Whole Blueberry and Isolated Polyphenol-Rich Fractions Modulate Specific Gut Microbes in an In Vitro Colon Model and in a Pilot Study in Human Consumers

 , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Faecal Inocula and the In Vitro Colon Model
2.2. Bacterial DNA Extraction for In Vitro and Human Study
2.3. Microbiome Profiling of In Vitro and Human Study Samples
2.4. Isolation of Enriched Fractions for Bioassay
2.5. Human Study Design
2.6. Plasma FRAP Assay
2.7. Plasma Glucose and CRP Measurements
2.8. Microbiota Composition and Statistical Analysis
3. Results
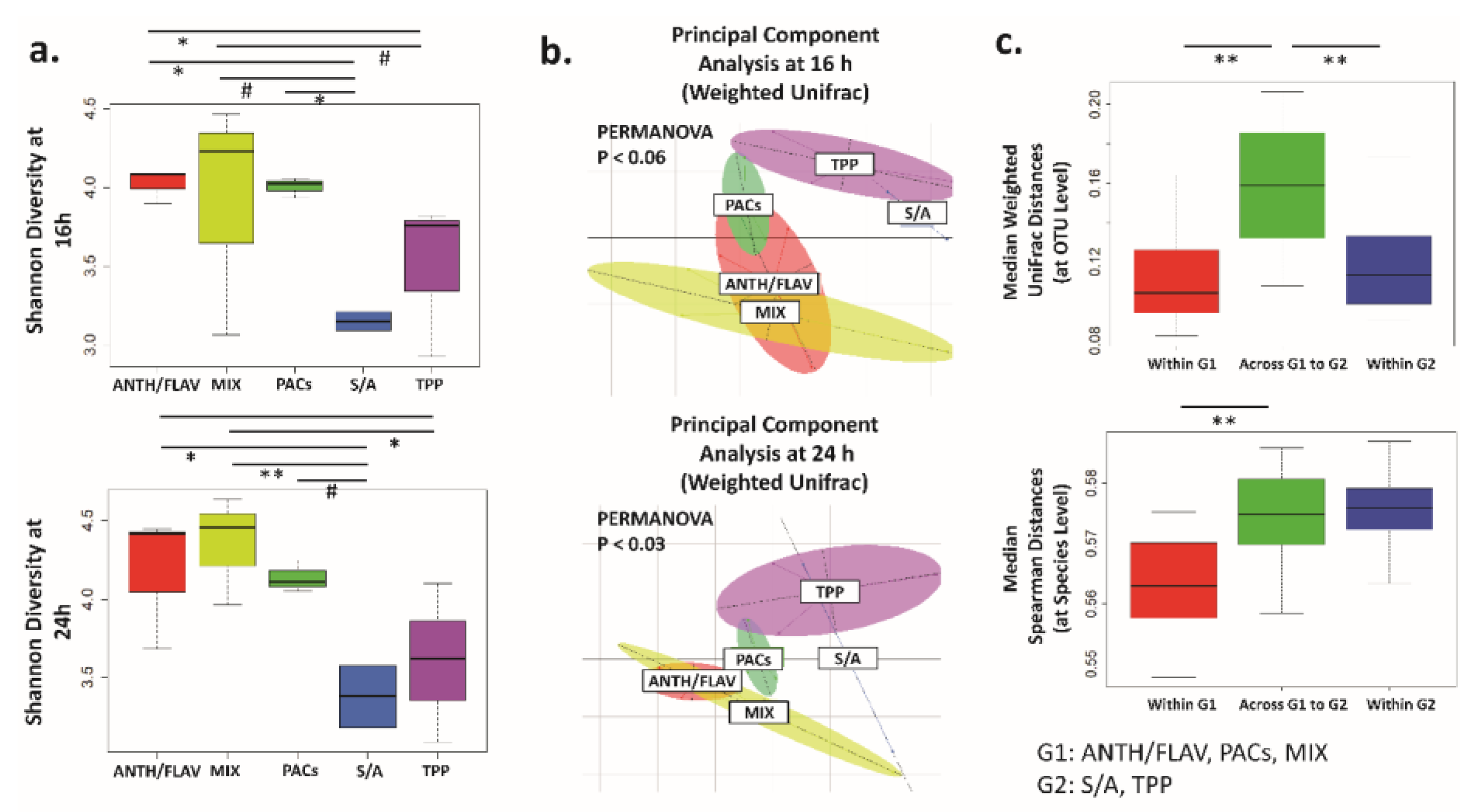
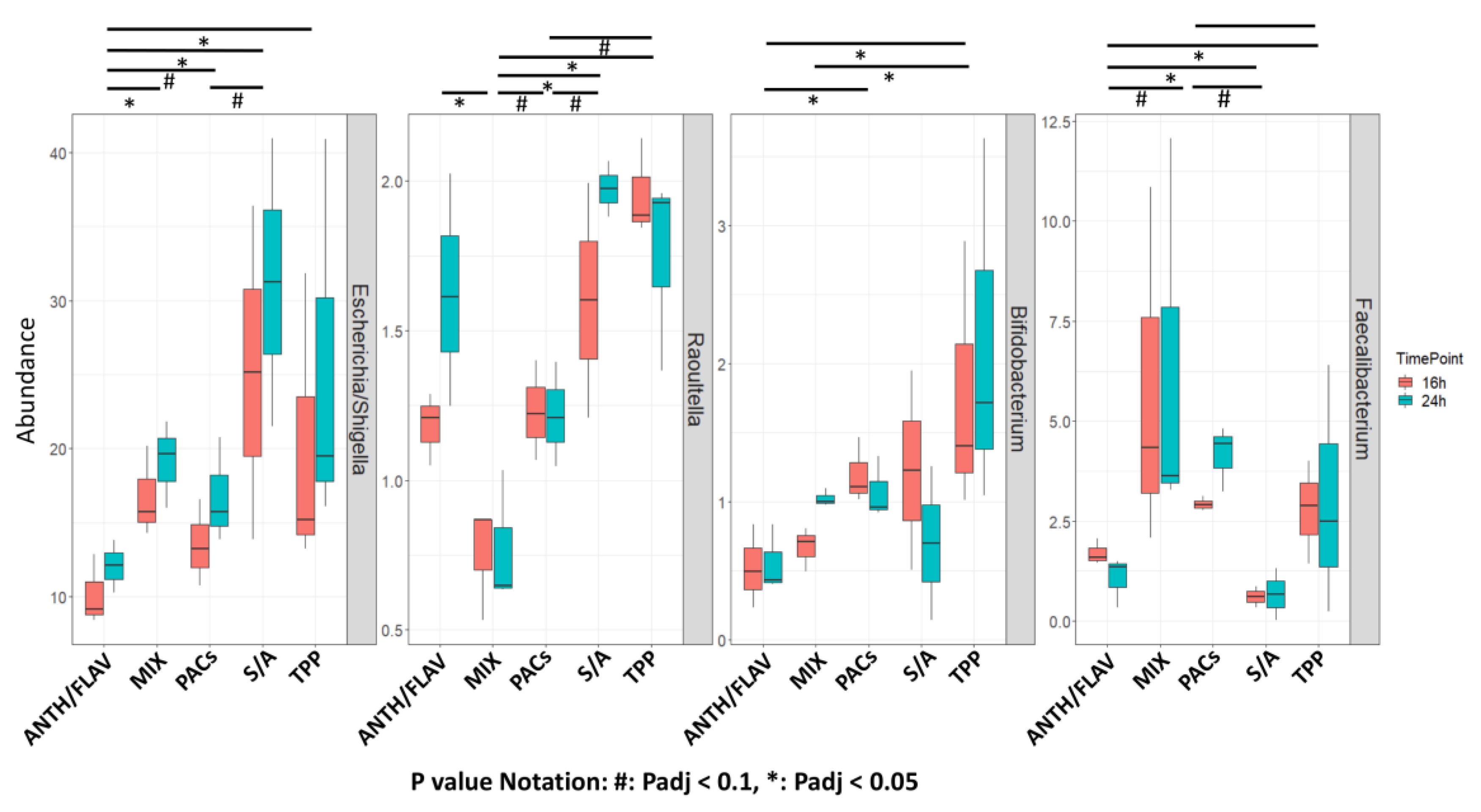
3.1. A Prebiotic MIX, PACs, or ANTH/FLAV Have a Similar Effect on Microbiota Structure in the In Vitro Colon Model
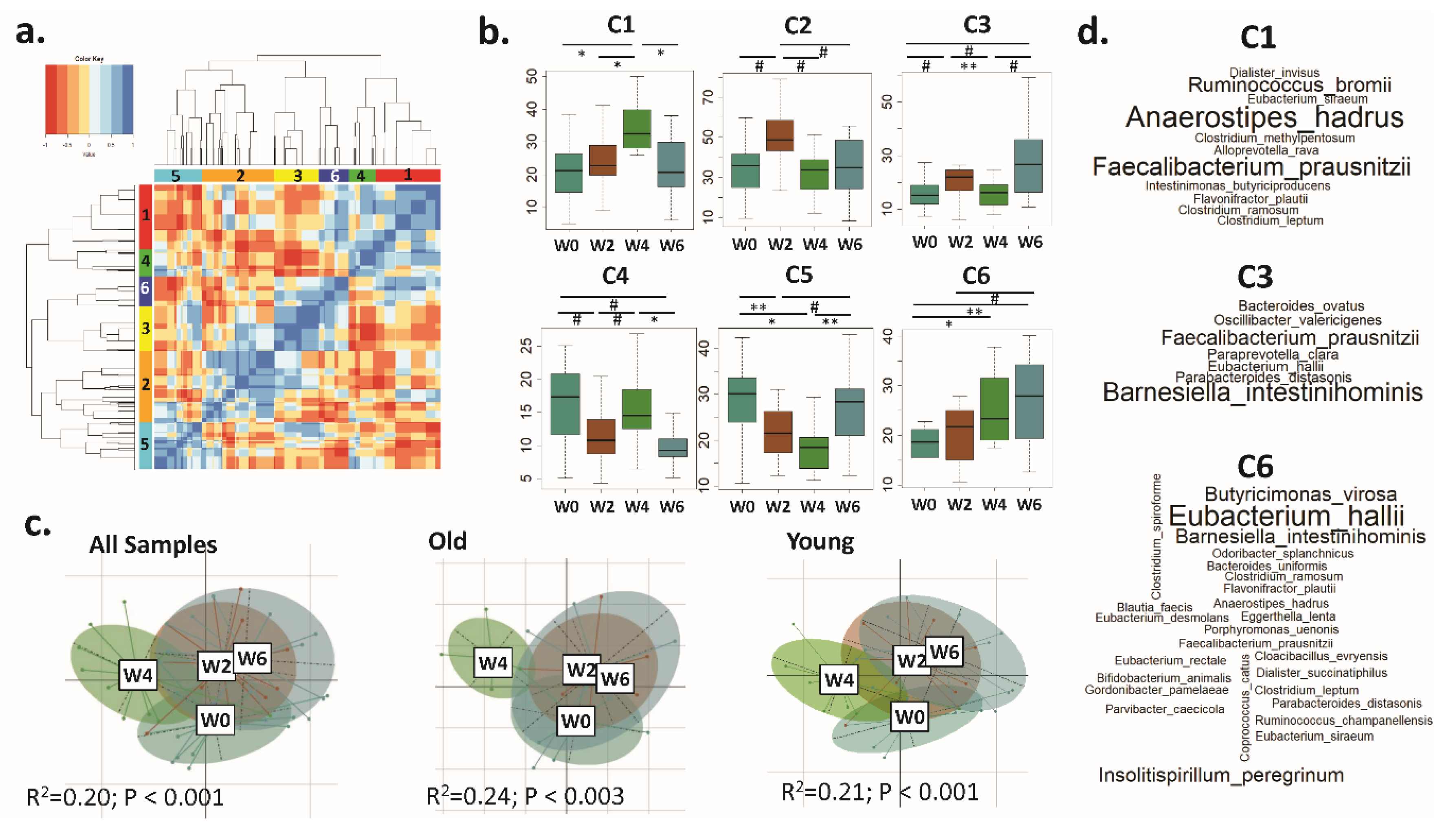
3.2. A Trend Towards Increased Microbiota α Diversity in Older Women Consuming BB
3.3. Distinct CAGs Represented by Health-Promoting Taxa Were Associated with BB Consumption at Each Time Point
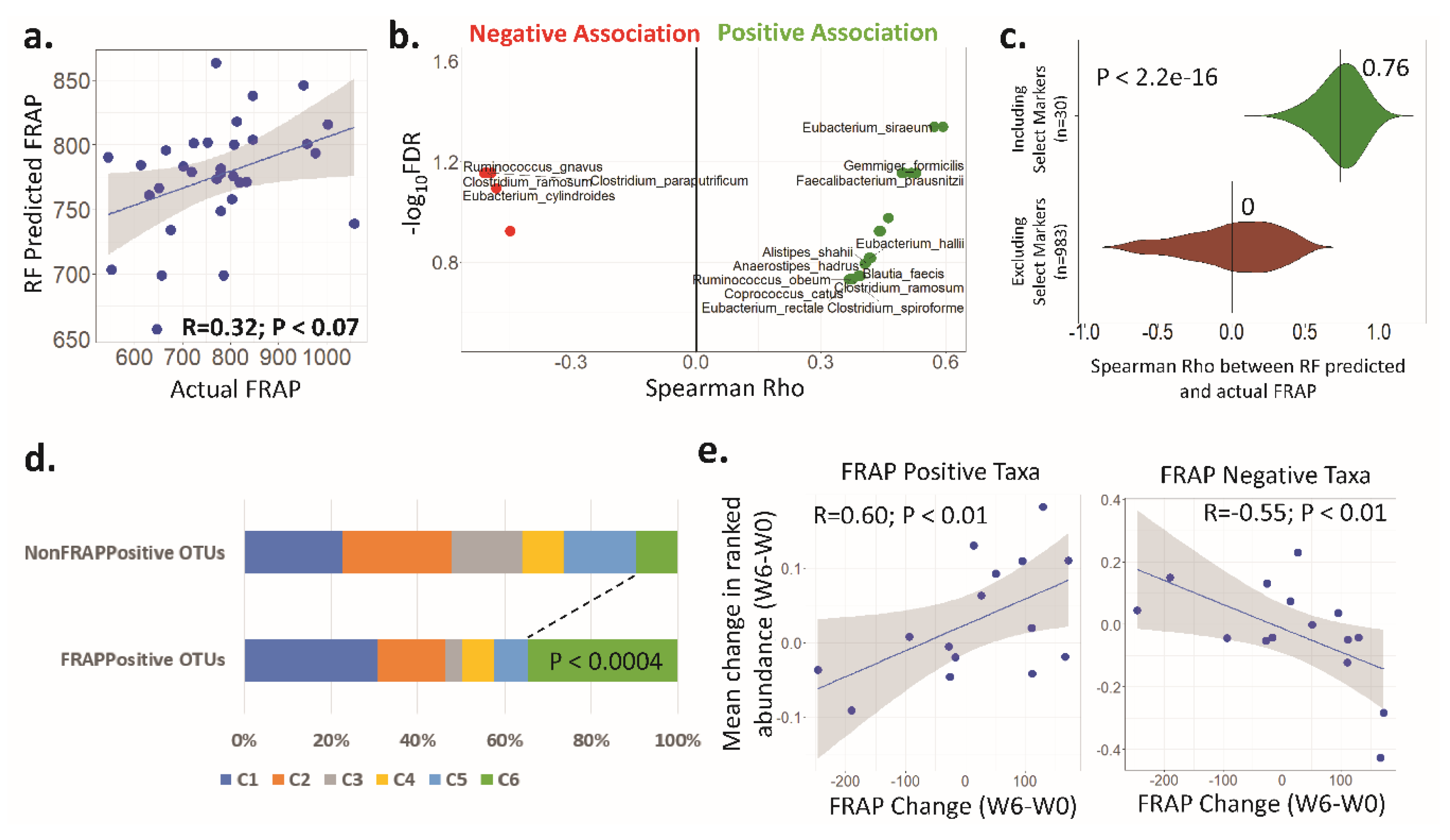
3.4. Antioxidant Activity (FRAP) Is Significantly Associated with the Faecal Microbiota
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sonnenburg, J.L.; Bäckhed, F. Diet-microbiota interactions as moderators of human metabolism. Nature 2016, 535, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Messer, J.S.; Liechty, E.R.; Vogel, O.A.; Chang, E.B. Evolutionary and ecological forces that shape the bacterial communities of the human gut. Mucosal Immunol. 2017, 10, 567–579. [Google Scholar] [CrossRef] [PubMed]
- Marchesi, J.R.; Adams, D.H.; Fava, F.; Hermes, G.D.; Hirschfield, G.M.; Hold, G.; Quraishi, M.N.; Kinross, J.; Smidt, H.; Tuohy, K.M.; et al. The gut microbiota and host health: A new clinical frontier. Gut 2016, 65, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Petersen, C.; Round, J.L. Defining dysbiosis and its influence on host immunity and disease. Cell. Microbiol. 2014, 16, 1024–1033. [Google Scholar] [CrossRef]
- Jeffery, I.B.; Lynch, D.B.; O’Toole, P.W. Composition and temporal stability of the gut microbiota in older persons. ISME J. 2016, 10, 170–182. [Google Scholar] [CrossRef]
- Thevaranjan, N.; Puchta, A.; Schulz, C.; Naidoo, A.; Szamosi, J.C.; Verschoor, C.P.; Loukov, D.; Schenck, L.P.; Jury, J.; Foley, K.P.; et al. Age-associated microbial dysbiosis promotes intestinal permeability, systemic inflammation, and macrophage dysfunction. Cell Host Microbe 2017, 21, 455–466.e4. [Google Scholar] [CrossRef]
- McCarville, J.L.; Caminero, A.; Verdu, E.F. Novel perspectives on therapeutic modulation of the gut microbiota. Therap. Adv. Gastroenterol. 2016, 9, 580–593. [Google Scholar] [CrossRef]
- Salazar, N.; Valdés-Varela, L.; González, S.; Gueimonde, M.; de Los Reyes-Gavilán, C.G. Nutrition and the gut microbiome in the elderly. Gut Microbes 2017, 8, 82–97. [Google Scholar] [CrossRef]
- O’Toole, P.W.; Marchesi, J.R.; Hill, C. Next-generation probiotics: The spectrum from probiotics to live biotherapeutics. Nat. Microbiol. 2017, 2, 17057. [Google Scholar] [CrossRef]
- Fraga, C.G.; Croft, K.D.; Kennedy, D.O.; Tomás-Barberán, F.A. The effects of polyphenols and other bioactives on human health. Food Funct. 2019, 10, 514–528. [Google Scholar] [CrossRef]
- Van Hul, M.; Cani, P.D. Targeting carbohydrates and polyphenols for a healthy microbiome and healthy weight. Curr. Nutr. Rep. 2019, 8, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Durazzo, A.; Lucarini, M.; Souto, E.B.; Cicala, C.; Caiazzo, E.; Izzo, A.A.; Novellino, E.; Santini, A. Polyphenols: A concise overview on the chemistry, occurrence, and human health. Phyther. Res. 2019, 33, 2221–2243. [Google Scholar] [CrossRef] [PubMed]
