Punicalagin Regulates Apoptosis-Autophagy Switch via Modulation of Annexin A1 in Colorectal Cancer



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Compounds
2.2. Cell Culture
2.3. Cell Viability Assay
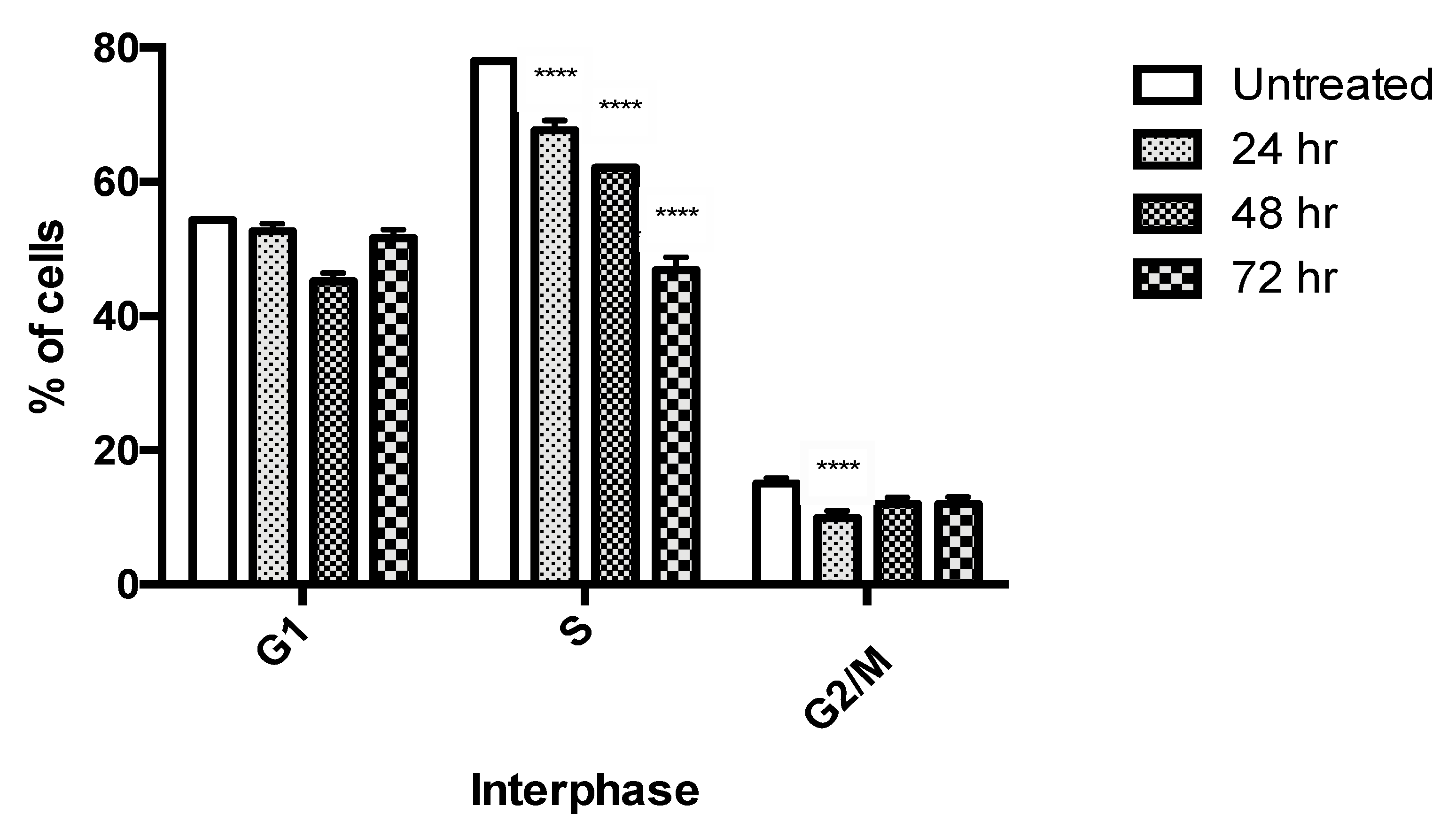
2.4. Cell Cycle Analysis by Flow Cytometry
