Uric Acid and Vascular Damage in Essential Hypertension: Role of Insulin Resistance

 , , ,
, , ,  , ,
, ,  , , , , and
, , , , and 
Abstract
1. Introduction
2. Material and Methods
2.1. Study Population
2.2. Blood Pressure Measurements
2.3. Laboratory Determinations
2.4. Pulse Wave Reflection and Central BP Measurements
2.5. Statistical Analysis
2.6. Ethical Approval
3. Results
3.1. Study Population
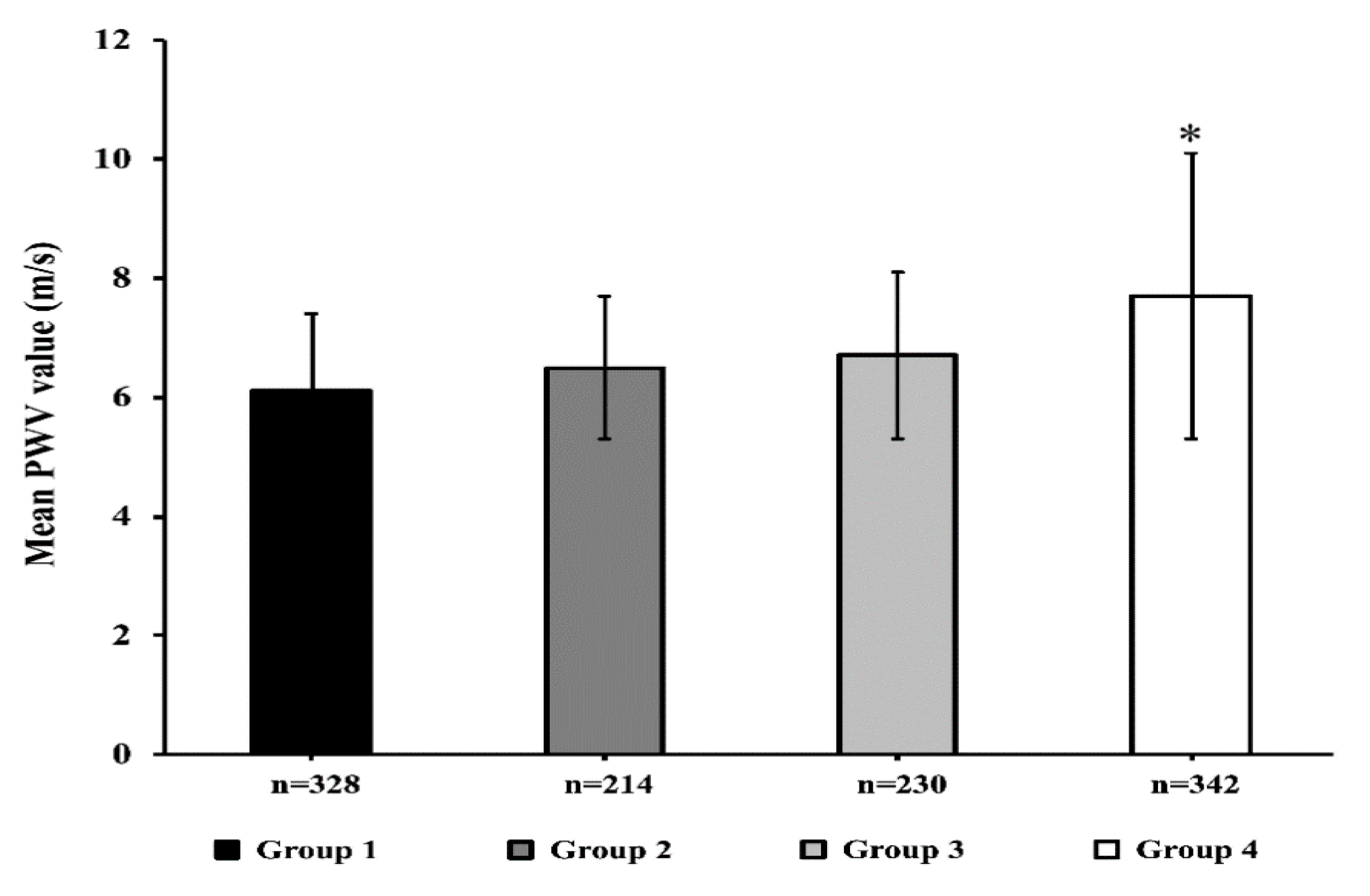
3.2. Hemodynamic Parameters
3.3. Correlational Analysis
3.4. Multivariate Analysis
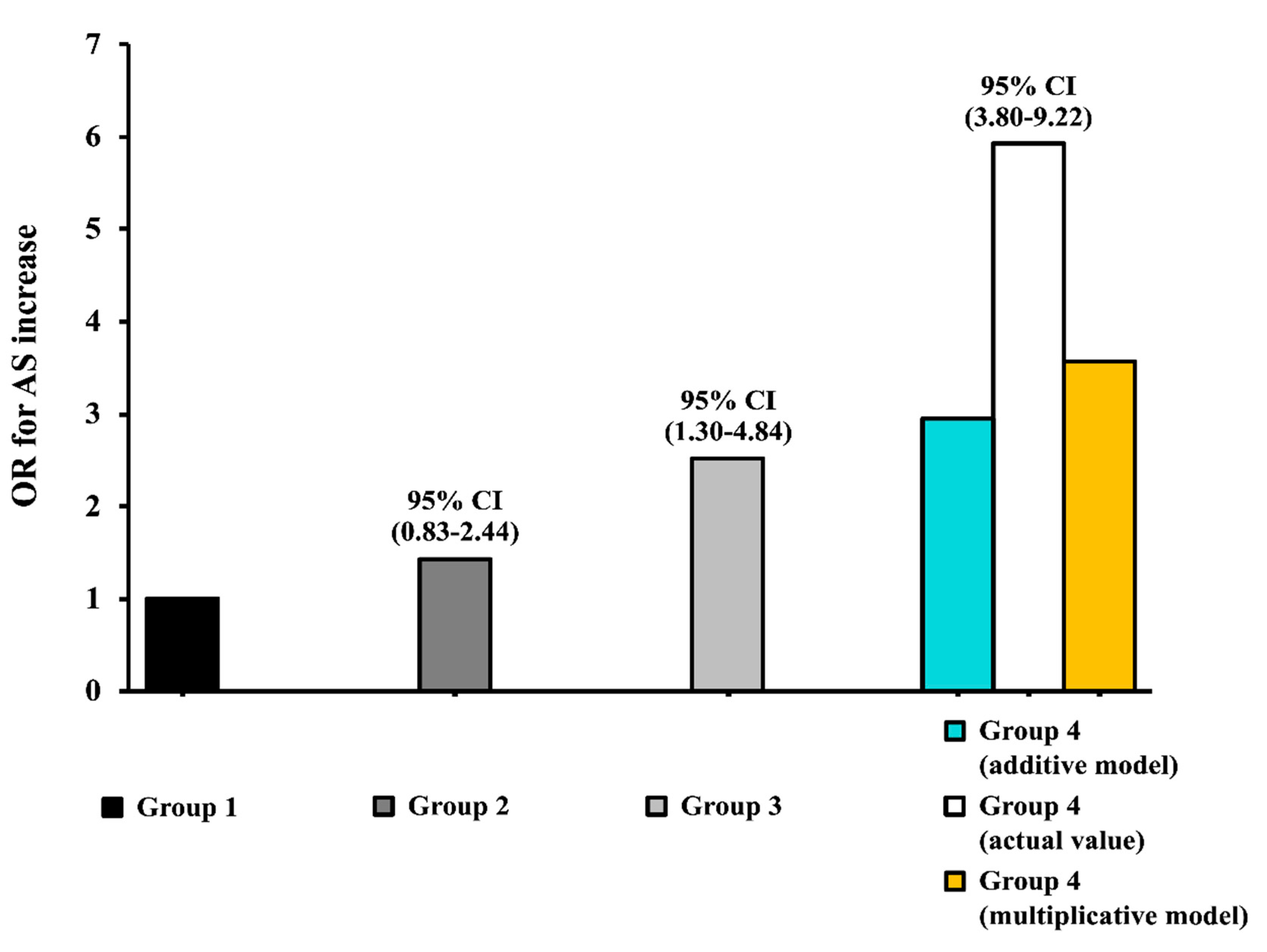
3.5. Interaction Analysis
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- So, A.; Thorens, B. Uric acid transport and disease. J. Clin. Investig. 2010, 120, 1791–1799. [Google Scholar] [CrossRef] [PubMed]
- Perticone, F.; Sciacqua, A.; Perticone, M.; Arturi, F.; Scarpino, P.E.; Quero, M.; Sesti, G. Serum uric acid and 1-h postload glucose in essential hypertension. Diabetes Care 2012, 35, 153–157. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Meshkani, R.; Zargari, M.; Larijani, B. The relationship between uric acid and metabolic syndrome in normal glucose tolerance and normal fasting glucose subjects. Acta Diabetol. 2011, 48, 79–88. [Google Scholar] [CrossRef]
- Bonora, E.; Targher, G.; Zenere, M.B.; Saggiani, F.; Cacciatori, V.; Tosi, F.; Travia, D.; Zenti, M.G.; Branzi, P.; Santi, L.; et al. Relationship of uric acid concentration to cardiovascular risk factors in young men. Role of obesity and central fat distribution. The Verona Young Men Atherosclerosis Risk Factors Study. Int. J. Obes. 1996, 20, 975–980. [Google Scholar]
- Perticone, F.; Maio, R.; Tassone, J.E.; Perticone, M.; Pascale, A.; Sciacqua, A.; Sesti, G. Interaction between uric acid and endothelial dysfunction predicts new onset of diabetes in hypertensive patients. Int. J. Cardiol. 2013, 167, 232–236. [Google Scholar] [CrossRef]
- Dehghan, A.; Van Hoek, M.; Sijbrands, E.J.G.; Hofman, A.; Witteman, J.C.M. High serum uric acid as a novel risk factor for type 2 diabetes. Diabetes Care 2008, 31, 361–362. [Google Scholar] [CrossRef]
