
A Systems Approach Dissociates Fructose-Induced Liver Triglyceride from Hypertriglyceridemia and Hyperinsulinemia in Male Mice

 , ,
, ,
Abstract
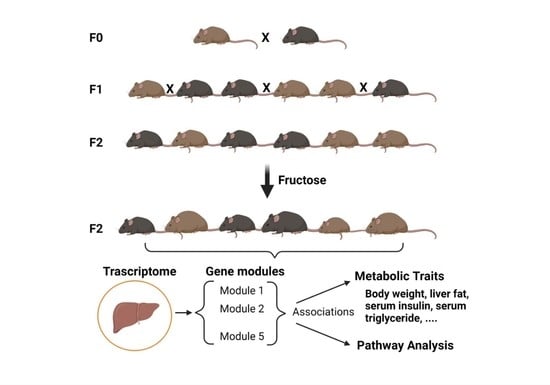
:
1. Introduction
2. Materials and Methods
2.1. Animals and Diets
2.2. Body Weight and Metabolic Parameters
2.3. Liver Triglyceride and Cholesterol Measurements
2.4. RNA Extraction and Quantitative PCR
2.5. SCRB-Seq, Gene Network Analysis, and Gene Set Enrichment Analysis
2.6. SNP Genotyping
2.7. Statistical Analysis
3. Results
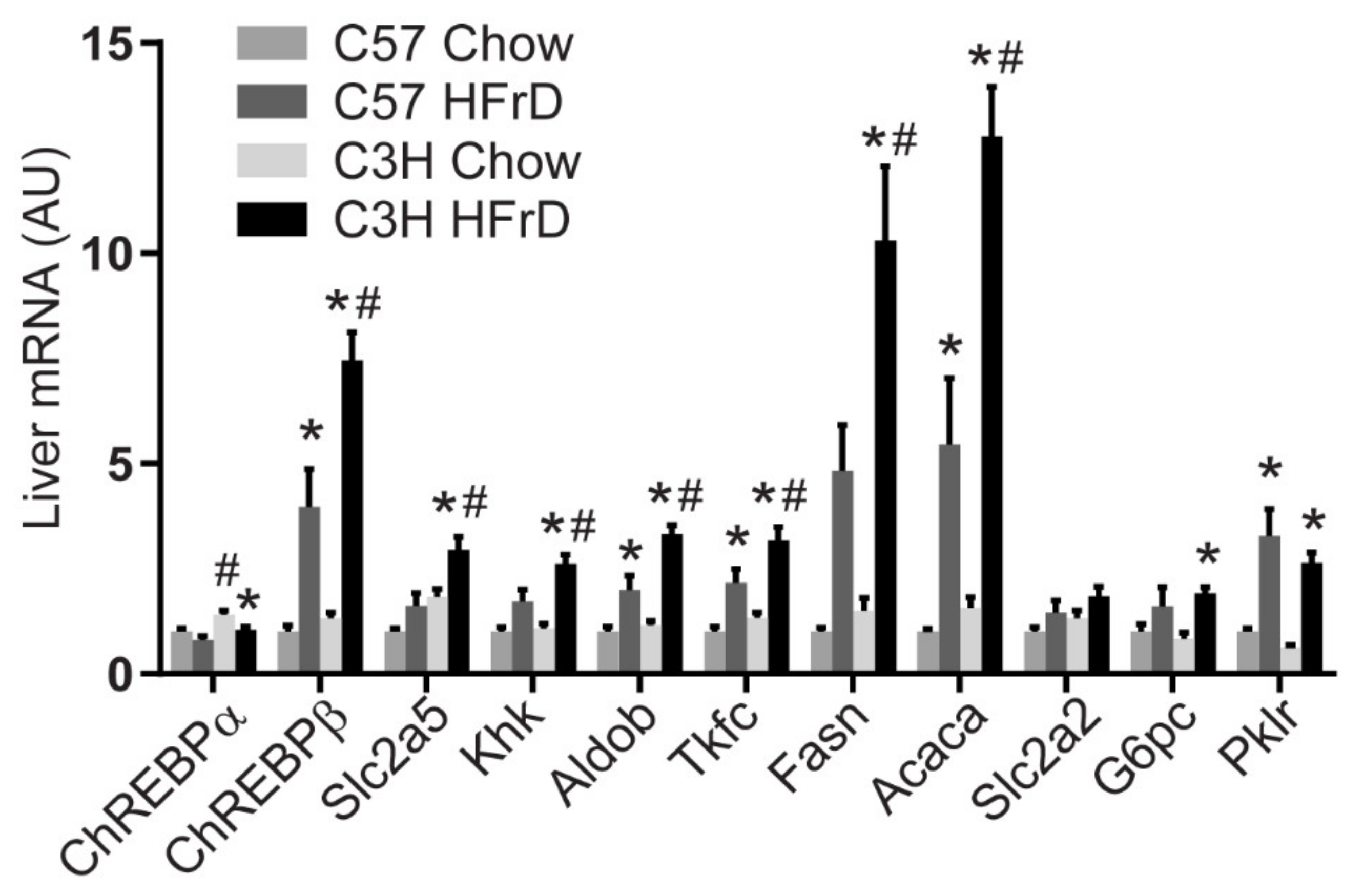
3.1. C3H and C57 Mice Are Sensitive and Resistant, Respectively to Fructose-Induced Hyperinsulinemia and Hypertriglyceridemia
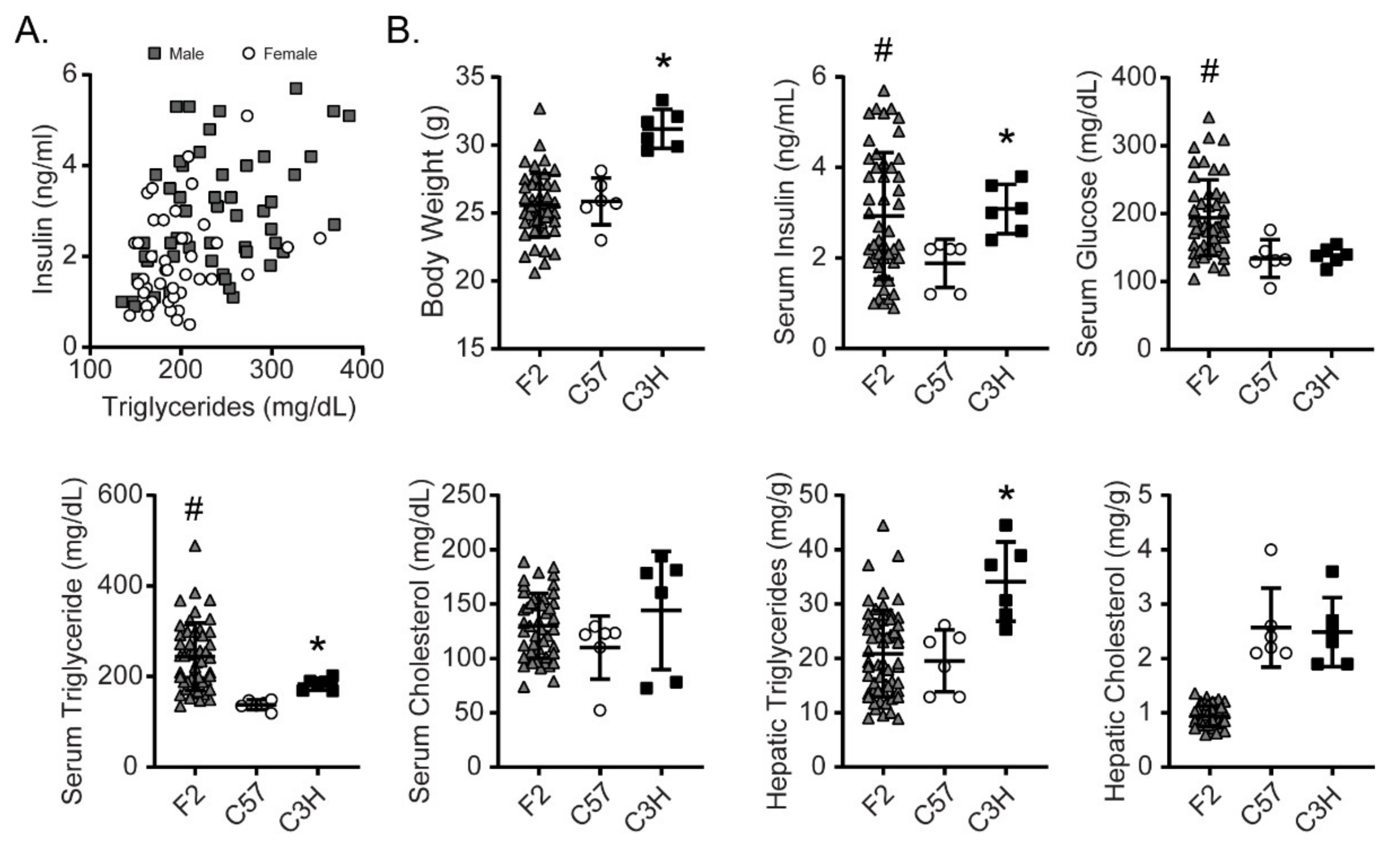
3.2. An F2 Cohort Demonstrates Larger Variance in Most Fructose-Affected Metabolic Traits Compared to the Parental Strains
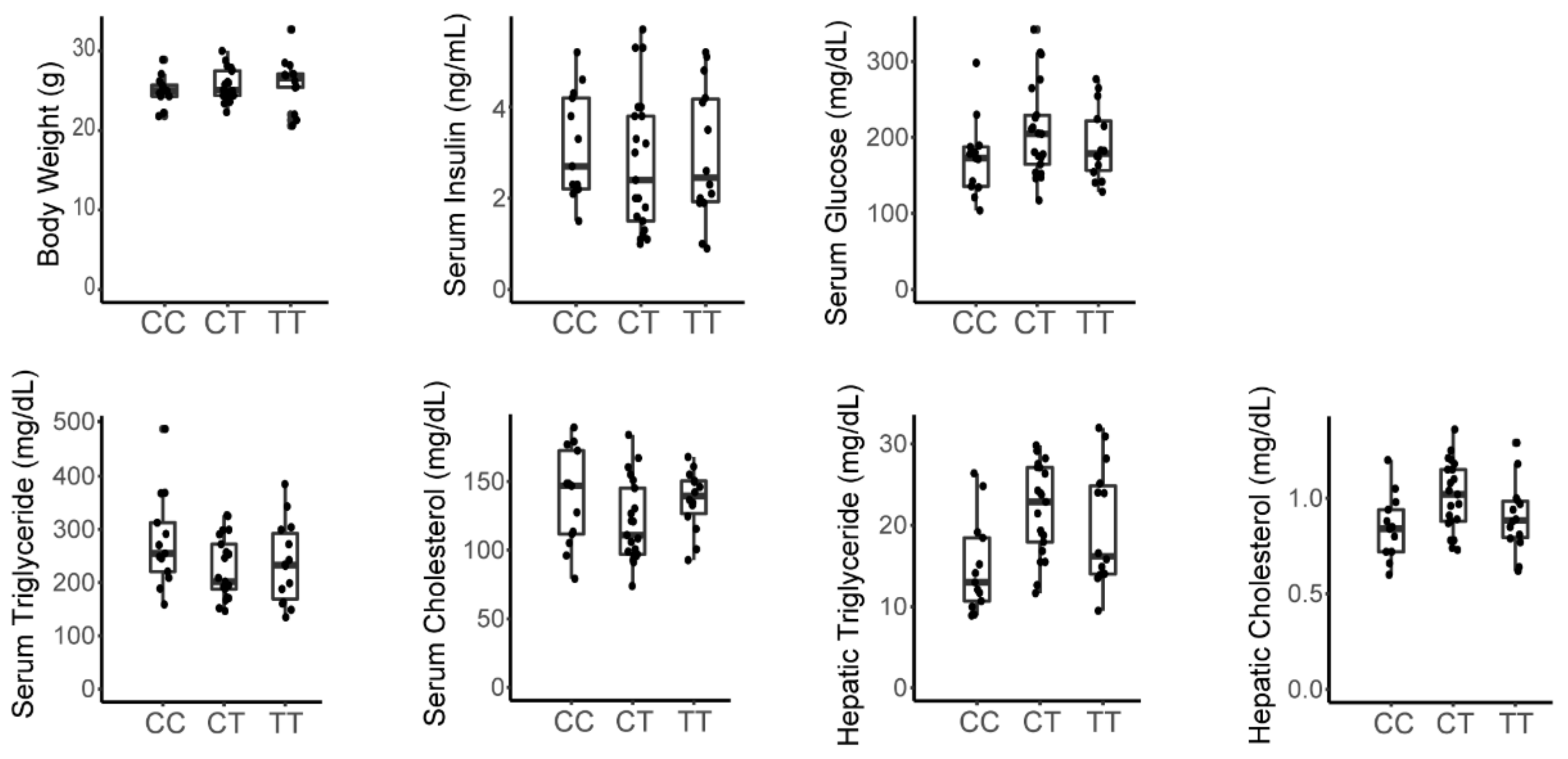
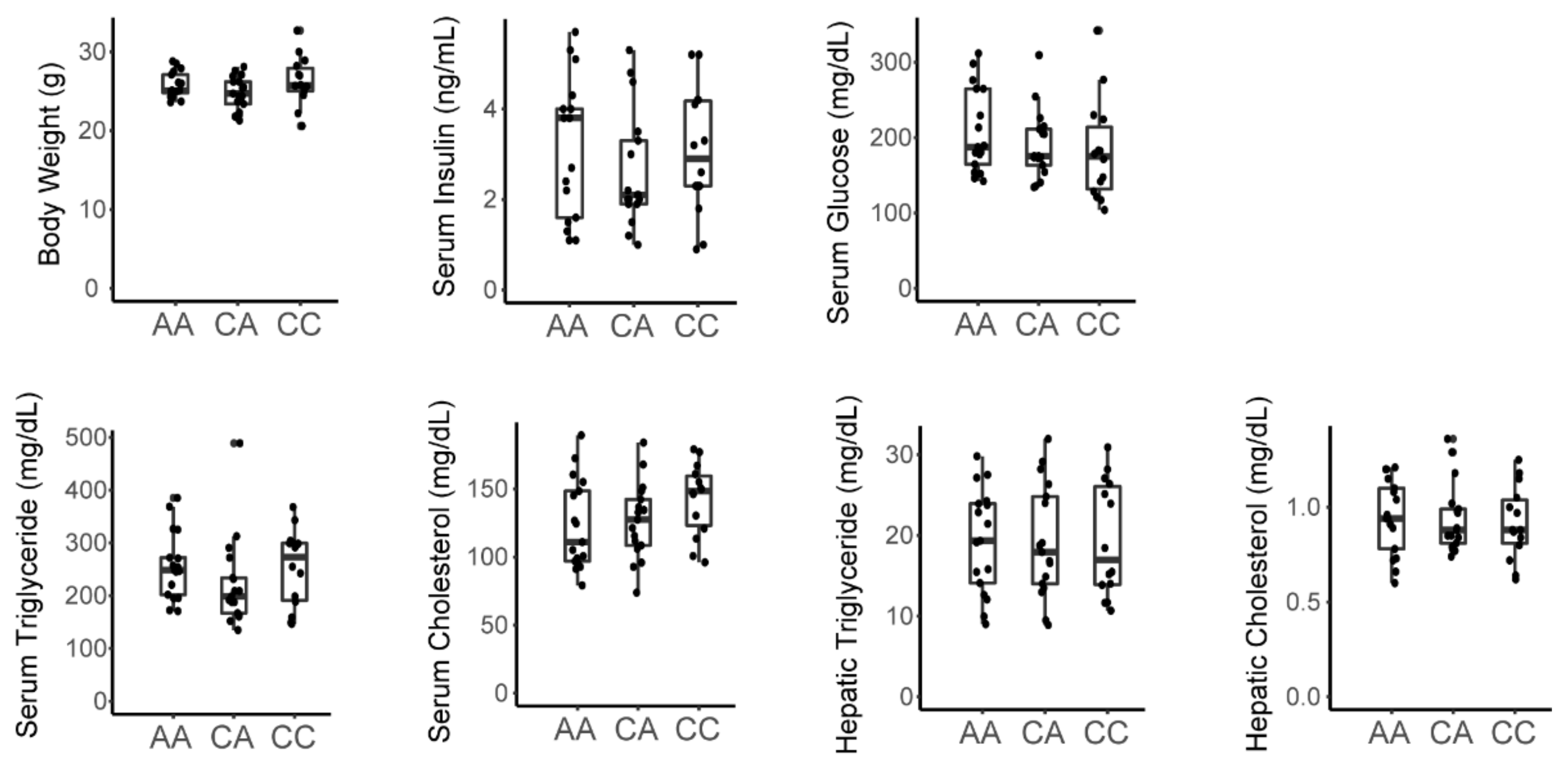
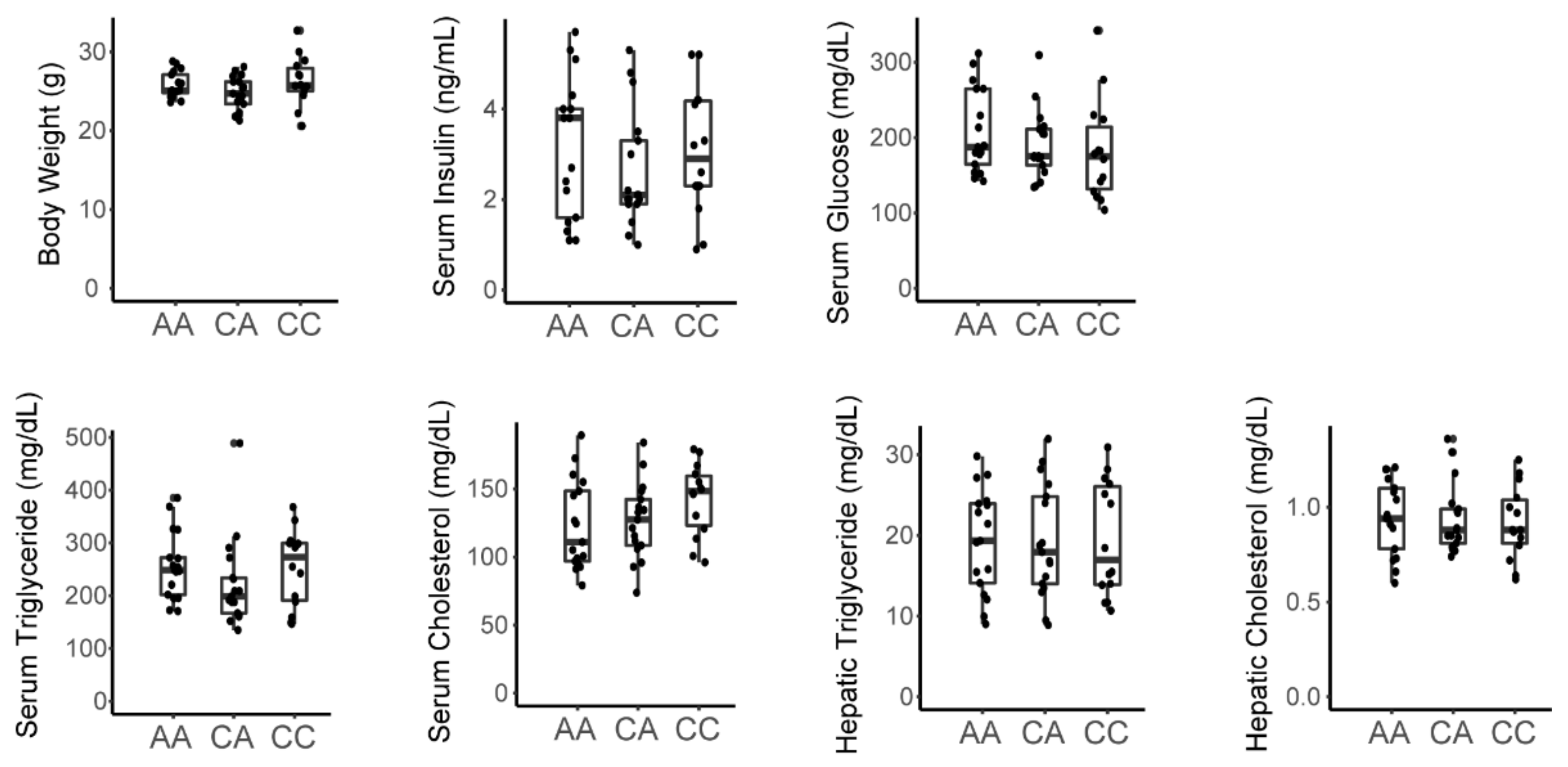
3.3. A Variant in the Srebp1 Locus Does Not Associate with Metabolic Phenotype in the F2 Cohort
3.4. The Inactivating Missense Variant in Tlr4 Present in C3H/HeJ Mice Does Not Associate with Metabolic Phenotype in the F2 Cohort
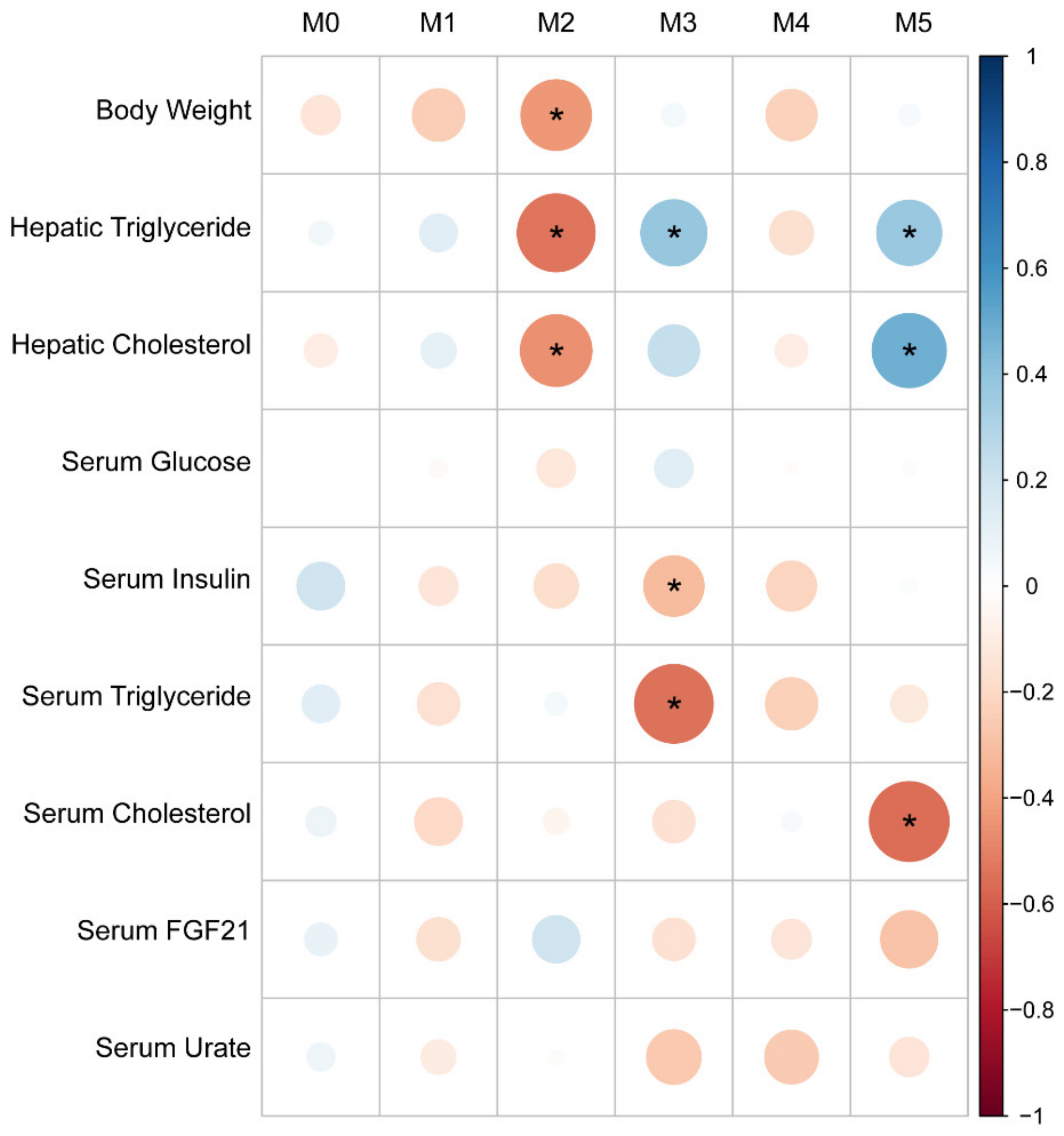
3.5. Distinct Hepatic Gene Programs Associate with Fructose-Induced Hypertriglyceridemia and Hyperinsulinemia Compared to Hepatic Triglyceride Levels
3.6. Gene Set Enrichment Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Eckel, R.H.; Alberti, K.G.M.M.; Grundy, S.M.; Zimmet, P.Z. The Metabolic Syndrome. Lancet 2010, 375, 181–183. [Google Scholar] [CrossRef]
- Esposito, K.; Chiodini, P.; Colao, A.; Lenzi, A.; Giugliano, D. Metabolic Syndrome and Risk of Cancer: A Systematic Review and Meta-Analysis. Diabetes Care 2012, 35, 2402–2411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herman, M.A. Metabolic Liver Disease–What’s in a Name? Nat. Rev. Endocrinol. 2021, 17, 79–80. [Google Scholar] [CrossRef] [PubMed]
- Moore, J.X.; Chaudhary, N.; Akinyemiju, T. Metabolic Syndrome Prevalence by Race/Ethnicity and Sex in the United States, National Health and Nutrition Examination Survey, 1988–2012. Prev. Chronic. Dis. 2017, 14, E24. [Google Scholar] [CrossRef] [Green Version]
- Hannou, S.A.; Haslam, D.E.; McKeown, N.M.; Herman, M.A. Fructose Metabolism and Metabolic Disease. J. Clin. Investig. 2018, 128, 545–555. [Google Scholar] [CrossRef]
