Role of Gut Microbiota and Their Metabolites on Atherosclerosis, Hypertension and Human Blood Platelet Function: A Review


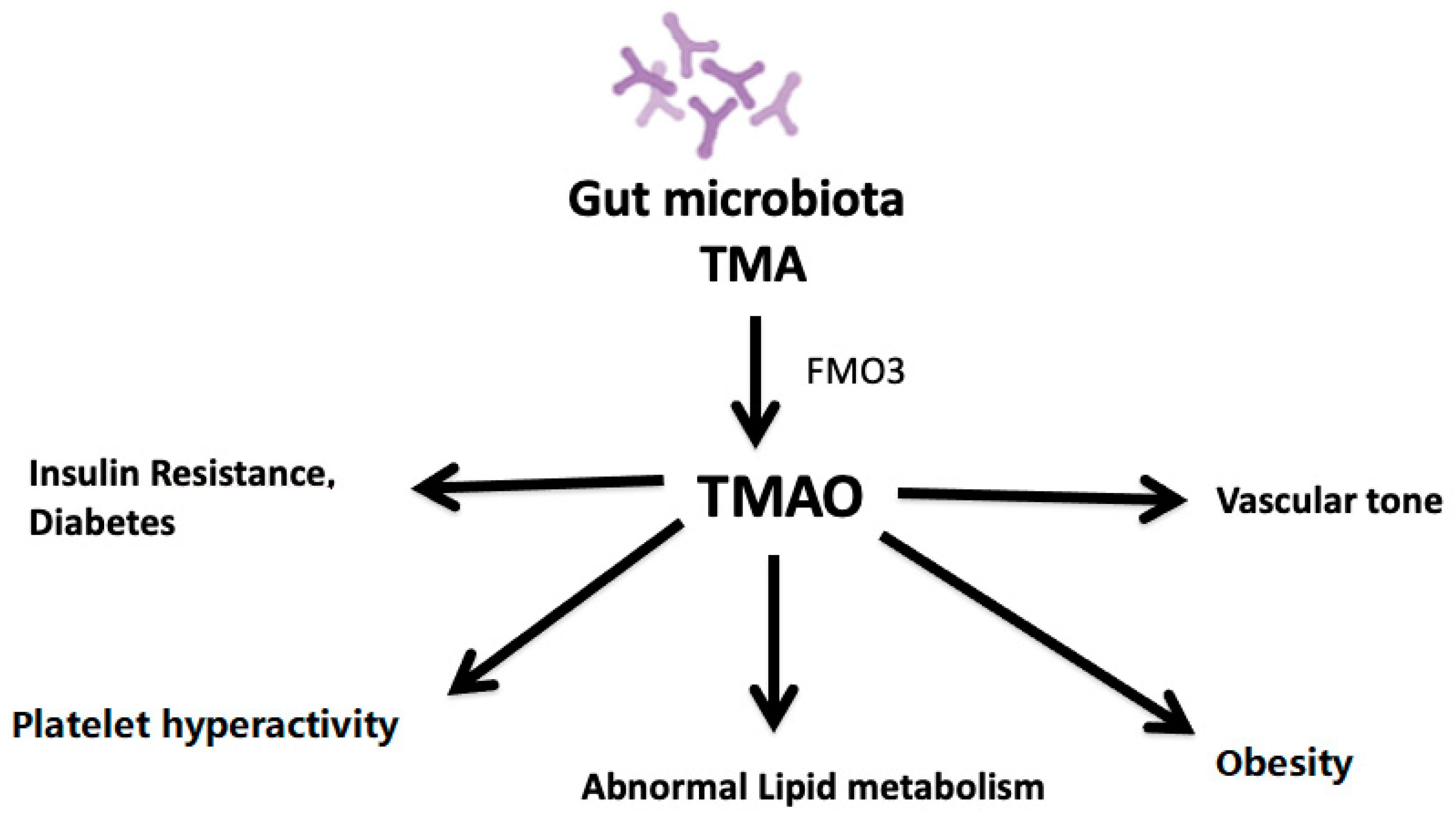
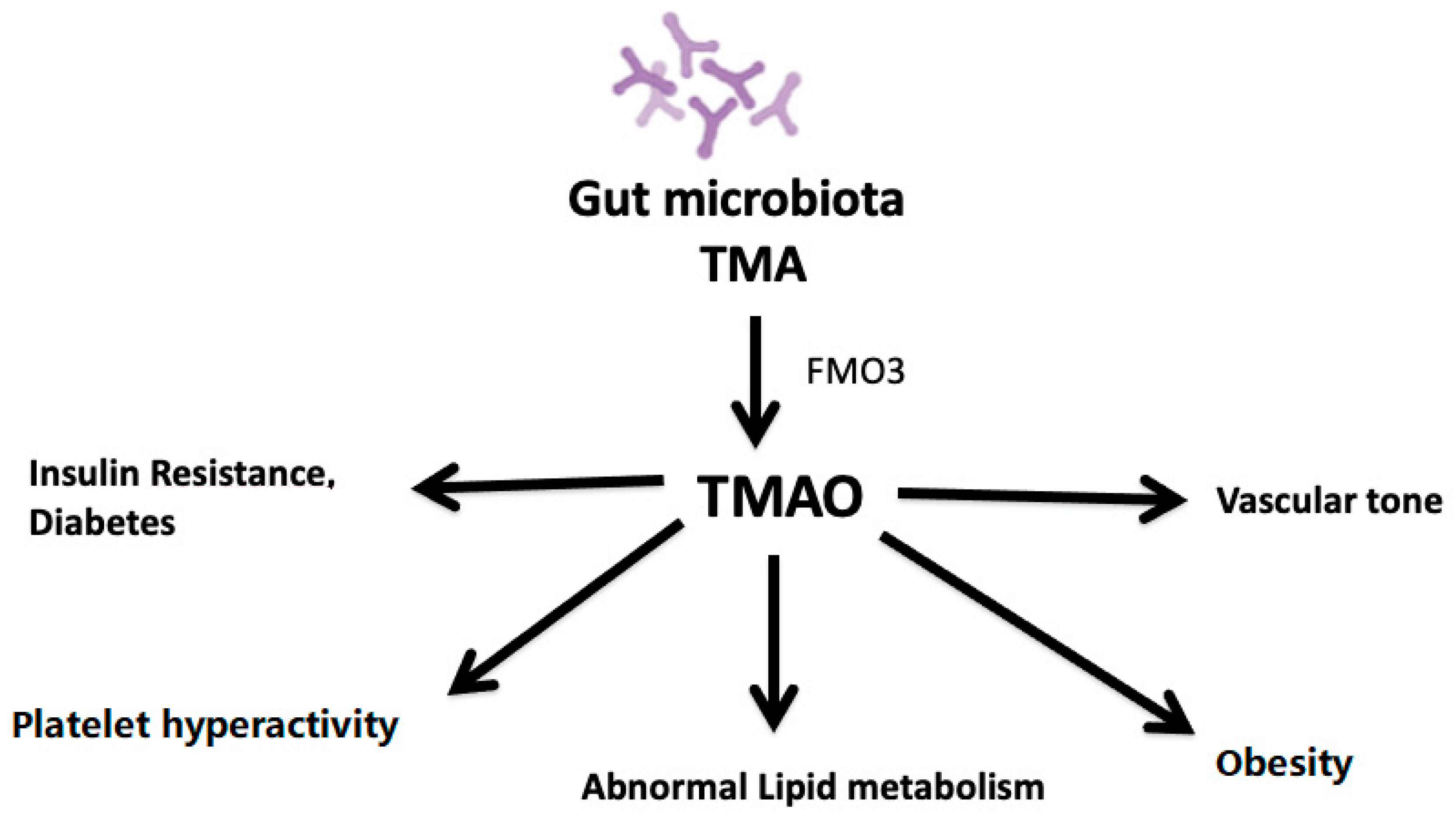{kind=link}
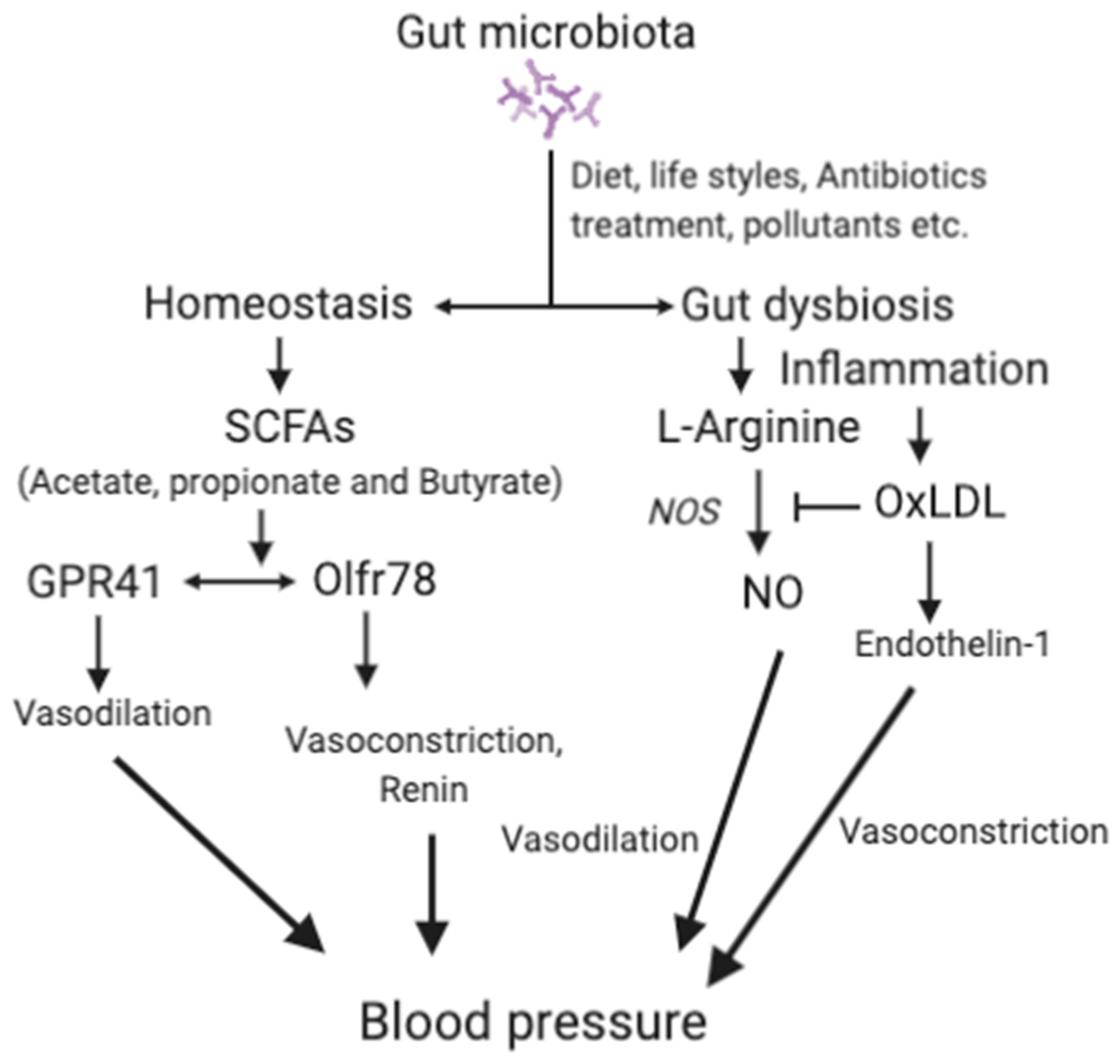
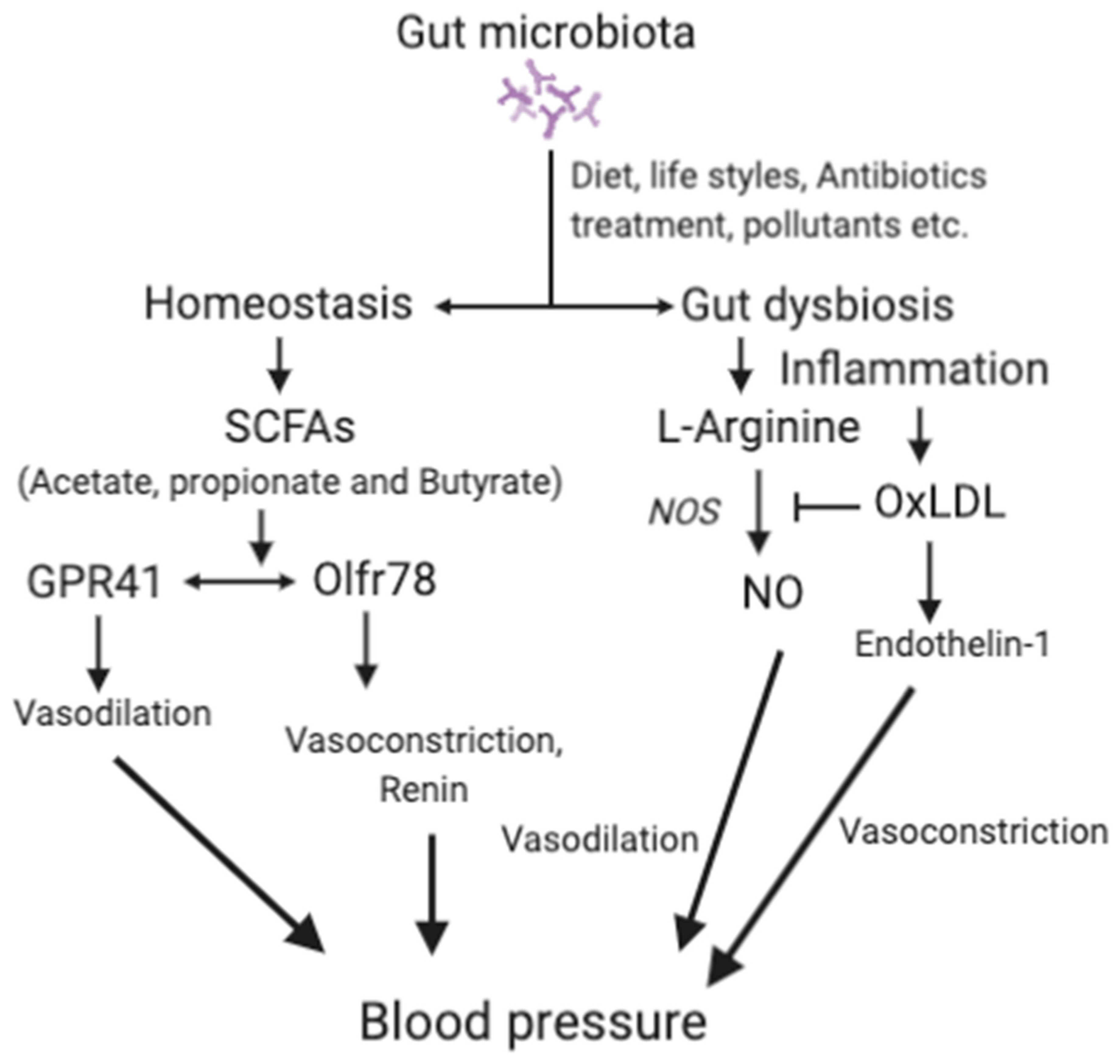{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Roles of the Gut Microbiota in the Atherosclerosis Process
3. Contribution of the Gut Microbiota to Hypertension
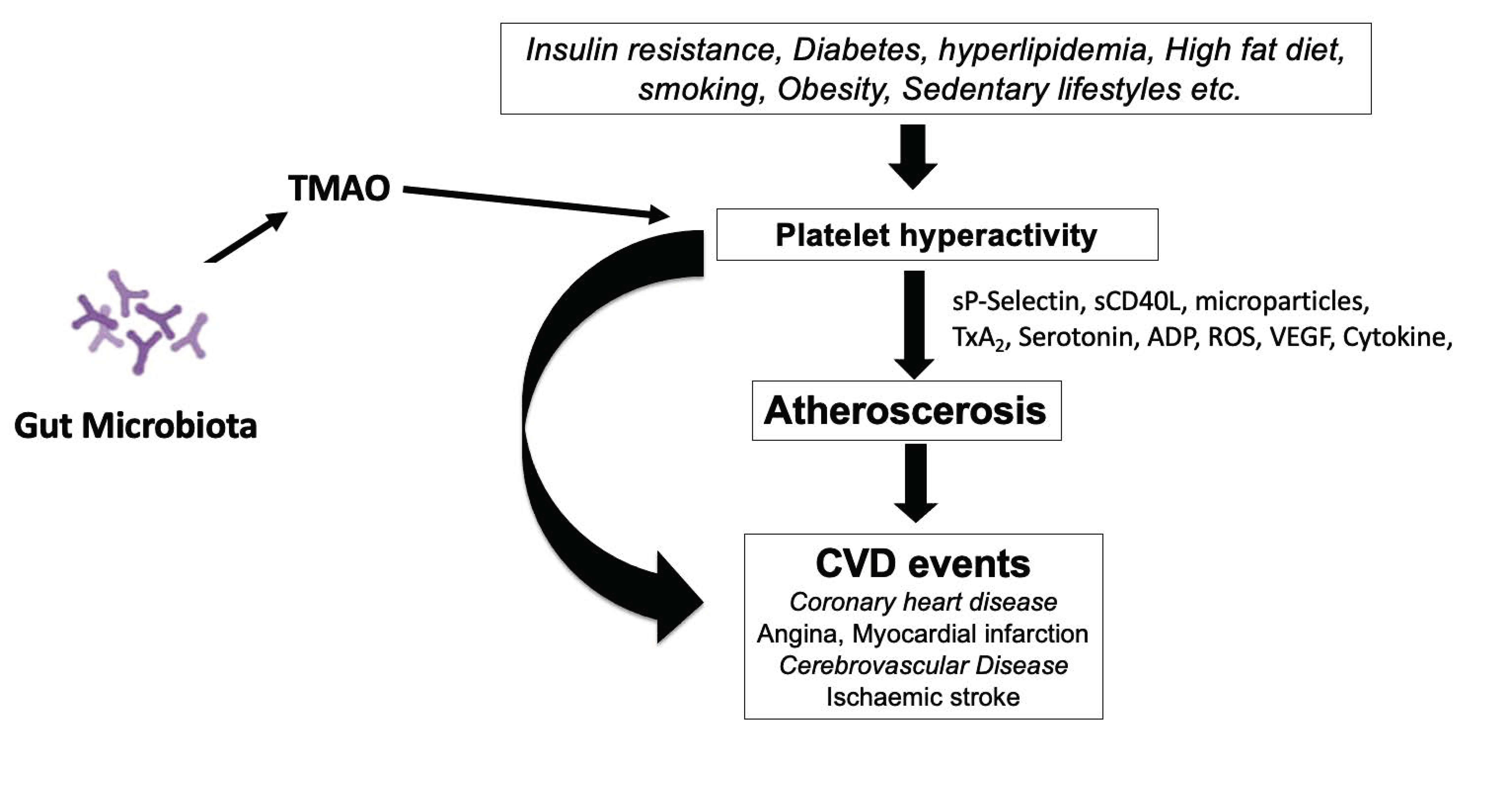
4. Platelet Hyperactivity is an Important Contributor to the Development of CVD
5. Conclusions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Cardiovascular disease | CVD |
| Trimethylamine-N-oxide | TMO |
| Glycoprotein IIb/IIIa | GPIIb/IIIa |
| von Willebrand factor | vWF |
| Tissue factor | TF |
| P-Selectin | CD62P |
| Short-chain fatty acids | SCFA |
| Peptidoglycan | PG |
References
- Kwan, G.F.; Mayosi, B.M.; Mocumbi, A.O.; Miranda, J.J.; Ezzati, M.; Jain, Y.; Robles, G.; Benjamin, E.J.; Subramanian, S.V.; Bukhman, G. Endemic Cardiovascular Diseases of the Poorest Billion. Circulation 2016, 133, 2561–2575. [Google Scholar] [CrossRef] [PubMed]
- Writing Group, M.; Mozaffarian, D.; Benjamin, E.J.; Go, A.S.; Arnett, D.K.; Blaha, M.J.; Cushman, M.; Das, S.R.; de Ferranti, S.; Despres, J.P.; et al. Heart Disease and Stroke Statistics-2016 Update: A Report From the American Heart Association. Circulation 2016, 133, e38–e360. [Google Scholar] [CrossRef]
- Zhu, W.; Gregory, J.C.; Org, E.; Buffa, J.A.; Gupta, N.; Wang, Z.; Li, L.; Fu, X.; Wu, Y.; Mehrabian, M.; et al. Gut Microbial Metabolite TMAO Enhances Platelet Hyperreactivity and Thrombosis Risk. Cell 2016, 165, 111–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Zhang, W.; Zuo, L.; Dong, J.; Zhu, W.; Li, Y.; Gu, L.; Gong, J.; Li, Q.; Li, N.; et al. Intestinal dysbacteriosis contributes to decreased intestinal mucosal barrier function and increased bacterial translocation. Lett. Appl. Microbiol. 2014, 58, 384–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willoughby, S.; Holmes, A.; Loscalzo, J. Platelets and cardiovascular disease. Eur. J. Cardiovasc. Nurs. 2002, 1, 273–288. [Google Scholar] [CrossRef]
- Michelson, A.D. Antiplatelet therapies for the treatment of cardiovascular disease. Nat. Rev. Drug Discov. 2010, 9, 154–169. [Google Scholar] [CrossRef]
- Jackson, S.P.; Nesbitt, W.S.; Westein, E. Dynamics of platelet thrombus formation. J. Thromb. Haemost. 2009, 7 (Suppl. 1), 17–20. [Google Scholar] [CrossRef]
- Wang, Z.; Klipfell, E.; Bennett, B.J.; Koeth, R.; Levison, B.S.; Dugar, B.; Feldstein, A.E.; Britt, E.B.; Fu, X.; Chung, Y.M.; et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 2011, 472, 57–63. [Google Scholar] [CrossRef] [Green Version]
- Lau, K.; Srivatsav, V.; Rizwan, A.; Nashed, A.; Liu, R.; Shen, R.; Akhtar, M. Bridging the Gap between Gut Microbial Dysbiosis and Cardiovascular Diseases. Nutrients 2017, 9, 859. [Google Scholar] [CrossRef] [Green Version]
- Anbazhagan, A.N.; Priyamvada, S.; Priyadarshini, M. Gut Microbiota in Vascular Disease: Therapeutic Target? Curr. Vasc. Pharm. 2017, 15, 291–295. [Google Scholar] [CrossRef]
- Santisteban, M.M.; Qi, Y.; Zubcevic, J.; Kim, S.; Yang, T.; Shenoy, V.; Cole-Jeffrey, C.T.; Lobaton, G.O.; Stewart, D.C.; Rubiano, A.; et al. Hypertension-Linked Pathophysiological Alterations in the Gut. Circ. Res. 2017, 120, 312–323. [Google Scholar] [CrossRef] [PubMed]
- Gui, T.; Shimokado, A.; Sun, Y.; Akasaka, T.; Muragaki, Y. Diverse roles of macrophages in atherosclerosis: From inflammatory biology to biomarker discovery. Mediat. Inflamm. 2012, 2012, 693083. [Google Scholar] [CrossRef] [PubMed]
- Drosos, I.; Tavridou, A.; Kolios, G. New aspects on the metabolic role of intestinal microbiota in the development of atherosclerosis. Metabolism 2015, 64, 476–481. [Google Scholar] [CrossRef] [PubMed]
- Gregory, J.C.; Buffa, J.A.; Org, E.; Wang, Z.; Levison, B.S.; Zhu, W.; Wagner, M.A.; Bennett, B.J.; Li, L.; DiDonato, J.A.; et al. Transmission of atherosclerosis susceptibility with gut microbial transplantation. J. Biol. Chem. 2015, 290, 5647–5660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jie, Z.; Xia, H.; Zhong, S.L.; Feng, Q.; Li, S.; Liang, S.; Zhong, H.; Liu, Z.; Gao, Y.; Zhao, H.; et al. The gut microbiome in atherosclerotic cardiovascular disease. Nat. Commun. 2017, 8, 845. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Barnes, G.T.; Yang, Q.; Tan, G.; Yang, D.; Chou, C.J.; Sole, J.; Nichols, A.; Ross, J.S.; Tartaglia, L.A.; et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Investig. 2003, 112, 1821–1830. [Google Scholar] [CrossRef]