- Krishnan, E.; Pandya, B.J.; Chung, L.; Hariri, A.; Dabbous, O. Hyperuricemia in young adults and risk of insulin resistance, prediabetes, and diabetes: A 15-year follow-up study. Am. J. Epidemiol. 2012, 176, 108–116. [Google Scholar] [CrossRef]
- Verdecchia, P.; Schillaci, G.; Reboldi, G.P.; Santeusanio, F.; Porcellati, C.; Brunetti, P. Relation between serum uric acid and risk of cardiovascular disease in essential hypertension: The PIUMA study. Hypertension 2000, 36, 1072–1078. [Google Scholar] [CrossRef]
- Niskanen, L.K.; Laaksonen, D.E.; Nyyssönen, K.; Alfthan, G.; Lakka, H.M.; Lakka, T.A.; Salonen, J.T. Uric acid level as a risk factor for cardiovascular and all-cause mortality in middle-aged men: A prospective cohort study. Arch. Intern. Med. 2004, 164, 1546–1551. [Google Scholar] [CrossRef] [PubMed]
- Perticone, M.; Tripepi, G.; Maio, R.; Cimellaro, A.; Addesi, D.; Baggetta, R.; Sciacqua, A.; Sesti, G.; Perticone, F. Risk reclassification ability of uric acid for cardiovascular outcomes in essential hypertension. Int. J. Cardiol. 2017, 243, 473–478. [Google Scholar] [CrossRef] [PubMed]
- Sautin, Y.Y.; Nakagawa, T.; Zharikov, S.; Johnson, R.J. Adverse effects of the classic antioxidant uric acid in adipocytes: NADPH oxidase-mediated oxidative/nitrosative stress. Am. J. Physiol.-Cell Physiol. 2007, 293. [Google Scholar] [CrossRef] [PubMed]
- Zoccali, C.; Maio, R.; Mallamaci, F.; Sesti, G.; Perticone, F. Uric acid and endothelial dysfunction in essential hypertension. J. Am. Soc. Nephrol. 2006, 17, 1466–1471. [Google Scholar] [CrossRef] [PubMed]
- Fiorentino, T.V.; Sesti, F.; Succurro, E.; Pedace, E.; Andreozzi, F.; Sciacqua, A.; Hribal, M.L.; Perticone, F.; Sesti, G. Higher serum levels of uric acid are associated with a reduced insulin clearance in non-diabetic individuals. Acta Diabetol. 2018, 55, 835–842. [Google Scholar] [CrossRef] [PubMed]
- Andreozzi, F.; Succurro, E.; Mancuso, M.R.; Perticone, M.; Sciacqua, A.; Perticone, F.; Sesti, G. Metabolic and cardiovascular risk factors in subjects with impaired fasting glucose: The 100 versus 110 mg/dL threshold. Diabetes Metab. Res. Rev. 2007, 23, 547–550. [Google Scholar] [CrossRef] [PubMed]
