesterol efflux in murine macrophages. J. Nutr. 2011, 141, 1931–1937. [Google Scholar] [CrossRef] [Green Version]
- Toney, A.M.; Fan, R.; Xian, Y.; Chaidez, V.; Ramer-Tait, A.E.; Chung, S. Urolithin A, a Gut Metabolite, Improves Insulin Sensitivity Through Augmentation of Mitochondrial Function and Biogenesis. Obesity 2019, 27, 612–620. [Google Scholar] [CrossRef]
- D’Amico, D.; Andreux, P.A.; Valdés, P.; Singh, A.; Rinsch, C.; Auwerx, J. Impact of the Natural Compound Urolithin A on Health, Disease, and Aging. Trends Mol. Med. 2021, 27, 687–699. [Google Scholar] [CrossRef]
- Webb, M. A Novel Mitophagy Assay for Skeletal Myotubes. Open Access J. Neurol. Neurosurg. 2017, 4. [Google Scholar] [CrossRef]
- Zhao, C.; Sakaguchi, T.; Fujita, K.; Ito, H.; Nishida, N.; Nagatomo, A.; Tanaka-Azuma, Y.; Katakura, Y. Pomegranate-Derived Polyphenols Reduce Reactive Oxygen Species Production via SIRT3-Mediated SOD2 Activation. Oxid. Med. Cell. Longev. 2016, 2016, 2927131. [Google Scholar] [CrossRef] [Green Version]
- Boakye, Y.D.; Groyer, L.; Heiss, E.H. An increased autophagic flux contributes to the anti-inflammatory potential of urolithin A in macrophages. Biochim. Biophys. Acta (BBA) Gen. Subj. 2018, 1862, 61–70. [Google Scholar] [CrossRef]
- Tan, S.; Wong, E. Mitophagy Transcriptome: Mechanistic Insights into Polyphenol-Mediated Mitophagy. Oxid. Med. Cell. Longev. 2017, 2017, 9028435. [Google Scholar] [CrossRef]
- Song, D.; Ma, J.; Chen, L.; Guo, C.; Zhang, Y.; Chen, T.; Zhang, S.; Zhu, Z.; Tian, L.; Niu, P. FOXO3 promoted mitophagy via nuclear retention induced by manganese chloride in SH-SY5Y cells. Met. Integr. Biometal Sci. 2017, 9, 1251–1259. [Google Scholar] [CrossRef]
- Palikaras, K.; Daskalaki, I.; Markaki, M.; Tavernarakis, N. Mitophagy and age-related pathologies: Development of new therapeutics by targeting mitochondrial turnover. Pharmacol. Ther. 2017, 178, 157–174. [Google Scholar] [CrossRef]
- Pillai, V.B.; Sundaresan, N.R.; Kim, G.; Gupta, M.; Rajamohan, S.B.; Pillai, J.B.; Samant, S.; Ravindra, P.V.; Isbatan, A.; Gupta, M.P. Exogenous NAD blocks cardiac hypertrophic response via activation of the SIRT3-LKB1-AMP-activated kinase pathway. J. Bol. Chem. 2010, 285, 3133–3144. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, N.; Das, A.; Biswas, N.; Gnyawali, S.; Singh, K.; Gorain, M.; Polcyn, C.; Khanna, S.; Roy, S.; Sen, C.K. Urolithin A augments angiogenic pathways in skeletal muscle by bolstering NAD+ and SIRT1. Sci. Rep. 2020, 10, 20184. [Google Scholar] [CrossRef]
- Sebastián, D.; Sorianello, E.; Segalés, J.; Irazoki, A.; Ruiz-Bonilla, V.; Sala, D.; Planet, E.; Berenguer-Llergo, A.; Muñoz, J.P.; Sánchez-Feutrie, M.; et al. Mfn2 deficiency links age-related sarcopenia and impaired autophagy to activation of an adaptive mitophagy pathway. EMBO J. 2016, 35, 1677–1693. [Google Scholar] [CrossRef] [PubMed]
- Qiao, H.; Ren, H.; Du, H.; Zhang, M.; Xiong, X.; Lv, R. Liraglutide repairs the infarcted heart: The role of the SIRT1/Parkin/mitophagy pathway. Mol. Med. Rep. 2018, 17, 3722–3734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egan, D.F.; Shackelford, D.B.; Mihaylova, M.M.; Gelino, S.; Kohnz, R.A.; Mair, W.; Vasquez, D.S.; Joshi, A.; Gwinn, D.M.; Taylor, R.; et al. Phosphorylation of ULK1 (hATG1) by AMP-Activated Protein Kinase Connects Energy Sensing to Mitophagy. Science 2011, 331, 456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, Z.; Huang, J.; Xu, B.; Ou, Z.; Zhang, L.; Lin, X.; Ye, X.; Kong, X.; Long, D.; Sun, X.; et al. Urolithin A attenuates memory impairment and neuroinflammation in APP/PS1 mice. J. Neuroinflamm. 2019, 16, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raben, N.; Puertollano, R. TFEB and TFE3: Linking Lysosomes to Cellular Adaptation to Stress. Annu. Rev. Cell Dev. Biol. 2016, 32, 255–278. [Google Scholar] [CrossRef] [PubMed]