- Stanhope, K.L.; Schwarz, J.M.; Keim, N.L.; Griffen, S.C.; Bremer, A.A.; Graham, J.L.; Hatcher, B.; Cox, C.L.; Dyachenko, A.; Zhang, W.; et al. Consuming Fructose-Sweetened, Not Glucose-Sweetened, Beverages Increases Visceral Adiposity and Lipids and Decreases Insulin Sensitivity in Overweight/Obese Humans. J. Clin. Investig. 2009, 119, 1322–1334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reaven, G.M. Banting Lecture 1988. Role of Insulin Resistance in Human Disease. Diabetes 1988, 37, 1595–1607. [Google Scholar] [CrossRef]
- Abdelmalek, M.F.; Suzuki, A.; Guy, C.; Unalp-Arida, A.; Colvin, R.; Johnson, R.J.; Diehl, A.M. Nonalcoholic Steatohepatitis Clinical Research Network Increased Fructose Consumption Is Associated with Fibrosis Severity in Patients with Nonalcoholic Fatty Liver Disease. Hepatology 2010, 51, 1961–1971. [Google Scholar] [CrossRef] [Green Version]
- Jang, C.; Hui, S.; Lu, W.; Cowan, A.J.; Morscher, R.J.; Lee, G.; Liu, W.; Tesz, G.J.; Birnbaum, M.J.; Rabinowitz, J.D. The Small Intestine Converts Dietary Fructose into Glucose and Organic Acids. Cell Metab. 2018, 27, 351–361.e3. [Google Scholar] [CrossRef] [Green Version]
- Jang, C.; Wada, S.; Yang, S.; Gosis, B.; Zeng, X.; Zhang, Z.; Shen, Y.; Lee, G.; Arany, Z.; Rabinowitz, J.D. The Small Intestine Shields the Liver from Fructose-Induced Steatosis. Nat. Metab. 2020, 2, 586–593. [Google Scholar] [CrossRef]
- Andres-Hernando, A.; Orlicky, D.J.; Kuwabara, M.; Ishimoto, T.; Nakagawa, T.; Johnson, R.J.; Lanaspa, M.A. Deletion of Fructokinase in the Liver or in the Intestine Reveals Differential Effects on Sugar-Induced Metabolic Dysfunction. Cell Metab. 2020, 32, 117–127.e3. [Google Scholar] [CrossRef] [PubMed]
- Softic, S.; Gupta, M.K.; Wang, G.-X.; Fujisaka, S.; O’Neill, B.T.; Rao, T.N.; Willoughby, J.; Harbison, C.; Fitzgerald, K.; Ilkayeva, O.; et al. Divergent Effects of Glucose and Fructose on Hepatic Lipogenesis and Insulin Signaling. J. Clin. Investig. 2018, 128, 1199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.; Astapova, I.I.; Flier, S.N.; Hannou, S.A.; Doridot, L.; Sargsyan, A.; Kou, H.H.; Fowler, A.J.; Liang, G.; Herman, M.A. Intestinal, but Not Hepatic, ChREBP Is Required for Fructose Tolerance. JCI Insight 2017, 2, e96703. [Google Scholar] [CrossRef] [Green Version]
- Erion, D.M.; Popov, V.; Hsiao, J.J.; Vatner, D.; Mitchell, K.; Yonemitsu, S.; Nagai, Y.; Kahn, M.; Gillum, M.P.; Dong, J.; et al. The Role of the Carbohydrate Response Element-Binding Protein in Male Fructose-Fed Rats. Endocrinology 2013, 154, 36–44. [Google Scholar] [CrossRef]