- Williams, B.; Mancia, G.; Spiering, W.; Agabiti Rosei, E.; Azizi, M.; Burnier, M.; Clement, D.; Coca, A.; De Simone, G.; Dominiczak, A.; et al. 2018 ESC/ESH Guidelines for the Management of Arterial Hypertension: The Task Force for the Management of Arterial Hypertension of the European Society of Cardiology and the European Society of Hypertension: The Task Force for the Management of Arterial Hypertension of the European Society of Cardiology and the European Society of Hypertension. J. Hypertens. 2018, 36, 1956–2041. [Google Scholar]
- Levey, A.S.; Stevens, L.A.; Schmid, C.H.; Zhang, Y.; Castro, A.F.; Feldman, H.I.; Kusek, J.W.; Eggers, P.; Van Lente, F.; Greene, T.; et al. A new equation to estimate glomerular filtration rate. Ann. Intern. Med. 2009, 150, 604–612. [Google Scholar] [CrossRef]
- Perticone, M.; Maio, R.; Tassone, E.J.; Tripepi, G.; Di Cello, S.; Miceli, S.; Caroleo, B.; Sciacqua, A.; Licata, A.; Sesti, G.; et al. Insulin-resistance HCV infection-related affects vascular stiffness in normotensives. Atherosclerosis 2015, 238, 108–112. [Google Scholar] [CrossRef]
- Chen, C.H.; Ting, C.T.; Nussbacher, A.; Nevo, E.; Kass, D.A.; Pak, P.; Wang, S.P.; Chang, M.S.; Yin, F.C.P. Validation of carotid artery tonometry as a means of estimating augmentation index of ascending aortic pressure. Hypertension 1996, 27, 168–175. [Google Scholar] [CrossRef]
- Covic, A.; Siriopol, D. Pulse wave velocity ratio: The new “gold standard” for measuring arterial stiffness. Hypertension 2015, 65, 289–290. [Google Scholar] [CrossRef]
- Tanaka, A.; Tomiyama, H.; Maruhashi, T.; Matsuzawa, Y.; Miyoshi, T.; Kabutoya, T. Physiological diagnostic criteria for vascular failure. Hypertension 2018, 5, 1060–1071. [Google Scholar] [CrossRef]
- Rothman, K.J.; Greenland, S.; Walker, A.M. Concepts of interaction. Am. J. Epidemiol. 1980, 112, 467–470. [Google Scholar] [CrossRef] [PubMed]
- De Mutsert, R.; Jager, K.J.; Zoccali, C.; Dekker, F.W. The effect of joint exposures: Examining the presence of interaction. Kidney Int. 2009, 75, 677–681. [Google Scholar] [CrossRef]