- Ding, S.; Chi, M.M.; Scull, B.P.; Rigby, R.; Schwerbrock, N.M.; Magness, S.; Jobin, C.; Lund, P.K. High-fat diet: Bacteria interactions promote intestinal inflammation which precedes and correlates with obesity and insulin resistance in mouse. PLoS ONE 2010, 5, e12191. [Google Scholar] [CrossRef] [Green Version]
- Kasahara, K.; Tanoue, T.; Yamashita, T.; Yodoi, K.; Matsumoto, T.; Emoto, T.; Mizoguchi, T.; Hayashi, T.; Kitano, N.; Sasaki, N.; et al. Commensal bacteria at the crossroad between cholesterol homeostasis and chronic inflammation in atherosclerosis. J. Lipid Res. 2017, 58, 519–528. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Tang, W.H.; Buffa, J.A.; Fu, X.; Britt, E.B.; Koeth, R.A.; Levison, B.S.; Fan, Y.; Wu, Y.; Hazen, S.L. Prognostic value of choline and betaine depends on intestinal microbiota-generated metabolite trimethylamine-N-oxide. Eur. Heart J. 2014, 35, 904–910. [Google Scholar] [CrossRef]
- Chen, W.Y.; Wang, M.; Zhang, J.; Barve, S.S.; McClain, C.J.; Joshi-Barve, S. Acrolein Disrupts Tight Junction Proteins and Causes Endoplasmic Reticulum Stress-Mediated Epithelial Cell Death Leading to Intestinal Barrier Dysfunction and Permeability. Am. J. Pathol. 2017, 187, 2686–2697. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Lin, S.; Vanhoutte, P.M.; Woo, C.W.; Xu, A. Akkermansia Muciniphila Protects Against Atherosclerosis by Preventing Metabolic Endotoxemia-Induced Inflammation in Apoe−/− Mice. Circulation 2016, 133, 2434–2446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karlsson, F.H.; Fak, F.; Nookaew, I.; Tremaroli, V.; Fagerberg, B.; Petranovic, D.; Backhed, F.; Nielsen, J. Symptomatic atherosclerosis is associated with an altered gut metagenome. Nat. Commun. 2012, 3, 1245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andraws, R.; Berger, J.S.; Brown, D.L. Effects of antibiotic therapy on outcomes of patients with coronary artery disease: A meta-analysis of randomized controlled trials. JAMA 2005, 293, 2641–2647. [Google Scholar] [CrossRef] [PubMed]
- Desai, M.S.; Seekatz, A.M.; Koropatkin, N.M.; Kamada, N.; Hickey, C.A.; Wolter, M.; Pudlo, N.A.; Kitamoto, S.; Terrapon, N.; Muller, A.; et al. A Dietary Fiber-Deprived Gut Microbiota Degrades the Colonic Mucus Barrier and Enhances Pathogen Susceptibility. Cell 2016, 167, 1339–1353.e1321. [Google Scholar] [CrossRef] [Green Version]
- Kholy, K.E.; Genco, R.J.; Van Dyke, T.E. Oral infections and cardiovascular disease. Trends Endocrinol. Metab. 2015, 26, 315–321. [Google Scholar] [CrossRef]
- Wiedermann, C.J.; Kiechl, S.; Dunzendorfer, S.; Schratzberger, P.; Egger, G.; Oberhollenzer, F.; Willeit, J. Association of endotoxemia with carotid atherosclerosis and cardiovascular disease: Prospective results from the Bruneck Study. J. Am. Coll. Cardiol. 1999, 34, 1975–1981. [Google Scholar] [CrossRef] [Green Version]
- Miller, M.A.; McTernan, P.G.; Harte, A.L.; Silva, N.F.; Strazzullo, P.; Alberti, K.G.; Kumar, S.; Cappuccio, F.P. Ethnic and sex differences in circulating endotoxin levels: A novel marker of atherosclerotic and cardiovascular risk in a British multi-ethnic population. Atherosclerosis 2009, 203, 494–502. [Google Scholar] [CrossRef]
- Mitra, S.; Drautz-Moses, D.I.; Alhede, M.; Maw, M.T.; Liu, Y.; Purbojati, R.W.; Yap, Z.H.; Kushwaha, K.K.; Gheorghe, A.G.; Bjarnsholt, T.; et al. In silico analyses of metagenomes from human atherosclerotic plaque samples. Microbiome 2015, 3, 38. [Google Scholar] [CrossRef] [Green Version]
- Cani, P.D.; Bibiloni, R.; Knauf, C.; Waget, A.; Neyrinck, A.M.; Delzenne, N.M.; Burcelin, R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes 2008, 57, 1470–1481. [Google Scholar] [CrossRef] [Green Version]
- Harris, K.; Kassis, A.; Major, G.; Chou, C.J. Is the gut microbiota a new factor contributing to obesity and its metabolic disorders? J. Obes. 2012, 2012, 879151. [Google Scholar] [CrossRef] [Green Version]
- Chacon, M.R.; Lozano-Bartolome, J.; Portero-Otin, M.; Rodriguez, M.M.; Xifra, G.; Puig, J.; Blasco, G.; Ricart, W.; Chaves, F.J.; Fernandez-Real, J.M. The gut mycobiome composition is linked to carotid atherosclerosis. Benef. Microbes 2018, 9, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.H.; Shah, P.K.; Faure, E.; Equils, O.; Thomas, L.; Fishbein, M.C.; Luthringer, D.; Xu, X.P.; Rajavashisth, T.B.; Yano, J.; et al. Toll-like receptor-4 is expressed by macrophages in murine and human lipid-rich atherosclerotic plaques and upregulated by oxidized LDL. Circulation 2001, 104, 3103–3108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edfeldt, K.; Swedenborg, J.; Hansson, G.K.; Yan, Z.Q. Expression of toll-like receptors in human atherosclerotic lesions: A possible pathway for plaque activation. Circulation 2002, 105, 1158–1161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barton, G.M.; Kagan, J.C. A cell biological view of Toll-like receptor function: Regulation through compartmentalization. Nat. Rev. Immunol. 2009, 9, 535–542. [Google Scholar] [CrossRef]