- Panche, A.N.; Diwan, A.D.; Chandra, S.R. Flavonoids: An overview. J. Nutr. Sci. 2016, 5, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Pojer, E.; Mattivi, F.; Johnson, D.; Stockley, C.S. The case for anthocyanin consumption to promote human health: A review. Compr. Rev. Food Sci. Food Saf. 2013, 12, 483–508. [Google Scholar] [CrossRef]
- Li, D.; Wang, P.; Luo, Y.; Zhao, M.; Chen, F. Health benefits of anthocyanins and molecular mechanisms: Update from recent decade. Crit. Rev. Food Sci. Nutr. 2017, 57, 1729–1741. [Google Scholar] [CrossRef] [PubMed]
- Chun, O.K.; Chung, S.J.; Song, W.O. Estimated dietary flavonoid intake and major food sources of U.S. adults. J. Nutr. 2007, 137, 1244–1252. [Google Scholar] [CrossRef] [PubMed]
- Chun, O.K.; Lee, S.G.; Wang, Y.; Vance, T.; Song, W.O. Estimated flavonoid intake of the elderly in the United States and around the world. J. Nutr. Gerontol. Geriatr. 2012, 31, 190–205. [Google Scholar] [CrossRef] [PubMed]
- Beking, K.; Vieira, A. An assessment of dietary flavonoid intake in the UK and Ireland. Int. J. Food Sci. Nutr. 2011, 62, 17–19. [Google Scholar] [CrossRef]
- Vogiatzoglou, A.; Mulligan, A.A.; Lentjes, M.A.H.; Luben, R.N.; Spencer, J.P.E.; Schroeter, H.; Khaw, K.T.; Kuhnle, G.G.C. Flavonoid intake in European adults (18 to 64 Years). PLoS ONE 2015, 10, e0128132. [Google Scholar] [CrossRef] [PubMed]
- Rowland, I.; Gibson, G.; Heinken, A.; Scott, K.; Swann, J.; Thiele, I.; Tuohy, K. Gut microbiota functions: Metabolism of nutrients and other food components. Eur. J. Nutr. 2018, 57, 1–24. [Google Scholar] [CrossRef]
- Dueñas, M.; Muñoz-González, I.; Cueva, C.; Jiménez-Girón, A.; Sánchez-Patán, F.; Santos-Buelga, C.; Moreno-Arribas, M.V.; Bartolomé, B. A survey of modulation of gut microbiota by dietary polyphenols. Biomed. Res. Int. 2015, 2015, 15. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yang, Y.; Wu, Z.; Weng, P. The modulatory effect of anthocyanins from purple sweet potato on human intestinal microbiota in vitro. J. Agric. Food Chem. 2016, 64, 2582–2590. [Google Scholar] [CrossRef] [PubMed]
- Lacombe, A.; Li, R.W.; Klimis-Zacas, D.; Kristo, A.S.; Tadepalli, S.; Krauss, E.; Young, R.; Wu, V.C.H. Lowbush wild blueberries have the potential to modify gut microbiota and xenobiotic metabolism in the rat colon. PLoS ONE 2013, 8, e67497. [Google Scholar] [CrossRef]
- Anhê, F.F.; Roy, D.; Pilon, G.; Dudonné, S.; Matamoros, S.; Varin, T.V.; Garofalo, C.; Moine, Q.; Desjardins, Y.; Levy, E.; et al. A polyphenol-rich cranberry extract protects from diet-induced obesity, insulin resistance and intestinal inflammation in association with increased Akkermansia spp. population in the gut microbiota of mice. Gut 2015, 64, 872–883. [Google Scholar] [CrossRef]
- Baldwin, J.; Collins, B.; Wolf, P.G.; Martinez, K.; Shen, W.; Chuang, C.C.; Zhong, W.; Cooney, P.; Cockrell, C.; Chang, E.; et al. Table grape consumption reduces adiposity and markers of hepatic lipogenesis and alters gut microbiota in butter fat-fed mice. J. Nutr. Biochem. 2016, 27, 123–135. [Google Scholar] [CrossRef]
- Fotschki, B.; Juśkiewicz, J.; Jurgoński, A.; Kołodziejczyk, K.; Milala, J.; Kosmala, M.; Zduńczyk, Z. Anthocyanins in strawberry polyphenolic extract enhance the beneficial effects of diets with fructooligosaccharides in the rat cecal environment. PLoS ONE 2016, 11, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Keirsey, K.I.; Kirkland, R.; Grunewald, Z.I.; Fischer, J.G.; de La Serre, C.B. Blueberry supplementation influences the gut microbiota, inflammation, and insulin resistance in high-fat-diet–fed rats. J. Nutr. 2018, 148, 209–219. [Google Scholar] [CrossRef]