- Ramirez, A.J.; Christen, A.I.; Sanchez, R.A. Serum Uric Acid Elevation Is Associated To Arterial Stiffness In Hypertensive Patients With Metabolic Disturbances. Curr. Hypertens. Rev. 2018, 14, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Canepa, M.; Viazzi, F.; Strait, J.B.; Ameri, P.; Pontremoli, R.; Brunelli, C.; Studenski, S.; Ferrucci, L.; Lakatta, E.G.; Alghatrif, M. Longitudinal Association between Serum Uric Acid and Arterial Stiffness: Results from the Baltimore Longitudinal Study of Aging. Hypertension 2017, 69, 228–235. [Google Scholar] [CrossRef] [PubMed]
- Vlachopoulos, C.; Xaplanteris, P.; Vyssoulis, G.; Bratsas, A.; Baou, K.; Tzamou, V.; Aznaouridis, K.; Dima, I.; Lazaros, G.; Stefanadis, C. Association of serum uric acid level with aortic stiffness and arterial wave reflections in newly diagnosed, never-treated hypertension. Am. J. Hypertens. 2011, 24, 33–39. [Google Scholar] [CrossRef]
- Mulè, G.; Riccobene, R.; Castiglia, A.; D’Ignoto, F.; Ajello, E.; Geraci, G.; Guarino, L.; Nardi, E.; Vaccaro, F.; Cerasola, G.; et al. Relationships between mild hyperuricaemia and aortic stiffness in untreated hypertensive patients. Nutr. Metab. Cardiovasc. Dis. 2014, 24, 744–750. [Google Scholar] [CrossRef]
- Nagano, S.; Takahashi, M.; Miyai, N.; Oka, M.; Utsumi, M.; Shiba, M.; Mure, K.; Takeshita, T.; Arita, M. Association of serum uric acid with subsequent arterial stiffness and renal function in normotensive subjects. Hypertens. Res. 2017, 40, 620–624. [Google Scholar] [CrossRef]
- Vlachopoulos, C.; Aznaouridis, K.; Stefanadis, C. Prediction of Cardiovascular Events and All-Cause Mortality With Arterial Stiffness. A Systematic Review and Meta-Analysis. J. Am. Coll. Cardiol. 2010, 55, 1318–1327. [Google Scholar] [CrossRef]
- Ding, X.H.; Wang, X.; Cao, R.; Yang, X.; Xiao, W.; Zhang, Y.; Bai, Y.; Wu, H.; Ye, P. A higher baseline plasma uric acid level is an independent predictor of arterial stiffness a community-based prospective study. Medicine (United States) 2017, 96, e9597. [Google Scholar] [CrossRef]
- Tassone, E.J.; Cimellaro, A.; Perticone, M.; Hribal, M.L.; Sciacqua, A.; Andreozzi, F.; Sesti, G.; Perticone, F. Uric acid impairs insulin signaling by promoting ENPP1 binding to insulin receptor in human umbilical vein endothelial cells. Front. Endocrinol. (Lausanne) 2018, 9, 98. [Google Scholar] [CrossRef]