- Guzzo, C.; Ayer, A.; Basta, S.; Banfield, B.W.; Gee, K. IL-27 enhances LPS-induced proinflammatory cytokine production via upregulation of TLR4 expression and signaling in human monocytes. J. Immunol. 2012, 188, 864–873. [Google Scholar] [CrossRef] [Green Version]
- Laman, J.D.; Schoneveld, A.H.; Moll, F.L.; van Meurs, M.; Pasterkamp, G. Significance of peptidoglycan, a proinflammatory bacterial antigen in atherosclerotic arteries and its association with vulnerable plaques. Am. J. Cardiol. 2002, 90, 119–123. [Google Scholar] [CrossRef]
- Philpott, D.J.; Sorbara, M.T.; Robertson, S.J.; Croitoru, K.; Girardin, S.E. NOD proteins: Regulators of inflammation in health and disease. Nat. Rev. Immunol. 2014, 14, 9–23. [Google Scholar] [CrossRef]
- Munford, R.S. Endotoxemia-menace, marker, or mistake? J. Leukoc. Biol. 2016, 100, 687–698. [Google Scholar] [CrossRef]
- Grayston, J.T.; Kronmal, R.A.; Jackson, L.A.; Parisi, A.F.; Muhlestein, J.B.; Cohen, J.D.; Rogers, W.J.; Crouse, J.R.; Borrowdale, S.L.; Schron, E.; et al. Azithromycin for the secondary prevention of coronary events. N. Engl. J. Med. 2005, 352, 1637–1645. [Google Scholar] [CrossRef] [Green Version]
- Lam, V.; Su, J.; Koprowski, S.; Hsu, A.; Tweddell, J.S.; Rafiee, P.; Gross, G.J.; Salzman, N.H.; Baker, J.E. Intestinal microbiota determine severity of myocardial infarction in rats. FASEB J. 2012, 26, 1727–1735. [Google Scholar] [CrossRef] [Green Version]
- Brown, J.M.; Hazen, S.L. The gut microbial endocrine organ: Bacterially derived signals driving cardiometabolic diseases. Annu. Rev. Med. 2015, 66, 343–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergeron, N.; Williams, P.T.; Lamendella, R.; Faghihnia, N.; Grube, A.; Li, X.; Wang, Z.; Knight, R.; Jansson, J.K.; Hazen, S.L.; et al. Diets high in resistant starch increase plasma levels of trimethylamine-N-oxide, a gut microbiome metabolite associated with CVD risk. Br. J. Nutr. 2016, 116, 2020–2029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Shimizu, Y.; Kimura, I. Gut microbial metabolite short-chain fatty acids and obesity. Biosci. Microbiota Food Health 2017, 36, 135–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Randrianarisoa, E.; Lehn-Stefan, A.; Wang, X.; Hoene, M.; Peter, A.; Heinzmann, S.S.; Zhao, X.; Konigsrainer, I.; Konigsrainer, A.; Balletshofer, B.; et al. Relationship of Serum Trimethylamine N-Oxide (TMAO) Levels with early Atherosclerosis in Humans. Sci. Rep. 2016, 6, 26745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Roberts, A.B.; Buffa, J.A.; Levison, B.S.; Zhu, W.; Org, E.; Gu, X.; Huang, Y.; Zamanian-Daryoush, M.; Culley, M.K.; et al. Non-lethal Inhibition of Gut Microbial Trimethylamine Production for the Treatment of Atherosclerosis. Cell 2015, 163, 1585–1595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koeth, R.A.; Wang, Z.; Levison, B.S.; Buffa, J.A.; Org, E.; Sheehy, B.T.; Britt, E.B.; Fu, X.; Wu, Y.; Li, L.; et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat. Med. 2013, 19, 576–585. [Google Scholar] [CrossRef] [Green Version]
- Li, X.S.; Obeid, S.; Klingenberg, R.; Gencer, B.; Mach, F.; Raber, L.; Windecker, S.; Rodondi, N.; Nanchen, D.; Muller, O.; et al. Gut microbiota-dependent trimethylamine N-oxide in acute coronary syndromes: A prognostic marker for incident cardiovascular events beyond traditional risk factors. Eur. Heart J. 2017, 38, 814–824. [Google Scholar] [CrossRef]
- Brown, J.M.; Hazen, S.L. Microbial modulation of cardiovascular disease. Nat. Rev. Microbiol. 2018, 16, 171–181. [Google Scholar] [CrossRef]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [Green Version]
- Backhed, F.; Ley, R.E.; Sonnenburg, J.L.; Peterson, D.A.; Gordon, J.I. Host-bacterial mutualism in the human intestine. Science 2005, 307, 1915–1920. [Google Scholar] [CrossRef] [Green Version]
- Bennett, B.J.; de Aguiar Vallim, T.Q.; Wang, Z.; Shih, D.M.; Meng, Y.; Gregory, J.; Allayee, H.; Lee, R.; Graham, M.; Crooke, R.; et al. Trimethylamine-N-oxide, a metabolite associated with atherosclerosis, exhibits complex genetic and dietary regulation. Cell Metab. 2013, 17, 49–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brugere, J.F.; Borrel, G.; Gaci, N.; Tottey, W.; O’Toole, P.W.; Malpuech-Brugere, C. Archaebiotics: Proposed therapeutic use of archaea to prevent trimethylaminuria and cardiovascular disease. Gut Microbes 2014, 5, 5–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dridi, B.; Fardeau, M.L.; Ollivier, B.; Raoult, D.; Drancourt, M. Methanomassiliicoccus luminyensis gen. nov., sp. nov., a methanogenic archaeon isolated from human faeces. Int. J. Syst. Evol. Microbiol. 