- Bekiares, N.; Krueger, C.G.; Meudt, J.J.; Shanmuganayagam, D.; Reed, J.D. Effect of sweetened dried cranberry consumption on urinary proteome and fecal microbiome in healthy human subjects. Omi. A J. Integr. Biol. 2018, 22, 145–153. [Google Scholar] [CrossRef]
- Rodríguez-Morató, J.; Matthan, N.R.; Liu, J.; de la Torre, R.; Chen, C.-Y.O. Cranberries attenuate animal-based diet-induced changes in microbiota composition and functionality: A randomized crossover controlled feeding trial. J. Nutr. Biochem. 2018, 62, 76–86. [Google Scholar] [CrossRef]
- Gómez-Juaristi, M.; Sarria, B.; Martínez-López, S.; Clemente, L.B.; Mateos, R. Flavanol bioavailability in two cocoa products with different phenolic content. A comparative study in humans. Nutrients 2019, 11, 1441. [Google Scholar] [CrossRef]
- Fallani, M.; Amarri, S.; Uusijarvi, A.; Adam, R.; Khanna, S.; Aguilera, M.; Gil, A.; Vieites, J.M.; Norin, E.; Young, D.; et al. Determinants of the human infant intestinal microbiota after the introduction of first complementary foods in infant samples from five European centres. Microbiology 2011, 157, 1385–1392. [Google Scholar] [CrossRef] [PubMed]
- Kalt, W.; Cassidy, A.; Howard, L.R.; Krikorian, R.; Stull, A.J.; Tremblay, F.; Zamora-ros, R. Recent research on the health benefits of blueberries and their anthocyanins. Adv. Nutr. 2020, 11, 224–236. [Google Scholar] [CrossRef] [PubMed]
- Kalt, W.; McDonald, J.E.; Donner, H. Antioxidant capacity of processed lowbush blueberry products. Food Chem. Toxicol. 2000, 65, 390–393. [Google Scholar]
- Naczk, M.; Amarowicz, R.; Zadernowski, R.; Pegg, R.; Shahidi, F. Antioxidant activity of crude phenolic extracts from wild blueberry leaves. Pol. J. Food Nutr. Sci. 2003, 12, 166–169. [Google Scholar]
- Nackz, M.; Zadernowski, R.; Shahidi, F. Antioxidant Capacity of Phenolic Extracts from Selected Food By-Products; American Chemical Society: Washington, DC, USA, 2007; pp. 184–194. ISBN 0841274282. [Google Scholar]
- Krikorian, R.; Shidler, M.D.; Nash, T.A.; Kalt, W.; Vinqvist-Tymchuk, M.R.; Shukitt-Hale, B.; Joseph, J.A. Blueberry supplementation improves memory in older adults. J. Agric. Food Chem. 2010, 58, 3996–4000. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Wei, C.; Lan, W.; Zhao, Y.; Pan, Y.; Sun, X.; Wu, V.C.H. The effect of Chinese wild blueberry fractions on the growth and membrane integrity of various foodborne pathogens. J. Food Sci. 2020, 85, 1513–1522. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, M.; Oruna-Concha, M.J.; Kolida, S.; Walton, G.E.; Kallithraka, S.; Spencer, J.P.E.; Gibson, G.R.; De Pascual-Teresa, S. Metabolism of anthocyanins by human gut microflora and their influence on gut bacterial growth. J. Agric. Food Chem. 2012, 60, 3882–3890. [Google Scholar] [CrossRef] [PubMed]
- Ntemiri, A.; Chonchúir, F.N.; O’Callaghan, T.F.; Stanton, C.; Ross, R.P.; O’Toole, P.W. Glycomacropeptide sustains microbiota diversity and promotes specific taxa in an artificial colon model of elderly gut microbiota. J. Agric. Food Chem. 2017, 65, 1836–1846. [Google Scholar] [CrossRef]
- Duncan, S.H.; Holtrop, G.; Lobley, G.E.; Calder, A.G.; Stewart, C.S.; Flint, H.J. Contribution of acetate to butyrate formation by human faecal bacteria. Br. J. Nutr. 2004, 91, 915–923. [Google Scholar] [CrossRef]
