
The Effects of a Ketogenic Diet on Patients with Dihydrolipoamide Dehydrogenase Deficiency

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Dietary Intervention
2.1.1. Partial Ketogenic Diet
2.1.2. Parenteral Nutrition
2.2. Imaging Studies
2.3. Biochemical Studies
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Patel Mulchand, S.; Roche, T.E. Molecular Biology and Biochemistry of pyruvate dehydrogenase complexes. FASEB J. 1990, 4, 3224–3233. [Google Scholar] [CrossRef]
- Cameron, J.M.; Levandovskiy, V.; Mackay, N.; Raiman, J.; Renaud, D.L.; Clarke, J.T.; Feigenbaum, A.; Elpeleg, O.; Robinson, B.H. Novel Mutations in Dihydrolipoamide Dehydrogenase Deficiency in Two Cousins with Borderline-Normal PDH Complex Activity. Am. J. Med. Genet. Part A 2006, 140, 1542–1552. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.S.; Nemeria, N.S.; Furey, W.; Jordan, F. The pyruvate dehydrogenase complexes: Structure-based function and regulation. J. Biol. Chem. 2014, 289, 16615–16623. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.C.; Kim, H.; Arizmendi, C.; Kitano, A.; Patel, M.S. Identification of two missense mutations in a dihydrolipoamide dehydrogenase-deficient patient. Proc. Natl. Acad. Sci. USA 1993, 90, 5186–5190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinonez, S.C.; Leber, S.M.; Martin, D.M.; Thoene, J.G.; Bedoyan, J.K. Leigh syndrome in a girl with a novel DLD mutation causing E3 deficiency. Pediatr. Neurol. 2015, 48, 67–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, Y.S.; Kerr, D.S.; Craigen, W.J.; Tan, J.; Pan, Y.; Lusk, M.; Patel, M.S. Identification of two mutations in a compound heterozygous child with dihydrolipoamide dehydrogenase deficiency. Hum. Mol. Genet. 1996, 5, 1925–1930. [Google Scholar] [CrossRef] [Green Version]
- Cerna, L.; Wenchich, L.; Hansiková, H.; Kmoch, S.; Pešková, K.; Chrastina, P.; Brynda, J.; Zeman, J. Novel mutations in a boy with dihydrolipoamide dehydrogenase deficiency. Med. Sci. Monit. 2001, 7, 1319–1325. [Google Scholar] [PubMed]
- Odièvre, M.; Chretien, D.; Munnich, A.; Robinson, B.H.; Dumoulin, R.; Masmoudi, S.; Kadhom, N.; Rotig, A.; Rustin, P.; Bonnefont, J.-P. A Novel Mutation in the Dihydrolipoamide Dehydrogenase E3 Subunit Gene ( DLD ) Resulting in an Atypical Form of α -Ketoglutarate Dehydrogenase Deficiency. Hum. Mutat. 2005, 790, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Shany, E.; Saada, A.; Landau, D.; Shaag, A.; Hershkovitz, E.; Elpeleg, O.N. Lipoamide Dehydrogenase Deficiency Due to a Novel Mutation in the Interface Domain. Biochem. Biophys. Res. Commun. 1999, 262, 163–166. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.S.; Korman, S.H.; Lee, J.; Ghoshal, P.; Wu, Q.; Barash, V.; Kang, S.; Oh, S.; Kwon, M.; Gutman, A.; et al. Identification of a common mutation (Gly194Cys) in both Arab Moslem and Ashkenazi Jewish patients with dihydrolipoamide dehydrogenase (E3) deficiency: Possible beneficial effect of vitamin therapy. J. Inherit. Metab. Dis. 2003, 26, 816–818. [Google Scholar] [CrossRef]