2012, 62, 1902–1907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.L.; Yi, L.; Zhang, Y.; Zhou, X.; Ran, L.; Yang, J.; Zhu, J.D.; Zhang, Q.Y.; Mi, M.T. Resveratrol Attenuates Trimethylamine-N-Oxide (TMAO)-Induced Atherosclerosis by Regulating TMAO Synthesis and Bile Acid Metabolism via Remodeling of the Gut Microbiota. MBio 2016, 7, e02210–e02215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larrosa, M.; Yanez-Gascon, M.J.; Selma, M.V.; Gonzalez-Sarrias, A.; Toti, S.; Ceron, J.J.; Tomas-Barberan, F.; Dolara, P.; Espin, J.C. Effect of a low dose of dietary resveratrol on colon microbiota, inflammation and tissue damage in a DSS-induced colitis rat model. J. Agric. Food Chem. 2009, 57, 2211–2220. [Google Scholar] [CrossRef]
- Sun, G.; Yin, Z.; Liu, N.; Bian, X.; Yu, R.; Su, X.; Zhang, B.; Wang, Y. Gut microbial metabolite TMAO contributes to renal dysfunction in a mouse model of diet-induced obesity. Biochem. Biophys. Res. Commun. 2017, 493, 964–970. [Google Scholar] [CrossRef]
- Bu, J.; Wang, Z. Cross-Talk between Gut Microbiota and Heart via the Routes of Metabolite and Immunity. Gastroenterol. Res. Pr. 2018, 2018, 6458094. [Google Scholar] [CrossRef]
- Qi, J.; You, T.; Li, J.; Pan, T.; Xiang, L.; Han, Y.; Zhu, L. Circulating trimethylamine N-oxide and the risk of cardiovascular diseases: A systematic review and meta-analysis of 11 prospective cohort studies. J. Cell. Mol. Med. 2018, 22, 185–194. [Google Scholar] [CrossRef]
- Heianza, Y.; Ma, W.; Manson, J.E.; Rexrode, K.M.; Qi, L. Gut Microbiota Metabolites and Risk of Major Adverse Cardiovascular Disease Events and Death: A Systematic Review and Meta-Analysis of Prospective Studies. J. Am. Heart Assoc. 2017, 6, e004947. [Google Scholar] [CrossRef]
- Tang, W.H.; Wang, Z.; Levison, B.S.; Koeth, R.A.; Britt, E.B.; Fu, X.; Wu, Y.; Hazen, S.L. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N. Engl. J. Med. 2013, 368, 1575–1584. [Google Scholar] [CrossRef] [Green Version]
- Fu, Q.; Zhao, M.; Wang, D.; Hu, H.; Guo, C.; Chen, W.; Li, Q.; Zheng, L.; Chen, B. Coronary Plaque Characterization Assessed by Optical Coherence Tomography and Plasma Trimethylamine-N-oxide Levels in Patients With Coronary Artery Disease. Am. J. Cardiol. 2016, 118, 1311–1315. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Buffa, J.A.; Wang, Z.; Warrier, M.; Schugar, R.; Shih, D.M.; Gupta, N.; Gregory, J.C.; Org, E.; Fu, X.; et al. Flavin monooxygenase 3, the host hepatic enzyme in the metaorganismal trimethylamine N-oxide-generating pathway, modulates platelet responsiveness and thrombosis risk. J. Thromb. Haemost. 2018, 16, 1857–1872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, T.; Heaney, L.M.; Jones, D.J.; Ng, L.L. Trimethylamine N-oxide and Risk Stratification after Acute Myocardial Infarction. Clin. Chem. 2017, 63, 420–428. [Google Scholar] [CrossRef] [PubMed]
- Janeiro, M.H.; Ramirez, M.J.; Milagro, F.I.; Martinez, J.A.; Solas, M. Implication of Trimethylamine N-Oxide (TMAO) in Disease: Potential Biomarker or New Therapeutic Target. Nutrients 2018, 10, 1398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Jiao, X.; Ma, Y.; Liu, Y.; Zhang, L.; He, Y.; Chen, Y. Trimethylamine N-oxide induces inflammation and endothelial dysfunction in human umbilical vein endothelial cells via activating ROS-TXNIP-NLRP3 inflammasome. Biochem. Biophys. Res. Commun. 2016, 481, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Miao, J.; Morbid Obesity Study Group; Ling, A.V.; Manthena, P.V.; Gearing, M.E.; Graham, M.J.; Crooke, R.M.; Croce, K.J.; Esquejo, R.M.; Clish, C.B.; et al. Flavin-containing monooxygenase 3 as a potential player in diabetes-associated atherosclerosis. Nat. Commun. 2015, 6, 6498. [Google Scholar] [CrossRef] [PubMed]
- Shih, D.M.; Wang, Z.; Lee, R.; Meng, Y.; Che, N.; Charugundla, S.; Qi, H.; Wu, J.; Pan, C.; Brown, J.M.; et al. Flavin containing monooxygenase 3 exerts broad effects on glucose and lipid metabolism and atherosclerosis. J. Lipid Res. 2015, 56, 22–37. [Google Scholar] [CrossRef] [Green Version]
- Warrier, M.; Shih, D.M.; Burrows, A.C.; Ferguson, D.; Gromovsky, A.D.; Brown, A.L.; Marshall, S.; McDaniel, A.; Schugar, R.C.; Wang, Z.; et al. The TMAO-Generating Enzyme Flavin Monooxygenase 3 Is a Central Regulator of Cholesterol Balance. Cell Rep. 2015, 10, 326–338. [Google Scholar] [CrossRef] [Green Version]
- Koeth, R.A.; Levison, B.S.; Culley, M.K.; Buffa, J.A.; Wang, Z.; Gregory, J.C.; Org, E.; Wu, Y.; Li, L.; Smith, J.D.; et al. gamma-Butyrobetaine is a proatherogenic intermediate in gut microbial metabolism of L-carnitine to TMAO. Cell Metab. 2014, 20, 799–812. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Zheng, X.; Feng, M.; Li, D.; Zhang, H. Gut Microbiota-Dependent Metabolite Trimethylamine N-Oxide Contributes to Cardiac Dysfunction in Western Diet-Induced Obese Mice. Front. Physiol. 2017, 8, 139. [Google Scholar] [CrossRef]