- Tzounis, X. Flavanol monomer-induced changes to the human faecal microflora. Br. J. Nutr. 2008, 99, 782–792. [Google Scholar] [CrossRef]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2012, 41, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Howell, A.B.; Reed, J.D.; Krueger, C.G.; Winterbottom, R.; Cunningham, D.G.; Leahy, M. A-type cranberry proanthocyanidins and uropathogenic bacterial anti-adhesion activity. Phytochemistry 2005, 66, 2281–2291. [Google Scholar] [CrossRef] [PubMed]
- Youdim, K.A.; McDonald, J.; Kalt, W.; Joseph, J.A. Potential role of dietary flavonoids in reducing microvascular endothelium vulnerability to oxidative and inflammatory insults. J. Nutr. Biochem. 2002, 13, 282–288. [Google Scholar] [CrossRef]
- Schmidt, B.M.; Howell, A.B.; McEniry, B.; Knight, C.T.; Seigler, D.; Erdman, J.W.; Lila, M.A. Effective separation of potent antiproliferation and antiadhesion components from wild blueberry (Vaccinium angustifolium Ait.) fruits. J. Agric. Food Chem. 2004, 52, 6433–6442. [Google Scholar] [CrossRef] [PubMed]
- Yi, W.; Fischer, J.; Krewer, G.; Akoh, C.C. Phenolic compounds from blueberries can inhibit colon cancer cell proliferation and induce apoptosis. J. Agric. Food Chem. 2005, 53, 7320–7329. [Google Scholar] [CrossRef]
- Benzie, I.F.F.; Devaki, M. The Ferric Reducing/Antioxidant Power (FRAP) assay for non-enzymatic antioxidant capacity: Concepts, procedures, limitations and applications. In Measurement of Antioxidant Activity & Capacity Functional Food Science and Technology Series, 1st ed.; Apak, R., Capanoglu, E., Shahidi, F., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2018; ISBN 9781119135357. [Google Scholar]
- Ntemiri, A.; Ribière, C.; Stanton, C.; Ross, R.P.; O’Connor, E.M.; O’Toole, P.W. Retention of microbiota diversity by lactose-free milk in a mouse model of elderly gut microbiota. J. Agric. Food Chem. 2019, 67, 2098–2112. [Google Scholar] [CrossRef]
- Magoc, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Gonzalez Peña, A.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10. [Google Scholar] [CrossRef]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef]
- Allard, G.; Ryan, F.J.; Jeffery, I.B.; Claesson, M.J. SPINGO: A rapid species-classifier for microbial amplicon sequences. BMC Bioinform. 2015, 16, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Bittinger, K.; Bushman, F.D.; Desantis, T.Z.; Andersen, G.L.; Knight, R. PyNAST: A flexible tool for aligning sequences to a template alignment. Bioinformatics 2010, 26, 266–267. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, T.S.; Rampelli, S.; Jeffery, I.B.; Santoro, A.; Neto, M.; Capri, M.; Giampieri, E.; Jennings, A.; Candela, M.; Turroni, S.; et al. Mediterranean diet intervention alters the gut microbiome in older people reducing frailty and improving health status: The NU-AGE 1-year dietary intervention across five European countries. Gut 2020, 69, 1218–1228. [Google Scholar] [CrossRef] [PubMed]
- Macfarlane, G.T.; Macfarlane, S. Models for intestinal fermentation: Association between food components, delivery systems, bioavailability and functional interactions in the gut. Curr. Opin. Biotechnol. 2007, 18, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, M.; Eck, A.; Koenen, M.E.; Savelkoul, P.H.M.; Budding, A.E.; Venema, K. Evaluation of an optimal preparation of human standardized fecal inocula for in vitro fermentation studies. J. Microbiol. Methods 2015, 117, 78–84. [Google Scholar] [CrossRef]