- Lapierre, L.R.; Kumsta, C.; Sandri, M.; Ballabio, A.; Hansen, M. Transcriptional and epigenetic regulation of autophagy in aging. Autophagy 2015, 11, 867–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Füllgrabe, J.; Klionsky, D.J.; Joseph, B. The return of the nucleus: Transcriptional and epigenetic control of autophagy. Nat. Rev. Mol. Cell Biol. 2014, 15, 65–74. [Google Scholar] [CrossRef]
- Sardiello, M.; Palmieri, M.; di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; Di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.S.; et al. A gene network regulating lysosomal biogenesis and function. Science 2009, 325, 473–477. [Google Scholar] [CrossRef] [Green Version]
- Roczniak-Ferguson, A.; Petit, C.S.; Froehlich, F.; Qian, S.; Ky, J.; Angarola, B.; Walther, T.C.; Ferguson, S.M. The Transcription Factor TFEB Links mTORC1 Signaling to Transcriptional Control of Lysosome Homeostasis. Sci. Sign. 2012, 5, ra42. [Google Scholar] [CrossRef] [Green Version]
- Nezich, C.L.; Wang, C.; Fogel, A.I.; Youle, R.J. MiT/TFE transcription factors are activated during mitophagy downstream of Parkin and Atg5. J. Cell Biol. 2015, 210, 435–450. [Google Scholar] [CrossRef] [Green Version]
- Tan, S.; Yu, C.Y.; Sim, Z.W.; Low, Z.S.; Lee, B.; See, F.; Min, N.; Gautam, A.; Chu, J.J.H.; Ng, K.W.; et al. Pomegranate activates TFEB to promote autophagy-lysosomal fitness and mitophagy. Sci. Rep. 2019, 9, 727. [Google Scholar] [CrossRef] [Green Version]
- Palmieri, M.; Pal, R.; Nelvagal, H.R.; Lotfi, P.; Stinnett, G.R.; Seymour, M.L.; Chaudhury, A.; Bajaj, L.; Bondar, V.V.; Bremner, L.; et al. mTORC1-independent TFEB activation via Akt inhibition promotes cellular clearance in neurodegenerative storage diseases. Nat. Commun. 2017, 8, 14338. [Google Scholar] [CrossRef]
- Totiger, T.M.; Srinivasan, S.; Jala, V.R.; Lamichhane, P.; Dosch, A.R.; Gaidarski, A.A.; Joshi, C.; Rangappa, S.; Castellanos, J.; Vemula, P.K.; et al. Urolithin A, a novel natural compound to target PI3K/AKT/mTOR pathway in pancreatic cancer. Mol. Cancer Ther. 2018. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, W.; Kishi, H.; Yagasaki, K.; Ohhira, S. Urolithin A attenuates pro-inflammatory mediator production by suppressing PI3-K/Akt/NF-κB and JNK/AP-1 signaling pathways in lipopolysaccharide-stimulated RAW264 macrophages: Possible involvement of NADPH oxidase-derived reactive oxygen species. Eur. J. Pharmacol. 2018, 833, 411–424. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Mo, Y.; Li, Y.; Zhong, Y.; He, S.; Zhang, Y.; Tang, Y.; Fu, S.; Wang, X.; Chen, A. Urolithin A alleviates myocardial ischemia/reperfusion injury via PI3K/Akt pathway. Biochem. Biophys. Res. Commun. 2017, 486, 774–780. [Google Scholar] [CrossRef]
- Das, S.; Mitrovsky, G.; Vasanthi, H.R.; Das, D.K. Antiaging properties of a grape-derived antioxidant are regulated by mitochondrial balance of fusion and fission leading to mitophagy triggered by a signaling network of Sirt1-Sirt3-Foxo3-PINK1-PARKIN. Oxid. Med. Cell. Longev. 2014, 2014, 345105. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, K.M.; Pennington, J.D.; Bisht, K.S.; Aykin-Burns, N.; Kim, H.S.; Mishra, M.; Sun, L.; Nguyen, P.; Ahn, B.H.; Leclerc, J.; et al. SIRT3 interacts with the daf-16 homolog FOXO3a in the mitochondria, as well as increases FOXO3a dependent gene expression. Int. J. Biol. Sci. 2008, 4, 291–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soutar, M.P.M.; Kempthorne, L.; Miyakawa, S.; Annuario, E.; Melandri, D.; Harley, J.; O’Sullivan, G.A.; Wray, S.; Hancock, D.C.; Cookson, M.R.; et al. AKT signalling selectively regulates PINK1 mitophagy in SHSY5Y cells and human iPSC-derived neurons. Sci. Rep. 2018, 8, 8855. [Google Scholar] [CrossRef] [Green Version]