- Guasch-Ferre, M.; Hu, F.B.; Ruiz-Canela, M.; Bullo, M.; Toledo, E.; Wang, D.D.; Corella, D.; Gomez-Gracia, E.; Fiol, M.; Estruch, R.; et al. Plasma Metabolites From Choline Pathway and Risk of Cardiovascular Disease in the PREDIMED (Prevention With Mediterranean Diet) Study. J. Am. Heart Assoc. 2017, 6, e006524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Troseid, M.; Ueland, T.; Hov, J.R.; Svardal, A.; Gregersen, I.; Dahl, C.P.; Aakhus, S.; Gude, E.; Bjorndal, B.; Halvorsen, B.; et al. Microbiota-dependent metabolite trimethylamine-N-oxide is associated with disease severity and survival of patients with chronic heart failure. J. Intern. Med. 2015, 277, 717–726. [Google Scholar] [CrossRef] [PubMed]
- Mencarelli, A.; Renga, B.; Distrutti, E.; Fiorucci, S. Antiatherosclerotic effect of farnesoid X receptor. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H272–H281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyazaki-Anzai, S.; Masuda, M.; Levi, M.; Keenan, A.L.; Miyazaki, M. Dual activation of the bile acid nuclear receptor FXR and G-protein-coupled receptor TGR5 protects mice against atherosclerosis. PLoS ONE 2014, 9, e108270. [Google Scholar] [CrossRef] [Green Version]
- Ma, G.; Pan, B.; Chen, Y.; Guo, C.; Zhao, M.; Zheng, L.; Chen, B. Trimethylamine N-oxide in atherogenesis: Impairing endothelial self-repair capacity and enhancing monocyte adhesion. Biosci. Rep. 2017, 37, BSR20160244. [Google Scholar] [CrossRef] [Green Version]
- Senthong, V.; Li, X.S.; Hudec, T.; Coughlin, J.; Wu, Y.; Levison, B.; Wang, Z.; Hazen, S.L.; Tang, W.H. Plasma Trimethylamine N-Oxide, a Gut Microbe-Generated Phosphatidylcholine Metabolite, Is Associated With Atherosclerotic Burden. J. Am. Coll. Cardiol. 2016, 67, 2620–2628. [Google Scholar] [CrossRef]
- Senthong, V.; Wang, Z.; Li, X.S.; Fan, Y.; Wu, Y.; Tang, W.H.; Hazen, S.L. Intestinal Microbiota-Generated Metabolite Trimethylamine-N-Oxide and 5-Year Mortality Risk in Stable Coronary Artery Disease: The Contributory Role of Intestinal Microbiota in a COURAGE-Like Patient Cohort. J. Am. Heart Assoc. 2016, 5, e002816. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Li, Y.; Rimm, E.B.; Hu, F.B.; Albert, C.M.; Rexrode, K.M.; Manson, J.E.; Qi, L. Dietary phosphatidylcholine and risk of all-cause and cardiovascular-specific mortality among US women and men. Am. J. Clin. Nutr. 2016, 104, 173–180. [Google Scholar] [CrossRef]
- Dalmeijer, G.W.; Olthof, M.R.; Verhoef, P.; Bots, M.L.; van der Schouw, Y.T. Prospective study on dietary intakes of folate, betaine, and choline and cardiovascular disease risk in women. Eur. J. Clin. Nutr. 2008, 62, 386–394. [Google Scholar] [CrossRef]
- Meyer, K.A.; Benton, T.Z.; Bennett, B.J.; Jacobs, D.R., Jr.; Lloyd-Jones, D.M.; Gross, M.D.; Carr, J.J.; Gordon-Larsen, P.; Zeisel, S.H. Microbiota-Dependent Metabolite Trimethylamine N-Oxide and Coronary Artery Calcium in the Coronary Artery Risk Development in Young Adults Study (CARDIA). J. Am. Heart Assoc. 2016, 5, e003970. [Google Scholar] [CrossRef] [Green Version]
- Nagata, C.; Wada, K.; Tamura, T.; Konishi, K.; Kawachi, T.; Tsuji, M.; Nakamura, K. Choline and Betaine Intakes Are Not Associated with Cardiovascular Disease Mortality Risk in Japanese Men and Women. J. Nutr. 2015, 145, 1787–1792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, H.L.; Drazul-Schrader, D.; Sulpizio, A.C.; Koster, P.D.; Williamson, Y.; Adelman, S.J.; Owen, K.; Sanli, T.; Bellamine, A. L-Carnitine intake and high trimethylamine N-oxide plasma levels correlate with low aortic lesions in ApoE(−/−) transgenic mice expressing CETP. Atherosclerosis 2016, 244, 29–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bidulescu, A.; Chambless, L.E.; Siega-Riz, A.M.; Zeisel, S.H.; Heiss, G. Usual choline and betaine dietary intake and incident coronary heart disease: The Atherosclerosis Risk in Communities (ARIC) study. BMC Cardiovasc. Disord. 2007, 7, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parseus, A.; Sommer, N.; Sommer, F.; Caesar, R.; Molinaro, A.; Stahlman, M.; Greiner, T.U.; Perkins, R.; Backhed, F. Microbiota-induced obesity requires farnesoid X receptor. Gut 2017, 66, 429–437. [Google Scholar] [CrossRef] [Green Version]
- Russell, D.W. The enzymes, regulation, and genetics of bile acid synthesis. Annu. Rev. Biochem. 2003, 72, 137–174. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.; Huang, F.; Zhao, A.; Lei, S.; Zhang, Y.; Xie, G.; Chen, T.; Qu, C.; Rajani, C.; Dong, B.; et al. Bile acid is a significant host factor shaping the gut microbiome of diet-induced obese mice. BMC Biol. 2017, 15, 120. [Google Scholar] [CrossRef] [Green Version]