- Linden, A.G.; Li, S.; Choi, H.Y.; Fang, F.; Fukasawa, M.; Uyeda, K.; Hammer, R.E.; Horton, J.D.; Engelking, L.J.; Liang, G. Interplay between ChREBP and SREBP-1c Coordinates Postprandial Glycolysis and Lipogenesis in Livers of Mice. J. Lipid Res. 2018, 59, 475–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Surwit, R.S.; Feinglos, M.N.; Rodin, J.; Sutherland, A.; Petro, A.E.; Opara, E.C.; Kuhn, C.M.; Rebuffé-Scrive, M. Differential Effects of Fat and Sucrose on the Development of Obesity and Diabetes in C57BL/6J and A/J Mice. Metabolism 1995, 44, 645–651. [Google Scholar] [CrossRef]
- Surwit, R.S.; Kuhn, C.M.; Cochrane, C.; McCubbin, J.A.; Feinglos, M.N. Diet-Induced Type II Diabetes in C57BL/6J Mice. Diabetes 1988, 37, 1163–1167. [Google Scholar] [CrossRef] [PubMed]
- Nagata, R.; Nishio, Y.; Sekine, O.; Nagai, Y.; Maeno, Y.; Ugi, S.; Maegawa, H.; Kashiwagi, A. Single Nucleotide Polymorphism (-468 Gly to A) at the Promoter Region of SREBP-1c Associates with Genetic Defect of Fructose-Induced Hepatic Lipogenesis [Corrected]. J. Biol. Chem. 2004, 279, 29031–29042. [Google Scholar] [CrossRef] [Green Version]
- Folch, J.; Lees, M.; Sloane Stanley, G.H. A Simple Method for the Isolation and Purification of Total Lipides from Animal Tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar] [CrossRef]
- Soumillon, M.; Cacchiarelli, D.; Semrau, S.; van Oudenaarden, A.; Mikkelsen, T.S. Characterization of Directed Differentiation by High-Throughput Single-Cell RNA-Seq. bioRxiv 2014, 003236. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor Package for Differential Expression Analysis of Digital Gene Expression Data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemoine, G.G.; Scott-Boyer, M.-P.; Ambroise, B.; Périn, O.; Droit, A. GWENA: Gene Co-Expression Networks Analysis and Extended Modules Characterization in a Single Bioconductor Package. BMC Bioinform. 2021, 22, 267. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the Unification of Biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Han, H.; Cho, J.-W.; Lee, S.; Yun, A.; Kim, H.; Bae, D.; Yang, S.; Kim, C.Y.; Lee, M.; Kim, E.; et al. TRRUST v2: An Expanded Reference Database of Human and Mouse Transcriptional Regulatory Interactions. Nucleic Acids Res. 2018, 46, D380–D386. [Google Scholar] [CrossRef]
- Dushay, J.R.; Toschi, E.; Mitten, E.K.; Fisher, F.M.; Herman, M.A.; Maratos-Flier, E. Fructose Ingestion Acutely Stimulates Circulating FGF21 Levels in Humans. Mol. Metab. 2015, 4, 51–57. [Google Scholar] [CrossRef]