- Zoetendal, E.G.; Raes, J.; van den Bogert, B.; Arumugam, M.; Booijink, C.C.; Troost, F.J.; Bork, P.; Wels, M.; de Vos, W.M.; Kleerebezem, M. The human small intestinal microbiota is driven by rapid uptake and conversion of simple carbohydrates. ISME J. 2012, 6, 1415–1426. [Google Scholar] [CrossRef]
- Shao, Y.; Ding, R.; Xu, B.; Hua, R.; Shen, Q.; He, K.; Yao, Q. Alterations of gut microbiota after Roux-en-Y gastric bypass and sleeve gastrectomy in sprague-dawley rats. Obes. Surg. 2017, 27, 295–302. [Google Scholar] [CrossRef]
- Kawabata, K.; Yoshioka, Y.; Terao, J. Role of intestinal microbiota in the bioavailability and physiological functions of dietary polyphenols. Molecules 2019, 24, 370. [Google Scholar] [CrossRef]
- Pokusaeva, K.; Fitzgerald, G.F.; van Sinderen, D. Carbohydrate metabolism in Bifidobacteria. Genes Nutr. 2011, 6, 285–306. [Google Scholar] [CrossRef]
- Sánchez-Patán, F.; Barroso, E.; van de Wiele, T.; Jiménez-Girón, A.; Martín-Alvarez, P.J.; Moreno-Arribas, M.V.; Martínez-Cuesta, M.C.; Peláez, C.; Requena, T.; Bartolomé, B. Comparative in vitro fermentations of cranberry and grape seed polyphenols with colonic microbiota. Food Chem. 2015, 183, 273–282. [Google Scholar] [CrossRef]
- Guglielmetti, S.; Fracassetti, D.; Taverniti, V.; Del Bo’, C.; Vendrame, S.; Klimis-Zacas, D.; Arioli, S.; Riso, P.; Porrini, M. Differential modulation of human intestinal bifidobacterium populations after consumption of a wild blueberry (vaccinium angustifolium) drink. J. Agric. Food Chem. 2013, 61, 8134–8140. [Google Scholar] [CrossRef] [PubMed]
- Blum, J.; Gheller, B.; Hwang, S.; Bender, E.; Gheller, M.; Thalacker-Mercer, A. Consumption of a blueberry-enriched diet by women for 6 weeks alters determinants of human muscle progenitor cell function. J. Nutr. 2020, 1, 2412–2418. [Google Scholar] [CrossRef] [PubMed]
- Lusis, A.J.; Attie, A.D.; Reue, K. Metabolic syndrome: From epidemiology to systems biology. Nat. Rev. Genet. 2008, 9, 819–830. [Google Scholar] [CrossRef] [PubMed]
- Catrysse, L.; van Loo, G. Inflammation and the metabolic syndrome: The tissue-specific functions of NF-κB. Trends Cell Biol. 2017, 27, 417–429. [Google Scholar] [CrossRef] [PubMed]
- Christ, A.; Latz, E. The Western lifestyle has lasting effects on metaflammation. Nat. Rev. Immunol. 2019, 19, 267–268. [Google Scholar] [CrossRef] [PubMed]
- Le Chatelier, E.; Nielsen, T.; Qin, J.; Prifti, E.; Hildebrand, F.; Falony, G.; Almeida, M.; Arumugam, M.; Batto, J.-M.; Kennedy, S.; et al. Richness of human gut microbiome correlates with metabolic markers. Nature 2013, 500, 541–546. [Google Scholar] [CrossRef]
- Moeller, A.H. The shrinking human gut microbiome. Curr. Opin. Microbiol. 2017, 38, 30–35. [Google Scholar] [CrossRef]
- Rajilić-Stojanović, M.; de Vos, W.M. The first 1000 cultured species of the human gastrointestinal microbiota. FEMS Microbiol. Rev. 2014, 38, 996–1047. [Google Scholar] [CrossRef]
- Abell, G.C.J.; Cooke, C.M.; Bennett, C.N.; Conlon, M.A.; McOrist, A.L. Phylotypes related to Ruminococcus bromii are abundant in the large bowel of humans and increase in response to a diet high in resistant starch. FEMS Microbiol. Ecol. 2008, 66, 505–515. [Google Scholar] [CrossRef]
- Ze, X.; Duncan, S.H.; Louis, P.; Flint, H.J. Ruminococcus bromii is a keystone species for the degradation of resistant starch in the human colon. ISME J. 2012, 6, 1535–1543. [Google Scholar] [CrossRef]
- Cockburn, D.W.; Koropatkin, N.M. Polysaccharide degradation by the intestinal microbiota and its influence on human health and disease. J. Mol. Biol. 2016, 428, 3230–3252. [Google Scholar] [CrossRef] [PubMed]