- Sansaricq, C.; Pardo, S.; Balwani, M.; Grace, M.; Raymond, K. Biochemical and molecular diagnosis of lipoamide dehydrogenase deficiency in a North American Ashkenazi Jewish family. J. Inherit. Metab. Dis. 2005, 29, 203–204. [Google Scholar] [CrossRef] [PubMed]
- Shaag, A.; Saada, A.; Berger, I.; Mandel, H.; Joseph, A.; Feigenbaum, A.; Elpeleg, O.N. Molecular Basis of Lipoamide Dehydrogenase Deficiency in Ashkenazi Jews. Am. J. Med. Genet. 1999, 82, 177–182. [Google Scholar] [CrossRef]
- Aptowitzer, I.; Saada, A.; Faber, J.; Kleid, D.E.O. Liver disease in the Ashkenazi-Jewish lipoamide dehydrogenase deficiency. J. Pediatr. Gastroenterol. Nutr. 1997, 24, 599–601. [Google Scholar] [CrossRef]
- Carrozzo, R.; Torraco, A.; Fiermonte, G.; Martinelli, D.; Di Nottia, M.; Rizza, T.; Vozza, A.; Verrigni, D.; Diodato, D.; Parisi, G.; et al. Riboflavin responsive mitochondrial myopathy is a new phenotype of dihydrolipoamide dehydrogenase deficiency: The chaperon-like effect of vitamin B2. Mitochondrion 2014, 18, 49–57. [Google Scholar] [CrossRef]
- Quintana, E.; Pineda, M.; Font, A.; Vilaseca, M.A.; Tort, F.; Ribes, A.; Briones, P. Dihydrolipoamide dehydrogenase (DLD) deficiency in a Spanish patient with myopathic presentation due to a new mutation in the interface domain. J. Inherit. Metab. Dis. 2010, 33, S315–S319. [Google Scholar] [CrossRef]
- Quinonez, S.C.; Thoene, J.G. Dihydrolipoamide Dehydrogenase Deficiency Summary. GeneReviews 2014, 1–19. [Google Scholar] [PubMed]
- Swink, T.D.; Vining, E.P.F.J. The ketogenic diet: 1997. Adv. Pediatr. 1997, 44, 297–329. [Google Scholar]
- Neal, E.G.; Chaffe, H.; Schwartz, R.H.; Lawson, M.S.; Edwards, N.; Fitzsimmons, G.; Whitney, A.; Cross, J.H. The ketogenic diet for the treatment of childhood epilepsy: A randomised controlled trial. Lancet Neurol. 2008, 7, 500–506. [Google Scholar] [CrossRef]
- Falk, R.; Cederbaum, S.; Blass, J.; Gibson, G.; Kark, R.; Carrel, R. Ketonic diet in the management of pyruvate dehydrogenase deficiency. Pediatrics 1976, 58, 713–721. [Google Scholar] [PubMed]
- Reed, L.J. Multienzyme Complexes. Acc. Chem. Res. 1973, 4, 40–46. [Google Scholar]
- Masino, S.A.; Rho, J.M. Mechanisms of ketogenic diet action. Epilepsia 2010, 51 (Suppl. 5), 85. [Google Scholar] [CrossRef]
- Sofou, K.; Dahlin, M.; Hallböök, T.; Lindefeldt, M.; Viggedal, G.; Darin, N. Ketogenic diet in pyruvate dehydrogenase complex deficiency: Short- and long-term outcomes. J. Inherit. Metab. Dis. 2017, 40, 237–245. [Google Scholar] [CrossRef] [Green Version]