- Fisher, F.M.; Kim, M.; Doridot, L.; Cunniff, J.C.; Parker, T.S.; Levine, D.M.; Hellerstein, M.K.; Hudgins, L.C.; Maratos-Flier, E.; Herman, M.A. A Critical Role for ChREBP-Mediated FGF21 Secretion in Hepatic Fructose Metabolism. Mol. Metab. 2017, 6, 14–21. [Google Scholar] [CrossRef]
- Von Holstein-Rathlou, S.; BonDurant, L.D.; Peltekian, L.; Naber, M.C.; Yin, T.C.; Claflin, K.E.; Urizar, A.I.; Madsen, A.N.; Ratner, C.; Holst, B.; et al. FGF21 Mediates Endocrine Control of Simple Sugar Intake and Sweet Taste Preference by the Liver. Cell Metab. 2016, 23, 335–343. [Google Scholar] [CrossRef] [Green Version]
- Iizuka, K.; Takeda, J.; Horikawa, Y. Glucose Induces FGF21 MRNA Expression through ChREBP Activation in Rat Hepatocytes. FEBS Lett. 2009, 583, 2882–2886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talukdar, S.; Owen, B.M.; Song, P.; Hernandez, G.; Zhang, Y.; Zhou, Y.; Scott, W.T.; Paratala, B.; Turner, T.; Smith, A.; et al. FGF21 Regulates Sweet and Alcohol Preference. Cell Metab. 2016, 23, 344–349. [Google Scholar] [CrossRef] [Green Version]
- Flippo, K.H.; Potthoff, M.J. Metabolic Messengers: FGF21. Nat. Metab. 2021, 3, 309–317. [Google Scholar] [CrossRef]
- Haas, J.T.; Miao, J.; Chanda, D.; Wang, Y.; Zhao, E.; Haas, M.E.; Hirschey, M.; Vaitheesvaran, B.; Farese, R.V.; Kurland, I.J.; et al. Hepatic Insulin Signaling Is Required for Obesity-Dependent Expression of SREBP-1c MRNA but Not for Feeding-Dependent Expression. Cell Metab. 2012, 15, 873–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horton, J.D.; Goldstein, J.L.; Brown, M.S. SREBPs: Activators of the Complete Program of Cholesterol and Fatty Acid Synthesis in the Liver. J. Clin. Investig. 2002, 109, 1125–1131. [Google Scholar] [CrossRef]
- Keane, T.M.; Goodstadt, L.; Danecek, P.; White, M.A.; Wong, K.; Yalcin, B.; Heger, A.; Agam, A.; Slater, G.; Goodson, M.; et al. Mouse Genomic Variation and Its Effect on Phenotypes and Gene Regulation. Nature 2011, 477, 289–294. [Google Scholar] [CrossRef] [Green Version]
- Bergheim, I.; Weber, S.; Vos, M.; Krämer, S.; Volynets, V.; Kaserouni, S.; McClain, C.J.; Bischoff, S.C. Antibiotics Protect against Fructose-Induced Hepatic Lipid Accumulation in Mice: Role of Endotoxin. J. Hepatol. 2008, 48, 983–992. [Google Scholar] [CrossRef] [PubMed]