- Scott, K.P.; Martin, J.C.; Duncan, S.H.; Flint, H.J. Prebiotic stimulation of human colonic butyrate-producing bacteria and bifidobacteria, in vitro. FEMS Microbiol. Ecol. 2014, 87, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Vandeputte, D.; Falony, G.; Vieira-Silva, S.; Wang, J.; Sailer, M.; Theis, S.; Verbeke, K.; Raes, J. Prebiotic inulin-type fructans induce specific changes in the human gut microbiota. Gut 2017, 66, 1968–1974. [Google Scholar] [CrossRef] [PubMed]
- Aron-Wisnewsky, J.; Prifti, E.; Belda, E.; Ichou, F.; Kayser, B.D.; Dao, M.C.; Verger, E.O.; Hedjazi, L.; Bouillot, J.-L.; Chevallier, J.-M.; et al. Major microbiota dysbiosis in severe obesity: Fate after bariatric surgery. Gut 2019, 68, 70–82. [Google Scholar] [CrossRef]
- Zeevi, D.; Korem, T.; Zmora, N.; Israeli, D.; Rothschild, D.; Weinberger, A.; Ben-Yacov, O.; Lador, D.; Avnit-Sagi, T.; Lotan-Pompan, M.; et al. Personalized nutrition by prediction of glycemic responses. Cell 2015, 163, 1079–1095. [Google Scholar] [CrossRef] [PubMed]
- Koh, A.; De Vadder, F.; Kovatcheva-Datchary, P.; Bäckhed, F. From dietary fiber to host physiology: Short-chain fatty acids as key bacterial metabolites. Cell 2016, 165, 1332–1345. [Google Scholar] [CrossRef] [PubMed]
- Heyman-Lindén, L.; Kotowska, D.; Sand, E.; Bjursell, M.; Plaza, M.; Turner, C.; Holm, C.; Fåk, F.; Berger, K. Lingonberries alter the gut microbiota and prevent low-grade inflammation in high-fat diet fed mice. Food Nutr. Res. 2016, 60, 29993. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Indias, I.; Sánchez-Alcoholado, L.; Pérez-Martínez, P.; Andrés-Lacueva, C.; Cardona, F.; Tinahones, F.; Queipo-Ortuño, M.I. Red wine polyphenols modulate fecal microbiota and reduce markers of the metabolic syndrome in obese patients. Food Funct. 2016, 7, 1775–1787. [Google Scholar] [CrossRef]
- González-Sarrías, A.; Romo-Vaquero, M.; García-Villalba, R.; Cortés-Martín, A.; Selma, M.V.; Espín, J.C. The endotoxemia marker lipopolysaccharide-binding protein is reduced in overweight-obese subjects consuming pomegranate extract by modulating the gut microbiota: A randomized clinical trial. Mol. Nutr. Food Res. 2018, 62, 1800160. [Google Scholar] [CrossRef]
- Vrieze, A.; Van Nood, E.; Holleman, F.; Salojärvi, J.; Kootte, R.S.; Bartelsman, J.F.W.M.; Dallinga-Thie, G.M.; Ackermans, M.T.; Serlie, M.J.; Oozeer, R.; et al. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology 2012, 143, 913–916.e7. [Google Scholar] [CrossRef]
- Udayappan, S.; Manneras-Holm, L.; Chaplin-Scott, A.; Belzer, C.; Herrema, H.; Dallinga-Thie, G.M.; Duncan, S.H.; Stroes, E.S.G.; Groen, A.K.; Flint, H.J.; et al. Oral treatment with Eubacterium hallii improves insulin sensitivity in db/db mice. NPJ Biofilms Microbiomes 2016, 2, 16009. [Google Scholar] [CrossRef] [PubMed]
- Marín, L.; Miguélez, E.M.; Villar, C.J.; Lombó, F. Bioavailability of dietary polyphenols and gut microbiota metabolism: Antimicrobial properties. Biomed. Res. Int. 2015, 2015, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Ozdal, T.; Sela, D.A.; Xiao, J.; Boyacioglu, D.; Chen, F.; Capanoglu, E. The reciprocal interactions between polyphenols and gut microbiota and effects on bioaccessibility. Nutrients 2016, 8, 78. [Google Scholar] [CrossRef] [PubMed]
- Anhê, F.F.; Nachbar, R.T.; Varin, T.V.; Vilela, V.; Dudonné, S.; Pilon, G.; Fournier, M.; Lecours, M.-A.; Desjardins, Y.; Roy, D.; et al. A polyphenol-rich cranberry extract reverses insulin resistance and hepatic steatosis independently of body weight loss. Mol. Metab. 2017, 6, 1563–1573. [Google Scholar] [CrossRef] [PubMed]