- Zupec-Kania, B.A.; Spellman, E. An overview of the ketogenic diet for pediatric epilepsy. Nutr. Clin. Pract. 2008, 23, 589–596. [Google Scholar] [CrossRef]
- Zupec-Kania, B.A.; Aldaz, V.; Kostas, K.C. Enteral and Parenteral Applications of Ketogenic Diet Therapy: Experiences From Four Centers. Infant Child. Adolesc. Nutr. 2011, 3, 274–281. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.M.; Wang, H.S. Medium-chain triglyceride ketogenic diet, an effective treatment for drug-resistant epilepsy and a comparison with other ketogenic diets. Biomed. J. 2013, 36, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Kossoff, E.H.; Dorward, J.L. The modified Atkins diet. Epilepsia 2008, 49 (Suppl. 8), 37–41. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, H.H.; Thiele, E.A. Low-Glyecmic-Index Treatment: A liberalized ketogenic diet for treatment of intractable epilepsy. Neurology 2005, 65, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Rauchenzauner, M.; Klepper, J.; Leiendecker, B.; Luef, G.; Rostasy, K.; Ebenbichler, C. The Ketogenic Diet in Children with Glut1 Deficiency Syndrome and Epilepsy. J. Pediatrics 2008, 153, 716–718. [Google Scholar] [CrossRef]
- Wexler, I.D.; Hemalatha, S.G.; McConnell, J.; Buist, N.; Dahl, H.-H.M.; Berry, S.A.; Cederbaum, S.D.; Patel, M.S.; Kerr, D.S. Outcome of pyruvate dehydrogenase deficiency treated with ketogenic diets: Studies in patients with identical mutations. Neurology 1997, 49, 1655–1661. [Google Scholar] [CrossRef]
- Kim, J.A.; Yu, R.; Jo, W.; Ko, Y.H.; Lee, J.-S.; Kim, H.D.; Kang, H.-C. A modified Atkin’s diet for an infant with pyruvate dehydrogenase complex deficiency confirmed by PDHA1 gene mutation. Neurol. Asia 2014, 19, 327–329. [Google Scholar]
- Chida, R.; Shimura, M.; Nishimata, S.; Kashiwagi, Y.; Kawashima, H. Efficacy of ketogenic diet for pyruvate dehydrogenase complex deficiency. Pediatrics Int. 2018, 60, 1041–1042. [Google Scholar] [CrossRef]
- Martin, K.; Jackson, C.F.; Levy, R.G.; Cooper, P.N. Ketogenic diet and other dietary treatments for epilepsy. Cochrane Database Syst. Rev. 2016, 2, CD001903. [Google Scholar] [CrossRef] [Green Version]
- Robinson, B.H.; Taylor, J.; Kahler, S.G.; Kirkman, H.N. Lactic acidemia, neurologic deterioration and carbohydrate dependence in a girl with dihydrolipoyl dehydrogenase deficiency. Eur. J. Pediatr. 1981, 136, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Craigen, W.J. Leigh Disease with Deficiency of Lipoamide Dehydrogenase: Treatment Failure with Dichloroacetate. Pediatric Neurol. 1996, 14, 69–71. [Google Scholar] [CrossRef]