- Sesti, G.; Hribal, M.L.; Procopio, T.; Fiorentino, T.V.; Sciacqua, A.; Andreozzi, F.; Marini, M.A.; Perticone, F. Low circulating insulin-like growth factor-1 levels are associated with high serum uric acid in nondiabetic adult subjects. Nutr. Metab. Cardiovasc. Dis. 2014, 24, 1365–1372. [Google Scholar] [CrossRef] [PubMed]
- Mazzali, M.; Kanellis, J.; Han, L.; Feng, L.; Xia, Y.Y.; Chen, Q.; Kang, D.H.; Gordon, K.L.; Watanabe, S.; Nakagawa, T.; et al. Hyperuricemia induces a primary renal arteriolopathy in rats by a blood pressure-independent mechanism. Am. J. Physiol.-Ren. Physiol. 2002, 282, F991–F997. [Google Scholar] [CrossRef] [PubMed]
- Mazzali, M.; Hughes, J.; Kim, Y.G.; Jefferson, J.A.; Kang, D.H.; Gordon, K.L.; Lan, H.Y.; Kivlighn, S.; Johnson, R.J. Elevated uric acid increases blood pressure in the rat by a novel crystal-independent mechanism. Hypertension 2001, 38, 1101–1106. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.F.; Chuang, S.Y.; Cheng, H.M.; Sung, S.H.; Ting, C.T.; Lakatta, E.G.; Yin, F.C.P.; Chou, P.; Chen, C.H. Associations of serum uric acid levels with arterial wave reflections and central systolic blood pressure. Int. J. Cardiol. 2013, 168, 2057–2063. [Google Scholar] [CrossRef] [PubMed]
- Ishizaka, N.; Ishizaka, Y.; Toda, E.I.; Hashimoto, H.; Nagai, R.; Yamakado, M. Higher serum uric acid is associated with increased arterial stiffness in Japanese individuals. Atherosclerosis 2007, 192, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Ng, K.P.; Stringer, S.J.; Jesky, M.D.; Yadav, P.; Athwal, R.; Dutton, M.; Ferro, C.J.; Cockwell, P. Allopurinol is an independent determinant of improved arterial stiffness in chronic kidney disease: A cross-sectional study. PLoS ONE 2014, 9, e91961. [Google Scholar] [CrossRef]
- Doehner, W.; Jankowska, E.A.; Springer, J.; Lainscak, M.; Anker, S.D. Uric acid and xanthine oxidase in heart failure—Emerging data and therapeutic implications. Int. J. Cardiol. 2016, 213, 15–19. [Google Scholar] [CrossRef]