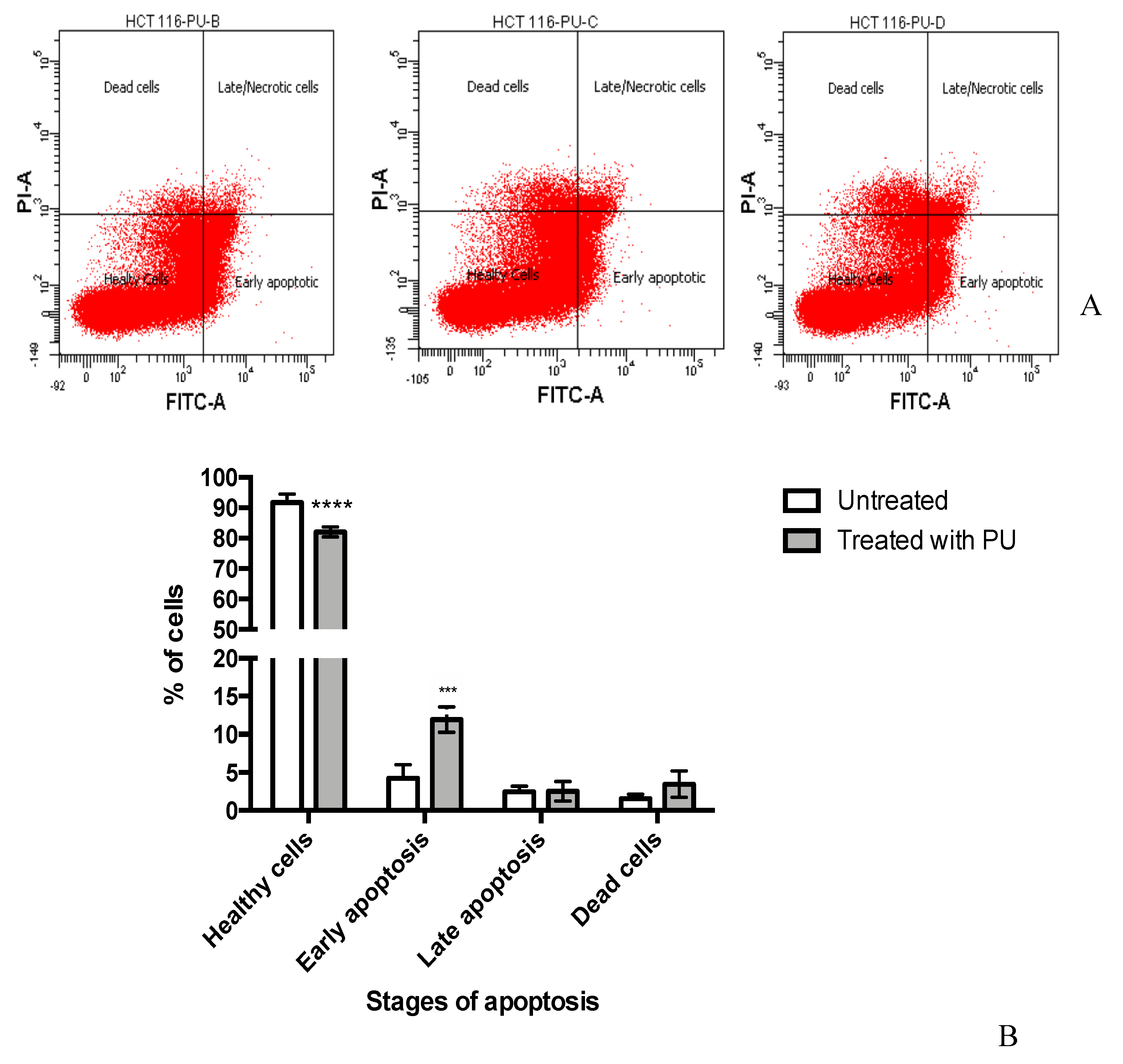
2.5. Annexin V by Flow Cytometry
2.6. Caspase 3/7, 8 and 9 Assay
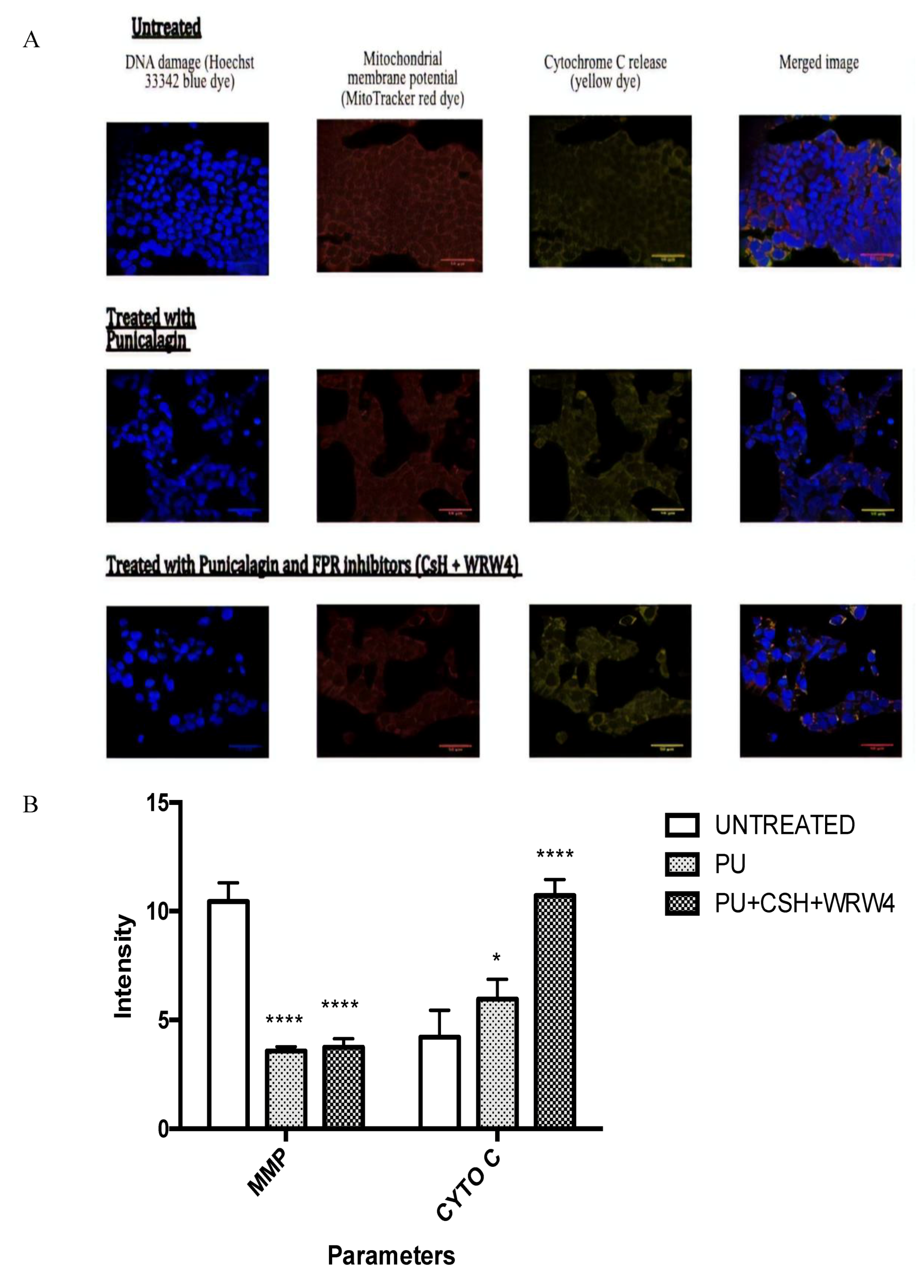
2.7. Confocal Analysis