- Lambertz, J.; Weiskirchen, S.; Landert, S.; Weiskirchen, R. Fructose: A Dietary Sugar in Crosstalk with Microbiota Contributing to the Development and Progression of Non-Alcoholic Liver Disease. Front. Immunol. 2017, 8, 1159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Todoric, J.; Di Caro, G.; Reibe, S.; Henstridge, D.C.; Green, C.R.; Vrbanac, A.; Ceteci, F.; Conche, C.; McNulty, R.; Shalapour, S.; et al. Fructose Stimulated de Novo Lipogenesis Is Promoted by Inflammation. Nat. Metab. 2020, 2, 1034–1045. [Google Scholar] [CrossRef]
- Hoshino, K.; Takeuchi, O.; Kawai, T.; Sanjo, H.; Ogawa, T.; Takeda, Y.; Takeda, K.; Akira, S. Pillars Article: Cutting Edge: Toll-Like Receptor 4 (TLR4)-Deficient Mice Are Hyporesponsive to Lipopolysaccharide: Evidence for TLR4 as the Lps Gene Product. J. Immunol. 1999, 162, 3749–3752. [Google Scholar]
- Poltorak, A.; He, X.; Smirnova, I.; Liu, M.Y.; Van Huffel, C.; Du, X.; Birdwell, D.; Alejos, E.; Silva, M.; Galanos, C.; et al. Defective LPS Signaling in C3H/HeJ and C57BL/10ScCr Mice: Mutations in Tlr4 Gene. Science 1998, 282, 2085–2088. [Google Scholar] [CrossRef] [Green Version]
- Langfelder, P.; Horvath, S. WGCNA: An R Package for Weighted Correlation Network Analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A Comprehensive Gene Set Enrichment Analysis Web Server 2016 Update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [Green Version]
- Deng, X.; Zhang, W.; O-Sullivan, I.; Williams, J.B.; Dong, Q.; Park, E.A.; Raghow, R.; Unterman, T.G.; Elam, M.B. FoxO1 Inhibits Sterol Regulatory Element-Binding Protein-1c (SREBP-1c) Gene Expression via Transcription Factors Sp1 and SREBP-1c. J. Biol. Chem. 2012, 287, 20132–20143. [Google Scholar] [CrossRef] [Green Version]
- Reed, B.D.; Charos, A.E.; Szekely, A.M.; Weissman, S.M.; Snyder, M. Genome-Wide Occupancy of SREBP1 and Its Partners NFY and SP1 Reveals Novel Functional Roles and Combinatorial Regulation of Distinct Classes of Genes. PLoS Genet. 2008, 4, e1000133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, C.C.; Vossio, S.; Rougemont, J.; Gruenberg, J. TFAP2 Transcription Factors Are Regulators of Lipid Droplet Biogenesis. eLife 2018, 7, e36330. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Jang, C.; Liu, J.; Uehara, K.; Gilbert, M.; Izzo, L.; Zeng, X.; Trefely, S.; Fernandez, S.; Carrer, A.; et al. Dietary Fructose Feeds Hepatic Lipogenesis via Microbiota-Derived Acetate. Nature 2020, 579, 586–591. [Google Scholar] [CrossRef] [PubMed]