- Hong, Y.S.; Kerr, D.S.; Liu, T.; Lusk, M.; Powell, B.R.; Patel, M.S. Deficiency of dihydrolipoamide dehydrogenase due to two mutant alleles (E340K and G101del). Analysis of a family and prenatal testing. Biochim. Biophys. Acta 1997, 1362, 160–168. [Google Scholar] [CrossRef] [Green Version]
- Saudubray, J.-M.; Baumgartner, M.; Walter, J. Inborn Metabolic Diseases; Springer: Heidelberg, Germany, 2016. [Google Scholar]
- Ambrus, A.; Adam-Vizi, V. Human dihydrolipoamide dehydrogenase (E3) deficiency: Novel insights into the structural basis and molecular pathomechanism. Neurochem. Int. 2018, 117, 5–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaubel, R.A.; Rustin, P.; Isaya, G. Mutations in the dimer interface of dihydrolipoamide dehydrogenase promote site-specific oxidative damages in yeast and human cells. J. Biol. Chem. 2011, 286, 40232–40245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz Herrero, J.; Cañedo Villarroya, E.; García Peñas, J.J.; García Alcolea, B.; Gómez Fernández, B.; Puerta Macfarland, L.A.; Pedrón Giner, C. Safety and Effectiveness of the Prolonged Treatment of Children with a Ketogenic Diet. Nutrients 2020, 12, 306. [Google Scholar] [CrossRef] [Green Version]
- Cai, Q.Y.; Zhou, Z.J.; Luo, R.; Gan, J.; Li, S.P.; Mu, D.; Wan, C.M. Safety and tolerability of the ketogenic diet used for the treatment of refractory childhood epilepsy: A systematic review of published prospective studies. World J. Pediatr. 2017, 13, 528–536. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.; Pyzik, P.L.; Turner, Z.; Rubenstein, J.E.; Kossoff, E.H. Long-term outcomes of children treated with the ketogenic diet in the past. Epilepsia 2010, 51, 1277–1282. [Google Scholar] [CrossRef]
- Martin-McGill, K.J.; Jackson, C.F.; Bresnahan, R.; Levy, R.G.; Cooper, P.N. Ketogenic diets for drug-resistant epilepsy. Cochrane Database Syst. Rev. 2020, 6, CD001903. [Google Scholar] [CrossRef]
- Kossoff, E.H.; Dorward, J.L.; Turner, Z.; Pyzik, P.L. Prospective study of the modified Atkins diet in combination with a ketogenic liquid supplement during the initial month. J. Child Neurol. 2011, 26, 147–151. [Google Scholar] [CrossRef]
- Sondhi, V.; Agarwala, A.; Chakrabarty, B.; Jauhari, P.; Lodha, R.; Pandey, R.M.; Toteja, G.S.; Paul, V.K.; Gulati, S. Dietary Therapy in Epilepsy Treatment (DIET-Trial): A Randomised Non-Inferiority Trial Comparing KD, MAD & LGIT for Drug Resistant Epilepsy (S35.006). Neurology 2018, 90, S35.006. [Google Scholar]
- Sakaguchi, Y.; Yoshino, M.; Aramaki, S.; Yoshida, I.; Yamashita, F.; Kuhara, T.; Matsumoto, I.; Hayashi, T. Dihydrolipoyl dehydrogenase deficiency: A therapeutic trial with branched-chain amino acid restriction. Eur. J. Nucl. Med. Mol. Imaging 1986, 145, 271–274. [Google Scholar] [CrossRef]