- Anhê, F.F.; Nachbar, R.T.; Varin, T.V.; Trottier, J.; Dudonné, S.; Le Barz, M.; Feutry, P.; Pilon, G.; Barbier, O.; Desjardins, Y.; et al. Treatment with camu camu ( Myrciaria dubia ) prevents obesity by altering the gut microbiota and increasing energy expenditure in diet-induced obese mice. Gut 2019, 68, 453–464. [Google Scholar] [CrossRef] [PubMed]
- Del Bo, C.; Martini, D.; Porrini, M.; Klimis-Zacas, D.; Riso, P. Berries and oxidative stress markers: An overview of human intervention studies. Food Funct. 2015, 6, 2890–2917. [Google Scholar] [CrossRef] [PubMed]
- Wankhade, U.D.; Zhong, Y.; Lazarenko, O.P.; Chintapalli, S.V.; Piccolo, B.D.; Chen, J.R.; Shankar, K. Sex-specific changes in gut microbiome composition following blueberry consumption in C57Bl/6J mice. Nutrients 2019, 11, 313. [Google Scholar] [CrossRef]
- De Freitas Carvalho, M.M.; Lage, N.N.; de Souza Paulino, A.H.; Pereira, R.R.; de Almeida, L.T.; da Silva, T.F.; de Brito Magalhães, C.L.; de Lima, W.G.; Silva, M.E.; Pedrosa, M.L.; et al. Effects of açai on oxidative stress, ER stress, and inflammation-related parameters in mice with high fat diet-fed induced NAFLD. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef]
- Frolinger, T.; Sims, S.; Smith, C.; Wang, J.; Cheng, H.; Faith, J.; Ho, L.; Hao, K.; Pasinetti, G.M. The gut microbiota composition affects dietary polyphenols-mediated cognitive resilience in mice by modulating the bioavailability of phenolic acids. Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef]
- Rodríguez-Daza, M.C.; Daoust, L.; Boutkrabt, L.; Pilon, G.; Varin, T.; Dudonné, S.; Levy, É.; Marette, A.; Roy, D.; Desjardins, Y. Wild blueberry proanthocyanidins shape distinct gut microbiota profile and influence glucose homeostasis and intestinal phenotypes in high-fat high-sucrose fed mice. Sci. Rep. 2020, 10, 1–16. [Google Scholar] [CrossRef]
- Pedersen, H.K.; Gudmundsdottir, V.; Nielsen, H.B.; Hyotylainen, T.; Nielsen, T.; Jensen, B.A.H.; Forslund, K.; Hildebrand, F.; Prifti, E.; Falony, G.; et al. Human gut microbes impact host serum metabolome and insulin sensitivity. Nature 2016, 535, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Kasai, C.; Sugimoto, K.; Moritani, I.; Tanaka, J.; Oya, Y.; Inoue, H.; Tameda, M.; Shiraki, K.; Ito, M.; Takei, Y.; et al. Comparison of the gut microbiota composition between obese and non-obese individuals in a Japanese population, as analyzed by terminal restriction fragment length polymorphism and next-generation sequencing. BMC Gastroenterol. 2015, 15, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Lagkouvardos, I.; Kläring, K.; Heinzmann, S.S.; Platz, S.; Scholz, B.; Engel, K.-H.; Schmitt-Kopplin, P.; Haller, D.; Rohn, S.; Skurk, T.; et al. Gut metabolites and bacterial community networks during a pilot intervention study with flaxseeds in healthy adult men. Mol. Nutr. Food Res. 2015, 59, 1614–1628. [Google Scholar] [CrossRef]
- Patrone, V.; Vajana, E.; Minuti, A.; Callegari, M.L.; Federico, A.; Loguercio, C.; Dallio, M.; Tolone, S.; Docimo, L.; Morelli, L. Postoperative changes in fecal bacterial communities and fermentation products in obese patients undergoing bilio-intestinal bypass. Front. Microbiol. 2016, 7, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Q.; Li, D.; He, Y.; Li, Y.; Yang, Z.; Zhao, X.; Liu, Y.; Wang, Y.; Sun, J.; Feng, X.; et al. Discrepant gut microbiota markers for the classification of obesity-related metabolic abnormalities. Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Guerra, A.; Etienne-Mesmin, L.; Livrelli, V.; Denis, S.; Blanquet-Diot, S.; Alric, M. Relevance and challenges in modeling human gastric and small intestinal digestion. Trends Biotechnol. 2012, 30, 591–600. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, T.S.B.; Raes, J.; Bork, P. The human gut microbiome: From association to modulation. Cell 2018, 172, 1198–1215. [Google Scholar] [CrossRef] [PubMed]