- Farese, R.V.; Zechner, R.; Newgard, C.B.; Walther, T.C. The Problem of Establishing Relationships between Hepatic Steatosis and Hepatic Insulin Resistance. Cell Metab. 2012, 15, 570–573. [Google Scholar] [CrossRef] [Green Version]
- Semova, I.; Biddinger, S.B. Triglycerides in Nonalcoholic Fatty Liver Disease: Guilty Until Proven Innocent. Trends Pharmacol. Sci. 2021, 42, 183–190. [Google Scholar] [CrossRef]
- Katz, L.S.; Baumel-Alterzon, S.; Scott, D.K.; Herman, M.A. Adaptive and Maladaptive Roles for ChREBP in the Liver and Pancreatic Islets. J. Biol. Chem. 2021, 296, 100623. [Google Scholar] [CrossRef]
- Ma, L.; Robinson, L.N.; Towle, H.C. ChREBP*Mlx Is the Principal Mediator of Glucose-Induced Gene Expression in the Liver. J. Biol. Chem. 2006, 281, 28721–28730. [Google Scholar] [CrossRef] [Green Version]
- Smith, G.I.; Shankaran, M.; Yoshino, M.; Schweitzer, G.G.; Chondronikola, M.; Beals, J.W.; Okunade, A.L.; Patterson, B.W.; Nyangau, E.; Field, T.; et al. Insulin Resistance Drives Hepatic de Novo Lipogenesis in Nonalcoholic Fatty Liver Disease. J. Clin. Investig. 2020, 130, 1453–1460. [Google Scholar] [CrossRef]
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of Fatty Acids Stored in Liver and Secreted via Lipoproteins in Patients with Nonalcoholic Fatty Liver Disease. J. Clin. Investig. 2005, 115, 1343–1351. [Google Scholar] [CrossRef] [Green Version]
- Kooner, J.S.; Chambers, J.C.; Aguilar-Salinas, C.A.; Hinds, D.A.; Hyde, C.L.; Warnes, G.R.; Gómez Pérez, F.J.; Frazer, K.A.; Elliott, P.; Scott, J.; et al. Genome-Wide Scan Identifies Variation in MLXIPL Associated with Plasma Triglycerides. Nat. Genet. 2008, 40, 149–151. [Google Scholar] [CrossRef] [PubMed]
- Kathiresan, S.; Melander, O.; Guiducci, C.; Surti, A.; Burtt, N.P.; Rieder, M.J.; Cooper, G.M.; Roos, C.; Voight, B.F.; Havulinna, A.S.; et al. Six New Loci Associated with Blood Low-Density Lipoprotein Cholesterol, High-Density Lipoprotein Cholesterol or Triglycerides in Humans. Nat. Genet. 2008, 40, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Willer, C.J.; Sanna, S.; Jackson, A.U.; Scuteri, A.; Bonnycastle, L.L.; Clarke, R.; Heath, S.C.; Timpson, N.J.; Najjar, S.S.; Stringham, H.M.; et al. Newly Identified Loci That Influence Lipid Concentrations and Risk of Coronary Artery Disease. Nat. Genet. 2008, 40, 161–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| C57BL/6J-Chow | C57BL/6J-Fructose | C3H/HeJ-Chow | C3H/HeJ-Fructose | |
|---|---|---|---|---|
| Body Weight (g) Body Weight Change (g) | 28 ± 0.7 0.6 ± 0.3 | 25.6 ± 0.6 −1.1 ± 0.5 * | 29.2 ± 1.0 2.9 ± 0.4 # | 32.2 ± 0.6 # 4.4 ± 0.3 *,# |