2.8. Proteome Profiler
2.9. ELISA for Annexin A1
2.10. Autophagy Flux Assay
2.11. Statistical Analysis
3. Results
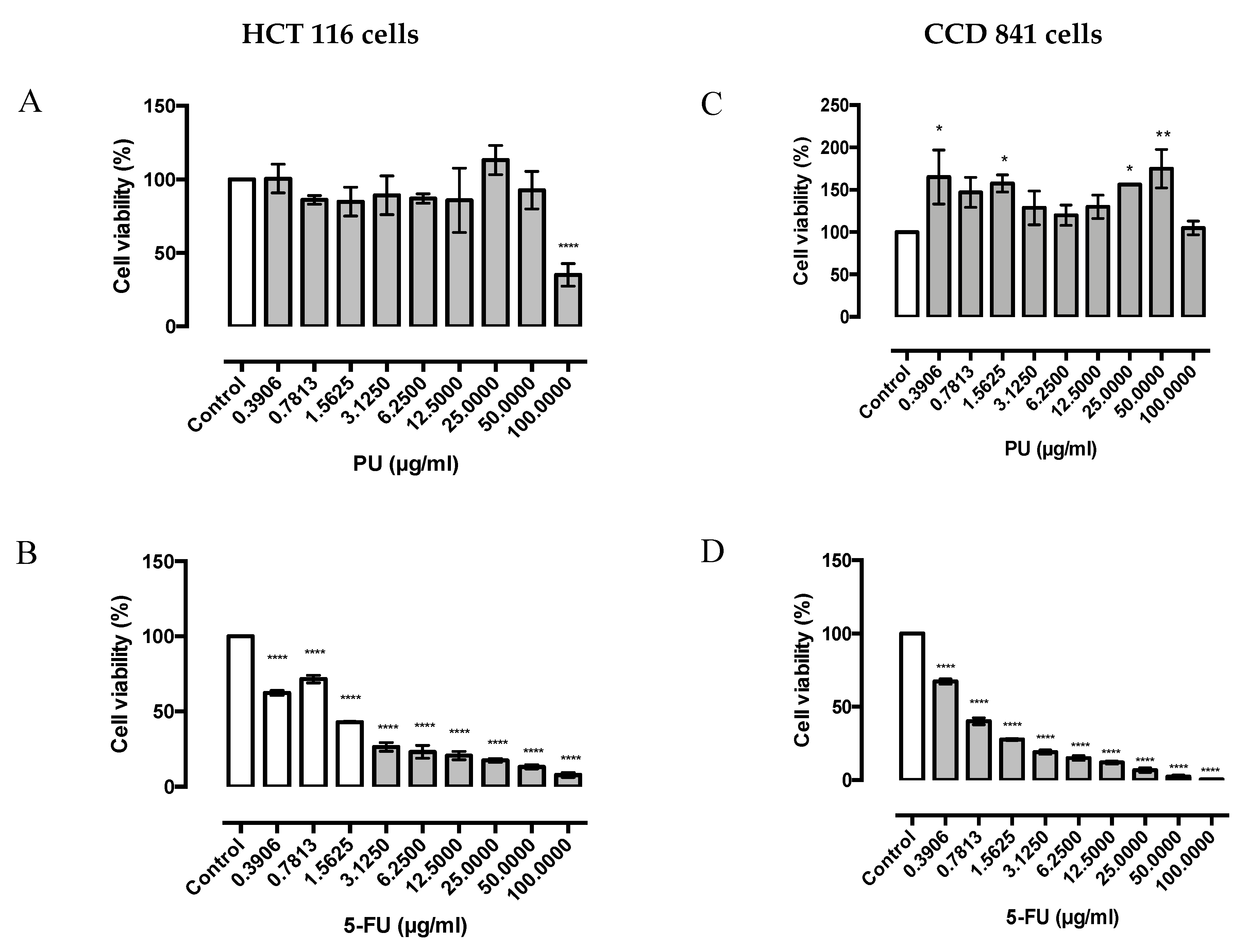