- Bonnefont, J.-P.; Chretien, D.; Rustin, P.; Robinson, B.; Vassault, A.; Aupetit, J.; Charpentier, C.; Rabier, D.; Saudubray, J.-M.; Munnich, A. Alpha-ketoglutarate dehydrogenase deficiency presenting as congenital lactic acidosis. J. Pediatr. 1992, 121, 255–258. [Google Scholar] [CrossRef]
- Grafakou, O.; Oexle, K.; van den Heuvel, L.; Smeets, R.; Trijbels, F.; Goebel, H.H.; Bosshard, N.; Superti-Furga, A.; Steinmann, B.; Smeitink, J. Leigh syndrome due to a compound heterozygousity of dihydrolipoamide dehydrogenase gene mutations: Description of the first E3 splice site mutation. Eur. J. Pediatr. 2003, 162, 714–718. [Google Scholar] [CrossRef]
- Brassier, A.; Ottolenghi, C.; Boutron, A.; Bertrand, A.M.; Valmary-Degano, S.; Cervoni, J.P.; Chrétien, D.; Arnoux, J.B.; Hubert, L.; Rabier, D.; et al. Dihydrolipoamide dehydrogenase deficiency: A still overlooked cause of recurrent acute liver faliure and Reye-like syndrome. Mol. Genet. Metab. 2013, 109, 28–32. [Google Scholar] [CrossRef] [PubMed]
- Elpeleg, O.N.; Christensen, E.; Hurvitz, H.; Branski, D. Recurrent, familial Reye-like syndrome with a new complex amino and organic aciduria. Eur. J. Nucl. Med. Mol. Imaging 1990, 149, 709–712. [Google Scholar] [CrossRef]

{kind=link}
| Patient No. | Age * | Weight * (Kg) | Height * (cm) | Kcal | Kcal/Kg | Protein | Protein/Kg | %Fat | Ketogenic Ratio |
|---|---|---|---|---|---|---|---|---|---|
| 12 | 3 y 3 m | 7.900 | 79.8 | 1284 | 162.5 | 27.7 | 3.5 | 67.9 | 1:1.06 |
| 7 | 4 y 2 m | 12.55 | 99 | 1362 | 108.5 | 41.8 | 3.3 | 61.5 | 1:1.4 |
| 16 | 3 y | 7.940 | 79 | 923.2 | 116.2 | 20.1 | 2.53 | 63.5 | 1:1.29 |
| 6 | 5 y | 10.900 | 95 | 1027 | 94.2 | 32.1 | 2.94 | 61.5 | 1:1.4 |
| Pt. No. | Gestational Age | Gender | Current Age/Death * (d/m/y) | First Presentation | Age at First Presentation | Treatment | No. of Hospitalizations (Per Year) |
|---|---|---|---|---|---|---|---|
| 1 h | 33 + 2 weeks | F | * 37 m | Metabolic acidosis | Birth | Started Ketocal at 2 y | 2–5 |
| 2 h | Term/SGA | F | * 29 m | Metabolic acidosis | Birth | Bicarbonate in crisis | 2 |
| 3 h | Term/SGA | M | * 3 d | Metabolic acidosis | Birth | Fluids | - |
| 4 h | Term | M | * 4 d | Metabolic acidosis | 2 d | Fluids | - |
| 5 h | Term | F | * 3 m | Metabolic acidosis | Birth | Fluids | 1 |
| k 6 | Late Preterm | M | 4.9 y | Metabolic acidosis and hypoglycemia | 1 d | Partial ketogenic diet and Bicarbonate | 2 |
| k 7 | Term | M | 4.1 y | Metabolic acidosis and hypoglycemia | 1.5 d | Partial ketogenic diet and Bicarbonate | 1–6 |
| 8 h | Term | M | * 3 d | Metabolic acidosis | Birth | - | - |
| 9 h | Term | F | 16 y | Metabolic acidosis and cutis | Birth | Canola oil | 1–2 |