| Serum Glucose (mg/dL) | 154 ± 11 | 134 ± 11 | 129 ± 11 | 138 ± 5 |
| Serum Insulin (ng/mL) | 2.5 ± 0.3 | 1.9 ± 0.2 | 1.3 ± 0.2 | 3.1 ± 0.2 *,# |
| Serum Triglyceride (mg/dL) | 137 ± 5 | 138 ± 5 | 149 ± 9 | 183 ± 5 *,# |
| Serum Cholesterol (mg/dL) | 93 ± 7 | 110 ± 12 | 129 ± 9 | 144 ± 22 |
| Hepatic Triglyceride (mg/g) | 6.2 ± 0.7 | 19.5 ± 2.3 * | 10.1 ± 1.5 | 34.1 ± 3.0 *,# |
| Hepatic Cholesterol (mg/g) | 2.1 ± 0.1 | 2.6 ± 0.3 | 2.4 ± 0.1 | 2.5 ± 0.3 |
| Body Weight | Serum Insulin | Serum Glucose | Serum Trig. | Serum Chol. | Hepatic Trig. | Hepatic Chol. | Serum FGF21 | |
|---|---|---|---|---|---|---|---|---|
| Body Weight | — | |||||||
| Serum Insulin | 0.45 ** | — | ||||||
| Serum Glucose | 0.40 ** | 0.26 | — | |||||
| Serum Triglyceride | 0.27 | 0.44 ** | −0.12 | — | ||||
| Serum Cholesterol | 0.26 | 0.04 | 0.06 | 0.29 * | — | |||
| Hepatic Triglyceride | 0.20 | 0.08 | 0.32 * | −0.21 | −0.23 | — | ||
| Hepatic Cholesterol | 0.08 | 0.08 | 0.29 * | −0.19 | −0.24 | 0.68 **** | — | |
| Serum FGF21 | −0.04 | 0.29 * | −0.25 | 0.43 ** | 0.32 * | −0.33 * | −0.24 | — |
| Serum Urate | 0.23 | 0.12 | −0.11 | 0.46 *** | 0.19 | −0.15 | −0.14 | −0.01 |
| Module 2 | Overlap | −log10 (p-Value) |
|---|---|---|
| Gene Ontology Bio Process: Upregulated | ||
| protein N-linked glycosylation via asparagine (GO:0018279) | 12/30 | 13.0 |
| peptidyl-asparagine modification (GO:0018196) | 12/31 | 12.8 |
| protein exit from endoplasmic reticulum (GO:0032527) | 11/24 | 12.7 |
| response to unfolded protein (GO:0006986) | 14/49 | 12.7 |
| response to endoplasmic reticulum stress (GO:0034976) | 19/110 | 12.7 |
| Gene Ontology Bio Process: Down Regulated | ||
| cholesterol efflux (GO:0033344) | 6/24 | 6.5 |
| high-density lipoprotein particle remodeling (GO:0034375) | 5/18 | 5.8 |
| cholesterol homeostasis (GO:0042632) | 8/71 | 5.7 |
| sterol homeostasis (GO:0055092) | 8/72 | 5.7 |
| cholesterol transport (GO:0030301) | 7/51 | 5.7 |
| TRRUST 2019: Upregulated | ||
| ATF6 human | 3/14 | 2.7 |
| XBP1 human | 3/19 | 2.3 |
| HSF1 human | 3/31 | 1.7 |
| PPARG human | 4/66 | 1.4 |
| HDAC9 human | 2/18 | 1.4 |
| MYC mouse | 3/49 | 1.2 |
| TRRUST 2019: Downregulated | ||
| HNF4A mouse | 5/36 | 4.2 |
| HNF4A human | 5/45 | 3.7 |
| NR1H4 human | 3/20 | 2.8 |
| ESRRA mouse | 2/8 | 2.4 |
| NR1H4 mouse | 2/11 | 2.1 |
| RORA mouse | 2/11 | 2.1 |
| Module 3 | Overlap | −log10 (p-Value) |
| Gene Ontology Bio Process: Upregulated | ||