- Nishizawa, T.; Taniura, T.; Nomura, S. Effects of febuxostat on platelet-derived microparticles and adiponectin in patients with hyperuricema. Blood Coagul. Fibrinolysis 2015, 26, 887–892. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| All | I Tertile | II Tertile | III Tertile | p | |
|---|---|---|---|---|---|
| Variables | (n = 1114) | (n = 371) | (n = 371) | (n = 372) | |
| Gender, m/f | 634/480 | 191/180 | 214/157 | 229/143 | 0.020 * |
| Age, years | 48.9 ± 11.5 | 48.1 ± 10.9 | 49.3 ± 11.9 | 48.9 ± 11.5 | 0.260 |
| BMI, Kg/m2 | 29.5 ± 5.6 | 29.2 ± 5.8 | 29.2 ± 5.2 | 30.1 ± 5.7 | 0.045 |
| Glucose, mg/dL | 92.7 ± 10.3 | 91.4 ± 9.9 | 92.1 ± 10.1 | 94.6 ± 10.7 | <0.0001 |
| Insulin, mU/mL | 13.2 ± 6.8 | 11.2 ± 5.3 | 13.1 ± 6.6 | 15.6 ± 7.6 | <0.0001 |
| HOMA | 3.1 ± 1.7 | 2.5 ± 1.3 | 2.9 ± 1.6 | 3.7 ± 1.9 | <0.0001 |
| Serum LDL, mg/dL | 123.6 ± 34.7 | 120.4 ± 35.3 | 125.5 ± 33.1 | 125.1 ± 35.5 | 0.085 |
| Serum HDL, mg/dL | 50.8 ± 13.4 | 52.1 ± 13.9 | 50.8 ± 13.6 | 49.3 ± 12.7 | 0.016 |
| Serum Triglycerides, mg/dL | 127.3 ± 57.6 | 123.7 ± 58.1 | 130.3 ± 58.2 | 127.9 ± 56.6 | 0.289 |
| e-GFR, mL/min/1.73 m2 | 106.2 ± 28.2 | 110.1 ± 31.6 | 105.4 ± 26.6 | 103.3 ± 25.8 | 0.005 |
| hs-CRP mg/dL | 3.0 ± 2.1 | 2.8 ± 1.9 | 2.9 ± 2.1 | 3.4 ± 2.4 | <0.0001 |
| Smokers, (n/%) | 310/27.8% | 95/25.6% | 104/28.0% | 111/29.8% | 0.434 * |
| All | I Tertile | II Tertile | III Tertile | p | |
|---|---|---|---|---|---|
| Variables | (n = 1114) | (n = 371) | (n = 371) | (n = 372) | |
| HR, bpm | 67.5 ± 20.9 | 67.5 ± 11.5 | 67.1 ± 12.5 | 68.7 ± 33.1 | 0.591 |
| SBP, mmHg | 143.9 ± 14.1 | 142.4 ± 13.5 | 143.4 ± 13.9 | 145.9 ± 14.3 | 0.002 |
| DBP, mmHg | 89.3 ± 8.2 | 90,1 ± 8,2 | 88.6 ± 8.1 | 87.9 ± 9.3 | 0.064 |
| PP, mmHg | 54 ± 15.4 | 53 ± 13.5 | 53 ± 15.2 | 56 ± 15.6 | 0.004 |
| Central-SBP, mmHg | 131 ± 13.4 | 129 ± 12.9 | 131 ± 14.5 | 132 ± 12.7 | 0.025 |
| Central-DBP, mmHg | 88 ± 9.4 | 89 ± 9.2 | 88 ± 9.4 | 87 ± 9.3 | 0.008 |
| Central-PP, mmHg | 42 ± 14.3 | 40 ± 14.8 | 42 ± 14.1 | 44 ± 13.6 | <0.0001 |
| AP, mmHg | 11.1 ± 7.1 | 9.1 ± 6.2 | 10.8 ± 6.7 | 13.2 ± 7.5 | <0.0001 |
| AI, % | 25.6 ± 12.9 | 22.1 ± 12.3 | 25.4 ± 12.9 | 29.2 ± 12.7 | <0.0001 |