- Guertin, D.A.; Stevens, D.M.; Thoreen, C.C.; Burds, A.A.; Kalaany, N.Y.; Moffat, J.; Brown, M.; Fitzgerald, K.J.; Sabatini, D.M. Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Dev. Cell 2006, 11, 859–871. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
| Compound | In Vitro Model | In Vivo Model | Dose/ Duration | Neuroprotective Roles | Reference |
|---|---|---|---|---|---|
| Ellagic acid (EA), Urolithins A, B, C and D | Human umbilical vein endothelial cells (HUVECs) | - | 10 µM | Anti-atherogenic effects | Mele et al., 2016 [190] |
| Punicalagin | Primary mixed glial cells, organotypic hippocampal slice cultures | - | 5–40 µM | Anti-inflammatory effects | Olajide et al., 2014 [191] |
| Urolithin A/B | - | C. elegans | 10 µg/mL | Anti-Aβ fibrillation effects | Yuan et al., 2016 [171] |
| Pomegranate (freeze dried) | SK-N-SH cells | - | 50–200 ppm | Anti-inflammatory effects Inhibition of BACE1 and Aβ | Velagapudi et al., 2016 [192] |
| Pomegranate- (pulp hydroalcoholic extract juice, pulp aqueous extract) | PC12 | - | 800 μg/mL 6 h/12 h | Antioxidant activity Anti-apoptogenic activity | Forouzanfar et al., 2013 [193] |
| Pomegranate peel extract | - | Male C57Bl/6 mice | 800 mg/kg/day for 35 days | Reduction of Aβ plaque density, lipid peroxidation Anti-inflammatory effects Increase in the expression of neurotrophin BDNF | Morzelle et al., 2016 [194] |
| Pomegranate extract | PC12 | mice | 800 mg/kg/day | Antioxidant effects Inhibition of Aβ-induced learning and memory deficiency | Choi et al., 2015 [195] |
| Pomegranate (freeze dried) | - | Transgenic mice APPsw/Tg2576 | 4% fruit diet | Antioxidant activity | Subash et al., 2014 [196] |
| Urolithin A/B | MCF7 | - | 40 µM | Estrogenic and anti-estrogenic activity | Larrosa et al., 2006 [140] |
| Urolithin A/B | SK-N-MC | - | 10 μM | Antiglycative activity | Verzelloni et al., 2011 [197] |
| Urolithins A/B | BV2 murine microglia and SH-SY5Y non-contact, co-culture model | - | 10 μM | Anti-neuroinflammatory activity | DaSilva et al., 2017 [198] |
| Urolithins A/B | BV2 murine microglia | - | 3–30 μM | Anti-neuroinflammatory activity | Xu et al., 2018 [199] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jayatunga, D.P.W.; Hone, E.; Khaira, H.; Lunelli, T.; Singh, H.; Guillemin, G.J.; Fernando, B.; Garg, M.L.; Verdile, G.; Martins, R.N. Therapeutic Potential of Mitophagy-Inducing Microflora Metabolite, Urolithin A for Alzheimer’s Disease. Nutrients 2021, 13, 3744. https://doi.org/10.3390/nu13113744
Jayatunga DPW, Hone E, Khaira H, Lunelli T, Singh H, Guillemin GJ, Fernando B, Garg ML, Verdile G, Martins RN. Therapeutic Potential of Mitophagy-Inducing Microflora Metabolite, Urolithin A for Alzheimer’s Disease. Nutrients. 2021; 13(11):3744. https://doi.org/10.3390/nu13113744
Chicago/Turabian StyleJayatunga, Dona Pamoda W., Eugene Hone, Harjot Khaira, Taciana Lunelli, Harjinder Singh, Gilles J. Guillemin, Binosha Fernando, Manohar L. Garg, Giuseppe Verdile, and Ralph N. Martins. 2021. "Therapeutic Potential of Mitophagy-Inducing Microflora Metabolite, Urolithin A for Alzheimer’s Disease" Nutrients 13, no. 11: 3744. https://doi.org/10.3390/nu13113744
APA StyleJayatunga, D. P. W., Hone, E., Khaira, H., Lunelli, T., Singh, H., Guillemin, G. J., Fernando, B., Garg, M. L., Verdile, G., & Martins, R. N. (2021). Therapeutic Potential of Mitophagy-Inducing Microflora Metabolite, Urolithin A for Alzheimer’s Disease. Nutrients, 13(11), 3744. https://doi.org/10.3390/nu13113744