- Jones, M.L.; Martoni, C.J.; Ganopolsky, J.G.; Labbe, A.; Prakash, S. The human microbiome and bile acid metabolism: Dysbiosis, dysmetabolism, disease and intervention. Expert Opin. Biol. Ther. 2014, 14, 467–482. [Google Scholar] [CrossRef]
- Lefebvre, P.; Cariou, B.; Lien, F.; Kuipers, F.; Staels, B. Role of bile acids and bile acid receptors in metabolic regulation. Physiol. Rev. 2009, 89, 147–191. [Google Scholar] [CrossRef] [Green Version]
- Ridlon, J.M.; Harris, S.C.; Bhowmik, S.; Kang, D.J.; Hylemon, P.B. Consequences of bile salt biotransformations by intestinal bacteria. Gut Microbes 2016, 7, 22–39. [Google Scholar] [CrossRef] [Green Version]
- Jones, M.L.; Martoni, C.J.; Parent, M.; Prakash, S. Cholesterol-lowering efficacy of a microencapsulated bile salt hydrolase-active Lactobacillus reuteri NCIMB 30242 yoghurt formulation in hypercholesterolaemic adults. Br. J. Nutr. 2012, 107, 1505–1513. [Google Scholar] [CrossRef] [Green Version]
- Tremaroli, V.; Backhed, F. Functional interactions between the gut microbiota and host metabolism. Nature 2012, 489, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Hansson, G.K.; Robertson, A.K.; Soderberg-Naucler, C. Inflammation and atherosclerosis. Annu. Rev. Pathol. 2006, 1, 297–329. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Chiang, J.Y. Bile acids as metabolic regulators. Curr. Opin. Gastroenterol. 2015, 31, 159–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pols, T.W.; Nomura, M.; Harach, T.; Lo Sasso, G.; Oosterveer, M.H.; Thomas, C.; Rizzo, G.; Gioiello, A.; Adorini, L.; Pellicciari, R.; et al. TGR5 activation inhibits atherosclerosis by reducing macrophage inflammation and lipid loading. Cell Metab. 2011, 14, 747–757. [Google Scholar] [CrossRef] [Green Version]
- Staudinger, J.L.; Goodwin, B.; Jones, S.A.; Hawkins-Brown, D.; MacKenzie, K.I.; LaTour, A.; Liu, Y.; Klaassen, C.D.; Brown, K.K.; Reinhard, J.; et al. The nuclear receptor PXR is a lithocholic acid sensor that protects against liver toxicity. Proc. Natl. Acad. Sci. USA 2001, 98, 3369–3374. [Google Scholar] [CrossRef] [Green Version]
- Zhou, C.; King, N.; Chen, K.Y.; Breslow, J.L. Activation of PXR induces hypercholesterolemia in wild-type and accelerates atherosclerosis in apoE deficient mice. J. Lipid Res. 2009, 50, 2004–2013. [Google Scholar] [CrossRef] [Green Version]
- Sui, Y.; Xu, J.; Rios-Pilier, J.; Zhou, C. Deficiency of PXR decreases atherosclerosis in apoE-deficient mice. J. Lipid Res. 2011, 52, 1652–1659. [Google Scholar] [CrossRef] [Green Version]
- Levi, M. Role of Bile Acid-Regulated Nuclear Receptor FXR and G Protein-Coupled Receptor TGR5 in Regulation of Cardiorenal Syndrome (Cardiovascular Disease and Chronic Kidney Disease). Hypertension 2016, 67, 1080–1084. [Google Scholar] [CrossRef] [Green Version]
- Townsend, M.K.; Aschard, H.; De Vivo, I.; Michels, K.B.; Kraft, P. Genomics, Telomere Length, Epigenetics, and Metabolomics in the Nurses’ Health Studies. Am. J. Public Health 2016, 106, 1663–1668. [Google Scholar] [CrossRef]
- Jonsson, A.L.; Backhed, F. Role of gut microbiota in atherosclerosis. Nat. Rev. Cardiol. 2017, 14, 79–87. [Google Scholar] [CrossRef]
- Yamashiro, K.; Tanaka, R.; Urabe, T.; Ueno, Y.; Yamashiro, Y.; Nomoto, K.; Takahashi, T.; Tsuji, H.; Asahara, T.; Hattori, N. Gut dysbiosis is associated with metabolism and systemic inflammation in patients with ischemic stroke. PLoS ONE 2017, 12, e0171521. [Google Scholar] [CrossRef]
- Honour, J. The possible involvement of intestinal bacteria in steroidal hypertension. Endocrinology 1982, 110, 285–287. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Santisteban, M.M.; Rodriguez, V.; Li, E.; Ahmari, N.; Carvajal, J.M.; Zadeh, M.; Gong, M.; Qi, Y.; Zubcevic, J.; et al. Gut dysbiosis is linked to hypertension. Hypertension 2015, 65, 1331–1340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luscher, T.F.; Barton, M. Biology of the endothelium. Clin. Cardiol. 1997, 20, II-3-10. [Google Scholar] [CrossRef] [PubMed]
- Karbach, S.H.; Schonfelder, T.; Brandao, I.; Wilms, E.; Hormann, N.; Jackel, S.; Schuler, R.; Finger, S.; Knorr, M.; Lagrange, J.; et al. Gut Microbiota Promote Angiotensin II-Induced Arterial Hypertension and Vascular Dysfunction. J. Am. Heart Assoc. 2016, 5, e003698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Kaoutari, A.; Armougom, F.; Gordon, J.I.; Raoult, D.; Henrissat, B. The abundance and variety of carbohydrate-active enzymes in the human gut microbiota. Nat. Rev. Microbiol. 2013, 11, 497–504. [Google Scholar] [CrossRef]