| PWV, m/s | 6.8 ± 1.9 | 6.2 ± 1.3 | 6.4 ± 1.1 | 7.8 ± 2.5 | <0.0001 |
| PWV | ||
|---|---|---|
| R | p | |
| Age, years | 0.108 | <0.0001 |
| BMI, Kg/m2 | 0.025 | 0.206 |
| SBP, mmHg | 0.158 | <0.0001 |
| DBP, mmHg | −0.048 | 0.055 |
| Serum HDL, mg/dL | −0.084 | 0.003 |
| Serum LDL, mg/dL | 0.077 | 0.005 |
| Serum Triglycerides, mg/dL | 0.013 | 0.330 |
| e-GFR, mL/min/1.73 m2 | −0.024 | 0.212 |
| hs-CRP, mg/dL | 0.159 | <0.0001 |
| HOMA | 0.391 | <0.0001 |
| Uric Acid, mg/dL | 0.453 | <0.0001 |
| R2 | p | |
|---|---|---|
| Uric Acid, mg/dL | 20.5 | <0.0001 |
| HOMA | 6.3 | <0.0001 |
| SBP, mmHg | 0.9 | <0.0001 |
| Age, years | 0.5 | 0.008 |
| e-GFR mL/min/1.73 m2 | 0.7 | 0.001 |
| hs-CRP, mg/dL | 0.3 | 0.045 |
| OR | 95% CI | |
|---|---|---|
| Uric Acid, 1 mg/dL | 1.80 | 1.55–2.08 |
| Homa, 1 U | 1.47 | 1.29–1.66 |
| Age, 10 years | 1.26 | 1.01–1.59 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cassano, V.; Crescibene, D.; Hribal, M.L.; Pelaia, C.; Armentaro, G.; Magurno, M.; Toscani, A.; Miceli, S.; Andreozzi, F.; Maio, R.; et al. Uric Acid and Vascular Damage in Essential Hypertension: Role of Insulin Resistance. Nutrients 2020, 12, 2509. https://doi.org/10.3390/nu12092509
Cassano V, Crescibene D, Hribal ML, Pelaia C, Armentaro G, Magurno M, Toscani A, Miceli S, Andreozzi F, Maio R, et al. Uric Acid and Vascular Damage in Essential Hypertension: Role of Insulin Resistance. Nutrients. 2020; 12(9):2509. https://doi.org/10.3390/nu12092509
Chicago/Turabian StyleCassano, Velia, Daniele Crescibene, Marta Letizia Hribal, Corrado Pelaia, Giuseppe Armentaro, Marcello Magurno, Alfredo Toscani, Sofia Miceli, Francesco Andreozzi, Raffaele Maio, and et al. 2020. "Uric Acid and Vascular Damage in Essential Hypertension: Role of Insulin Resistance" Nutrients 12, no. 9: 2509. https://doi.org/10.3390/nu12092509
APA StyleCassano, V., Crescibene, D., Hribal, M. L., Pelaia, C., Armentaro, G., Magurno, M., Toscani, A., Miceli, S., Andreozzi, F., Maio, R., Perticone, M., Sesti, G., Perticone, F., & Sciacqua, A. (2020). Uric Acid and Vascular Damage in Essential Hypertension: Role of Insulin Resistance. Nutrients, 12(9), 2509. https://doi.org/10.3390/nu12092509