| 10 h | Term/SGA | M | 14.2 y | Metabolic acidosis and hypertonus | Birth | - | 2–5 |
| 11 | Term | M | 4.3 y | Metabolic acidosis | Birth | Bicarbonate | 1 |
| k 12 | Term | F | 3.2 y | Metabolic acidosis | Birth | Partial ketogenic diet and Bicarbonate | 4–6 |
| 13 | Term | M | 3.5 y | Metabolic acidosis | 18 m | Bicarbonate | 4 |
| 14 | Term/SGA | F | 1.5 y | Metabolic acidosis | Birth | Bicarbonate | 4 |
| 15 | Term/SGA | F | 1.5 y | Metabolic acidosis | Birth | Bicarbonate | 4 |
| k 16 | Term | M | 2.8 y | Metabolic acidosis and seizures | Birth | Partial ketogenic diet and Bicarbonate | 2–3 |
| Pt. No. | Current Age/Death * (D)-(d/m/y) | GER | Liver Disease | Hypoglycemia | Most Advanced Development | Seizure | DD/ID/ PMD |
|---|---|---|---|---|---|---|---|
| 1 h | * 37 m | No | No | No | Smiled | No | Severe |
| 2 h | * 29 m | No | No | Repeated | Rolled both sides, said syllables | No | Severe |
| 3 h | * 3 d | No | No | Yes | NA | No | NA |
| 4 h | * 4 d | No | No | Yes | NA | No | NA |
| 5 h | * 3 m | No | No | Yes | Hypertonus, no spontaneous smiles | No | Severe |
| k 6 | 4.9 y | No | During crisis | Yes | Sat, rolled, said five words but regressed at 46 months and now does not say a word and no eye contact | No | Severe |
| k 7 | 4.1 y | Severe | During crisis | Yes | Rolls both sides, says syllables, sat with support, said 7 words but regressed at 48 months and now does not say a word and only smiles | Yes | Severe |
| 8 h | * 3 d | No | No | No | NA | No | NA |
| 9 h | 16 y | No | Mild during crisis | No | Severe spastic PMD, no communication, mastication movements | No | Severe |
| 10 h | 14.2 y | No | During crisis | During crisis | Severe spastic PMD, no communication | No | Severe |
| 11 | 4.3 y | No | No | During crisis | Smiles, says syllables, rolls and sits by himself | No | Moderate to severe |
| k 12 | 3.2 y | No | No | During crisis | Smiles; says syllables, rolls both sides, transfers objects, holds her bottle, sits with support | Yes | Moderate to severe |
| 13 | 3.5 y | No | No | No | Wandering eyes, no communication | Yes | Severe |
| 14 | 1.5 y | No | No | During crisis | Smiles, rolls both sides, says syllables | No | Moderate to severe |
| 15 | 1.5 y | No | No | During crisis | Smiles, rolls both sides, says syllables, sits with support | No | Moderate to severe |
| k 16 | 2.8 y | No | No | No | Smiles, rolls both sides, says syllables | Yes | Severe |
| Pt. No. | Urine Organic Acids | Highest Lactate (Norm: 0.5–2.2 mmol/L) | Pyruvate (Norm: 0.03–0.08 mmol/L) | Lactate/Pyruvate Ratio |
|---|---|---|---|---|
| 1 h | NA | 19 | NA | NA |
| 2 h | Elevated lactate, 2-OH-Butyrate Elevated 3-OH-Butyrate > Acetoacetate | 7.6 | 1.98 | 3.88 |
| 3 h | NA | 17 | 2 | 8.5 |
| 4 h | NA | 18 | 1 | 18 |