- Canfora, E.E.; Jocken, J.W.; Blaak, E.E. Short-chain fatty acids in control of body weight and insulin sensitivity. Nat. Rev. Endocrinol. 2015, 11, 577–591. [Google Scholar] [CrossRef]
- Koh, A.; De Vadder, F.; Kovatcheva-Datchary, P.; Backhed, F. From Dietary Fiber to Host Physiology: Short-Chain Fatty Acids as Key Bacterial Metabolites. Cell 2016, 165, 1332–1345. [Google Scholar] [CrossRef] [Green Version]
- Rey, F.E.; Faith, J.J.; Bain, J.; Muehlbauer, M.J.; Stevens, R.D.; Newgard, C.B.; Gordon, J.I. Dissecting the in vivo metabolic potential of two human gut acetogens. J. Biol. Chem. 2010, 285, 22082–22090. [Google Scholar] [CrossRef] [Green Version]
- Duncan, S.H.; Barcenilla, A.; Stewart, C.S.; Pryde, S.E.; Flint, H.J. Acetate utilization and butyryl coenzyme A (CoA): Acetate-CoA transferase in butyrate-producing bacteria from the human large intestine. Appl. Environ. Microbiol. 2002, 68, 5186–5190. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Arango, L.F.; Barrett, H.L.; McIntyre, H.D.; Callaway, L.K.; Morrison, M.; Dekker Nitert, M.; Group, S.T. Increased Systolic and Diastolic Blood Pressure Is Associated With Altered Gut Microbiota Composition and Butyrate Production in Early Pregnancy. Hypertension 2016, 68, 974–981. [Google Scholar] [CrossRef] [PubMed]
- Marques, F.Z.; Nelson, E.; Chu, P.Y.; Horlock, D.; Fiedler, A.; Ziemann, M.; Tan, J.K.; Kuruppu, S.; Rajapakse, N.W.; El-Osta, A.; et al. High-Fiber Diet and Acetate Supplementation Change the Gut Microbiota and Prevent the Development of Hypertension and Heart Failure in Hypertensive Mice. Circulation 2017, 135, 964–977. [Google Scholar] [CrossRef] [PubMed]
- Pluznick, J.L. Renal and cardiovascular sensory receptors and blood pressure regulation. Am. J. Physiol. Ren. Physiol. 2013, 305, F439–F444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, J.K.; McKenzie, C.; Marino, E.; Macia, L.; Mackay, C.R. Metabolite-Sensing G Protein-Coupled Receptors-Facilitators of Diet-Related Immune Regulation. Annu. Rev. Immunol. 2017, 35, 371–402. [Google Scholar] [CrossRef]
- Furusawa, Y.; Obata, Y.; Fukuda, S.; Endo, T.A.; Nakato, G.; Takahashi, D.; Nakanishi, Y.; Uetake, C.; Kato, K.; Kato, T.; et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 2013, 504, 446–450. [Google Scholar] [CrossRef]
- Pluznick, J.L.; Protzko, R.J.; Gevorgyan, H.; Peterlin, Z.; Sipos, A.; Han, J.; Brunet, I.; Wan, L.X.; Rey, F.; Wang, T.; et al. Olfactory receptor responding to gut microbiota-derived signals plays a role in renin secretion and blood pressure regulation. Proc. Natl. Acad. Sci. USA 2013, 110, 4410–4415. [Google Scholar] [CrossRef] [Green Version]
- Natarajan, N.; Hori, D.; Flavahan, S.; Steppan, J.; Flavahan, N.A.; Berkowitz, D.E.; Pluznick, J.L. Microbial short chain fatty acid metabolites lower blood pressure via endothelial G protein-coupled receptor 41. Physiol. Genom. 2016, 48, 826–834. [Google Scholar] [CrossRef]
- Miyamoto, J.; Kasubuchi, M.; Nakajima, A.; Irie, J.; Itoh, H.; Kimura, I. The role of short-chain fatty acid on blood pressure regulation. Curr. Opin. Nephrol. Hypertens. 2016, 25, 379–383. [Google Scholar] [CrossRef]
- Chawla, A.; Nguyen, K.D.; Goh, Y.P. Macrophage-mediated inflammation in metabolic disease. Nat. Rev. Immunol. 2011, 11, 738–749. [Google Scholar] [CrossRef] [Green Version]
- Peluso, I.; Morabito, G.; Urban, L.; Ioannone, F.; Serafini, M. Oxidative stress in atherosclerosis development: The central role of LDL and oxidative burst. Endocr. Metab. Immune Disord. Drug Targets 2012, 12, 351–360. [Google Scholar] [CrossRef]
- Ma, F.X.; Zhou, B.; Chen, Z.; Ren, Q.; Lu, S.H.; Sawamura, T.; Han, Z.C. Oxidized low density lipoprotein impairs endothelial progenitor cells by regulation of endothelial nitric oxide synthase. J. Lipid Res. 2006, 47, 1227–1237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subah Packer, C. Estrogen protection, oxidized LDL, endothelial dysfunction and vasorelaxation in cardiovascular disease: New insights into a complex issue. Cardiovasc. Res. 2007, 73, 6–7. [Google Scholar] [CrossRef] [PubMed]
- Boulanger, C.; Luscher, T.F. Release of endothelin from the porcine aorta. Inhibition by endothelium-derived nitric oxide. J. Clin. Investig. 1990, 85, 587–590. [Google Scholar] [CrossRef] [PubMed]
- Kamo, T.; Akazawa, H.; Suda, W.; Saga-Kamo, A.; Shimizu, Y.; Yagi, H.; Liu, Q.; Nomura, S.; Naito, A.T.; Takeda, N.; et al. Dysbiosis and compositional alterations with aging in the gut microbiota of patients with heart failure. PLoS ONE 2017, 12, e0174099. [Google Scholar] [CrossRef] [PubMed]
- Roberts, D.E.; McNicol, A.; Bose, R. Mechanism of collagen activation in human platelets. J. Biol. Chem. 2004, 279, 19421–19430. [Google Scholar] [CrossRef] [Green Version]