| fatty acid beta-oxidation (GO:0006635) | 19/52 | 25.8 |
| fatty acid catabolic process (GO:0009062) | 18/70 | 21.3 |
| fatty acid oxidation (GO:0019395) | 16/59 | 19.4 |
| fatty acid beta-oxidation using acyl-CoA oxidase (GO:0033540) | 9/15 | 15.0 |
| long-chain fatty acid metabolic process (GO:0001676) | 14/83 | 13.9 |
| Gene Ontology Bio Process: Down Regulated | ||
| regulation of lipid metabolic process (GO:0019216) | 15/92 | 16.6 |
| cholesterol biosynthetic process (GO:0006695) | 10/35 | 13.9 |
| sterol biosynthetic process (GO:0016126) | 10/38 | 13.5 |
| secondary alcohol biosynthetic process (GO:1902653) | 9/34 | 12.2 |
| regulation of primary metabolic process (GO:0080090) | 13/130 | 11.6 |
| TRRUST 2019: Upregulated | ||
| PPARA mouse | 10/46 | 11.2 |
| PPARA human | 7/39 | 7.4 |
| PPARG mouse | 5/50 | 4.1 |
| PPARD mouse | 3/11 | 4.0 |
| FOXA2 mouse | 4/40 | 3.4 |
| NR0B2 human | 2/8 | 2.7 |
| TRRUST 2019: Downregulated | ||
| SREBF1 human | 7/27 | 9.5 |
| SREBF1 mouse | 6/36 | 7.0 |
| SREBF2 human | 4/20 | 5.1 |
| TFAP2A mouse | 3/10 | 4.5 |
| SP1 mouse | 9/270 | 4.2 |
| MLXIPL mouse | 3/15 | 3.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Doridot, L.; Hannou, S.A.; Krawczyk, S.A.; Tong, W.; Kim, M.-S.; McElroy, G.S.; Fowler, A.J.; Astapova, I.I.; Herman, M.A. A Systems Approach Dissociates Fructose-Induced Liver Triglyceride from Hypertriglyceridemia and Hyperinsulinemia in Male Mice. Nutrients 2021, 13, 3642. https://doi.org/10.3390/nu13103642
Doridot L, Hannou SA, Krawczyk SA, Tong W, Kim M-S, McElroy GS, Fowler AJ, Astapova II, Herman MA. A Systems Approach Dissociates Fructose-Induced Liver Triglyceride from Hypertriglyceridemia and Hyperinsulinemia in Male Mice. Nutrients. 2021; 13(10):3642. https://doi.org/10.3390/nu13103642
Chicago/Turabian StyleDoridot, Ludivine, Sarah A. Hannou, Sarah A. Krawczyk, Wenxin Tong, Mi-Sung Kim, Gregory S. McElroy, Alan J. Fowler, Inna I. Astapova, and Mark A. Herman. 2021. "A Systems Approach Dissociates Fructose-Induced Liver Triglyceride from Hypertriglyceridemia and Hyperinsulinemia in Male Mice" Nutrients 13, no. 10: 3642. https://doi.org/10.3390/nu13103642
APA StyleDoridot, L., Hannou, S. A., Krawczyk, S. A., Tong, W., Kim, M.-S., McElroy, G. S., Fowler, A. J., Astapova, I. I., & Herman, M. A. (2021). A Systems Approach Dissociates Fructose-Induced Liver Triglyceride from Hypertriglyceridemia and Hyperinsulinemia in Male Mice. Nutrients, 13(10), 3642. https://doi.org/10.3390/nu13103642