| 5 h | NA | 5.8 | 0.3 | 19.3 |
| k 6 | Elevated lactate, 2-OH-Butyrate Elevated 3-OH-Butyrate > Acetoacetate | 14.3 | 0.54 | 26.5 |
| k 7 | Massively elevated lactate and moderately dicarboxylic and 3-hydroxybutyric acids | 29.9 | 0.28 | 9.2 * |
| 8 h | NA | NA | NA | NA |
| 9 h | NA | 10.4 | 0.38 | 27.4 |
| 10 h | NA | 11.5 | 0.73 | 15.7 |
| 11 | NA | 5.7 | 0.07 | 81.1 |
| k 12 | Elevated lactate, 2-OH-Butyrate Elevated 3-OH-Butyrate > Acetoacetate | 9.5 | 0.1 | 95.5 |
| 13 | Massively elevated lactate and ketones and Krebs metabolites | 14.8 | 0.17 | 87 |
| 14 | NA | 5.6 | 0.06 | 93.2 |
| 15 | NA | 3.2 | 0.1 | 31.9 |
| k 16 | NA | 8.2 | 0.23 | 35.5 |
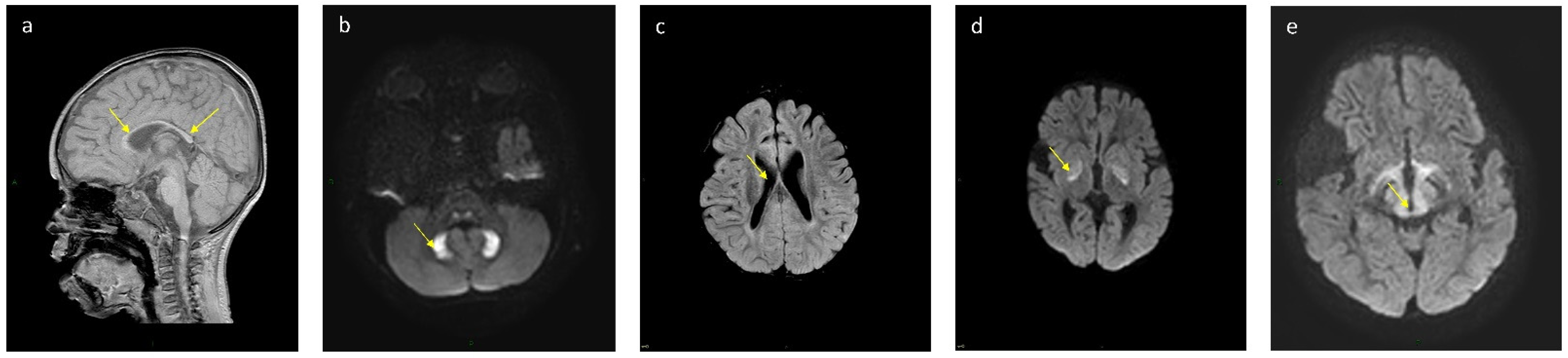
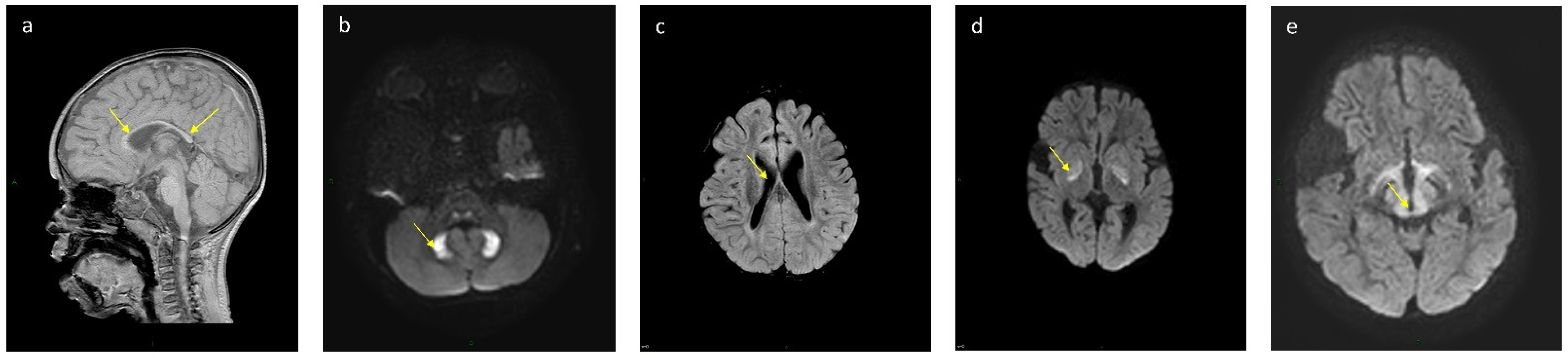
| Patient No. | Globus Pallidum | Dentate Nucleus | Corpus Callosum | Ventriculomegaly | Midbrain and Thalamus |
|---|---|---|---|---|---|
| k 16 | T2, Flair, DWI high signal, T1 low signal | Normal | Partial agenesis | Mild | Normal |
| 11 | T2, Flair, DWI high signal, T1 low signal | T2, Flair, DWI high signal, T1 low signal | Partial agenesis | No | Normal |
| k 12 | T2, Flair, DWI high signal, T1 low signal | Normal | Partial agenesis | Mild | Normal |
| 13 | T2, Flair, DWI high signal, T1 low signal | Normal | Partial agenesis | Moderate | Normal |
| k 6 | T2, Flair, DWI high signal, T1 low signal | Normal | Partial agenesis | Mild | Normal |
| k 7 | Normal | T2, Flair, DWI high signal, T1 low signal | Partial agenesis | Mild | T2, Flair, DWI high signal, T1 low signal |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Staretz-Chacham, O.; Pode-Shakked, B.; Kristal, E.; Abraham, S.Y.; Porper, K.; Wormser, O.; Shelef, I.; Anikster, Y. The Effects of a Ketogenic Diet on Patients with Dihydrolipoamide Dehydrogenase Deficiency. Nutrients 2021, 13, 3523. https://doi.org/10.3390/nu13103523
Staretz-Chacham O, Pode-Shakked B, Kristal E, Abraham SY, Porper K, Wormser O, Shelef I, Anikster Y. The Effects of a Ketogenic Diet on Patients with Dihydrolipoamide Dehydrogenase Deficiency. Nutrients. 2021; 13(10):3523. https://doi.org/10.3390/nu13103523
Chicago/Turabian StyleStaretz-Chacham, Orna, Ben Pode-Shakked, Eyal Kristal, Smadar Yaala Abraham, Keren Porper, Ohad Wormser, Ilan Shelef, and Yair Anikster. 2021. "The Effects of a Ketogenic Diet on Patients with Dihydrolipoamide Dehydrogenase Deficiency" Nutrients 13, no. 10: 3523. https://doi.org/10.3390/nu13103523
APA StyleStaretz-Chacham, O., Pode-Shakked, B., Kristal, E., Abraham, S. Y., Porper, K., Wormser, O., Shelef, I., & Anikster, Y. (2021). The Effects of a Ketogenic Diet on Patients with Dihydrolipoamide Dehydrogenase Deficiency. Nutrients, 13(10), 3523. https://doi.org/10.3390/nu13103523