- Jung, S.M.; Moroi, M. Activation of the platelet collagen receptor integrin alpha(2)beta(1): Its mechanism and participation in the physiological functions of platelets. Trends Cardiovasc. Med. 2000, 10, 285–292. [Google Scholar] [CrossRef]
- Bynagari, Y.S.; Nagy, B., Jr.; Tuluc, F.; Bhavaraju, K.; Kim, S.; Vijayan, K.V.; Kunapuli, S.P. Mechanism of activation and functional role of protein kinase Ceta in human platelets. J. Biol. Chem. 2009, 284, 13413–13421. [Google Scholar] [CrossRef] [Green Version]
- Davi, G.; Patrono, C. Platelet activation and atherothrombosis. N. Engl. J. Med. 2007, 357, 2482–2494. [Google Scholar] [CrossRef]
- El Haouari, M.; Rosado, J.A. Medicinal Plants with Antiplatelet Activity. Phytother. Res. 2016, 30, 1059–1071. [Google Scholar] [CrossRef]
- Elwood, P.C.; Beswick, A.D.; Sharp, D.S.; Yarnell, J.W.; Rogers, S.; Renaud, S. Whole blood impedance platelet aggregometry and ischemic heart disease. The Caerphilly Collaborative Heart Disease Study. Arteriosclerosis 1990, 10, 1032–1036. [Google Scholar] [CrossRef] [Green Version]
- Tran, H.; Anand, S.S. Oral antiplatelet therapy in cerebrovascular disease, coronary artery disease, and peripheral arterial disease. JAMA 2004, 292, 1867–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabbianelli, R.; Falcioni, G.; Dow, C.S.; Vince, F.P.; Swoboda, B. A new method to evaluate spontaneous platelet aggregation in type 2 diabetes by Cellfacts. Clin. Chim. Acta 2003, 329, 95–102. [Google Scholar] [CrossRef]
- Iwase, E.; Tawata, M.; Aida, K.; Ozaki, Y.; Kume, S.; Satoh, K.; Qi, R.; Onaya, T. A cross-sectional evaluation of spontaneous platelet aggregation in relation to complications in patients with type II diabetes mellitus. Metabolism 1998, 47, 699–705. [Google Scholar] [CrossRef]
- Duhamel, T.A.; Xu, Y.J.; Arneja, A.S.; Dhalla, N.S. Targeting platelets for prevention and treatment of cardiovascular disease. Expert Opin. Ther. Targets 2007, 11, 1523–1533. [Google Scholar] [CrossRef]
- Kjeldsen, S.E.; Gjesdal, K.; Eide, I.; Aakesson, I.; Amundsen, R.; Foss, O.P.; Leren, P. Increased beta-thromboglobulin in essential hypertension: Interactions between arterial plasma adrenaline, platelet function and blood lipids. Acta Med. Scand. 1983, 213, 369–373. [Google Scholar] [CrossRef]
- Hernandez Hernandez, R.; Carvajal, A.R.; Guerrero Pajuelo, J.; Armas de Hernandez, M.J.; Armas Padilla, M.C.; Barragan, O.; Boada Boada, J.J.; Roa, E. The effect of doxazosin on platelet aggregation in normotensive subjects and patients with hypertension: An in vitro study. Am. Heart J. 1991, 121, 389–394. [Google Scholar] [CrossRef]
- Ding, L.; Chang, M.; Guo, Y.; Zhang, L.; Xue, C.; Yanagita, T.; Zhang, T.; Wang, Y. Trimethylamine-N-oxide (TMAO)-induced atherosclerosis is associated with bile acid metabolism. Lipids Health Dis 2018, 17, 286. [Google Scholar] [CrossRef] [Green Version]
- Yazici, M.; Kaya, A.; Kaya, Y.; Albayrak, S.; Cinemre, H.; Ozhan, H. Lifestyle modification decreases the mean platelet volume in prehypertensive patients. Platelets 2009, 20, 58–63. [Google Scholar] [CrossRef]
- Dutta-Roy, A.K. Dietary components and human platelet activity. Platelets 2002, 13, 67–75. [Google Scholar] [CrossRef]
- Tsoupras, A.; Lordan, R.; Zabetakis, I. Thrombosis and COVID-19: The Potential Role of Nutrition. Front. Nutr. 2020, 7, 583080. [Google Scholar] [CrossRef]
- O’Kennedy, N.; Raederstorff, D.; Duttaroy, A.K. Fruitflow((R)): The first European Food Safety Authority-approved natural cardio-protective functional ingredient. Eur. J. Nutr. 2017, 56, 461–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Kennedy, N.; Crosbie, L.; Song, H.J.; Zhang, X.; Horgan, G.; Duttaroy, A.K. A randomised controlled trial comparing a dietary antiplatelet, the water-soluble tomato extract Fruitflow, with 75 mg aspirin in healthy subjects. Eur. J. Clin. Nutr. 2016, 71, 723–730. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duttaroy, A.K. Role of Gut Microbiota and Their Metabolites on Atherosclerosis, Hypertension and Human Blood Platelet Function: A Review. Nutrients 2021, 13, 144. https://doi.org/10.3390/nu13010144
Duttaroy AK. Role of Gut Microbiota and Their Metabolites on Atherosclerosis, Hypertension and Human Blood Platelet Function: A Review. Nutrients. 2021; 13(1):144. https://doi.org/10.3390/nu13010144
Chicago/Turabian StyleDuttaroy, Asim K. 2021. "Role of Gut Microbiota and Their Metabolites on Atherosclerosis, Hypertension and Human Blood Platelet Function: A Review" Nutrients 13, no. 1: 144. https://doi.org/10.3390/nu13010144
APA StyleDuttaroy, A. K. (2021). Role of Gut Microbiota and Their Metabolites on Atherosclerosis, Hypertension and Human Blood Platelet Function: A Review. Nutrients, 13(1), 144. https://doi.org/10.3390/nu13010144