- Edwards, C.A.; Havlik, J.; Cong, W.; Mullen, W.; Preston, T.; Morrison, D.J.; Combet, E. Polyphenols and health: Interactions between fibre, plant polyphenols and the gut microbiota. Nutr. Bull. 2017, 42, 356–360. [Google Scholar] [CrossRef]
- Duncan, S.H.; Flint, H.J. Probiotics and prebiotics and health in ageing populations. Maturitas 2013, 75, 44–50. [Google Scholar] [CrossRef]
- Cani, P.D. Human gut microbiome: Hopes, threats and promises. Gut 2018, 67, 1716–1725. [Google Scholar] [CrossRef]
- Cirstea, M.; Radisavljevic, N.; Finlay, B.B. Good bug, bad bug: Breaking through microbial stereotypes. Cell Host Microbe 2018, 23, 10–13. [Google Scholar] [CrossRef] [PubMed]
- De Filippis, F.; Pellegrini, N.; Laghi, L.; Gobbetti, M.; Ercolini, D. Unusual sub-genus associations of faecal Prevotella and Bacteroides with specific dietary patterns. Microbiome 2016, 4, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Anhê, F.F.; Choi, B.S.Y.; Dyck, J.R.B.; Schertzer, J.D.; Marette, A. Host–microbe interplay in the cardiometabolic benefits of dietary polyphenols. Trends Endocrinol. Metab. 2019, 30, 384–395. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Young (n = 10 a) | Old (n = 6) | |
|---|---|---|
| Age (yrs) | 28 ± 2 | 69 ± 2 |
| Weight (kg) | 64.31 ± 2.33 | 62.44 ± 3.84 |
| Height (cm) | 166.3 ± 1.5 | 161.3 ± 2.9 |
| BMI (kg/m2) | 23.3 ± 0.9 | 24.2 ± 2.0 |
| Glucose (mg/dL, Range: 74–106) | 89.5 ± 2.44 | 98.0 ± 1.7 |
| CRP (mg/L, Range: <1.1) | 1.21 ± 0.33 | 0.76 ± 0.28 |
| Clinical Indicator | OTU-Level Microbiota | CAG-Level Microbiota | ||
|---|---|---|---|---|
| R2 | p Value | R2 | p Value | |
| CRP | 0.03 | 0.95 | 0.02 | 0.81 |
| Glucose | 0.03 | 0.54 | 0.02 | 0.79 |
| FRAP | 0.04 | 0.01 | 0.06 | 0.05 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ntemiri, A.; Ghosh, T.S.; Gheller, M.E.; Tran, T.T.T.; Blum, J.E.; Pellanda, P.; Vlckova, K.; Neto, M.C.; Howell, A.; Thalacker-Mercer, A.; et al. Whole Blueberry and Isolated Polyphenol-Rich Fractions Modulate Specific Gut Microbes in an In Vitro Colon Model and in a Pilot Study in Human Consumers. Nutrients 2020, 12, 2800. https://doi.org/10.3390/nu12092800
Ntemiri A, Ghosh TS, Gheller ME, Tran TTT, Blum JE, Pellanda P, Vlckova K, Neto MC, Howell A, Thalacker-Mercer A, et al. Whole Blueberry and Isolated Polyphenol-Rich Fractions Modulate Specific Gut Microbes in an In Vitro Colon Model and in a Pilot Study in Human Consumers. Nutrients. 2020; 12(9):2800. https://doi.org/10.3390/nu12092800
Chicago/Turabian StyleNtemiri, Alexandra, Tarini S. Ghosh, Molly E. Gheller, Tam T. T. Tran, Jamie E. Blum, Paola Pellanda, Klara Vlckova, Marta C. Neto, Amy Howell, Anna Thalacker-Mercer, and et al. 2020. "Whole Blueberry and Isolated Polyphenol-Rich Fractions Modulate Specific Gut Microbes in an In Vitro Colon Model and in a Pilot Study in Human Consumers" Nutrients 12, no. 9: 2800. https://doi.org/10.3390/nu12092800
APA StyleNtemiri, A., Ghosh, T. S., Gheller, M. E., Tran, T. T. T., Blum, J. E., Pellanda, P., Vlckova, K., Neto, M. C., Howell, A., Thalacker-Mercer, A., & O’Toole, P. W. (2020). Whole Blueberry and Isolated Polyphenol-Rich Fractions Modulate Specific Gut Microbes in an In Vitro Colon Model and in a Pilot Study in Human Consumers. Nutrients, 12(9), 2800. https://doi.org/10.3390/nu12092800