3.1. PU Exhibits Selective Cytotoxicity on HCT 116
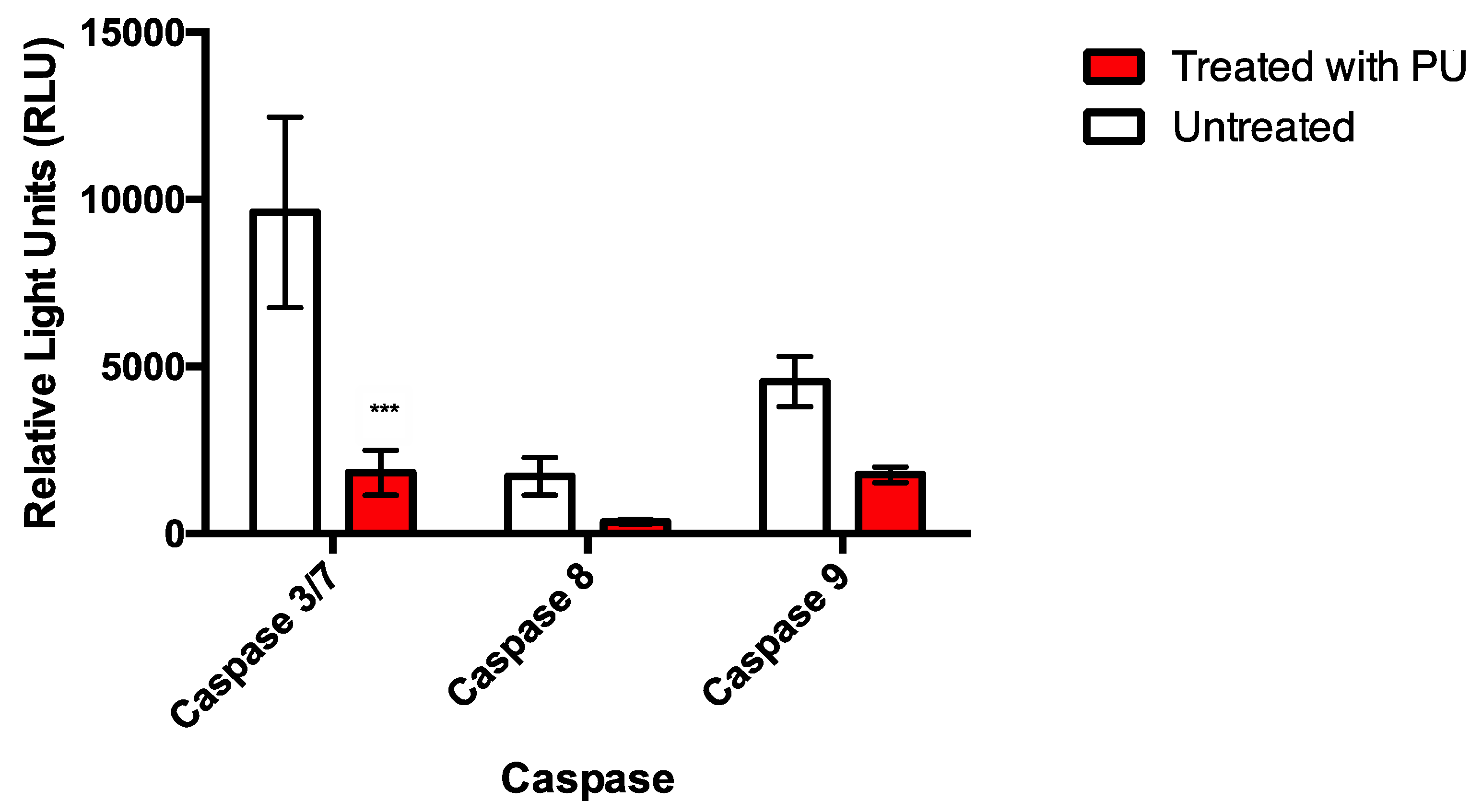
3.2. PU Exhibits Apoptotic Properties on HCT 116
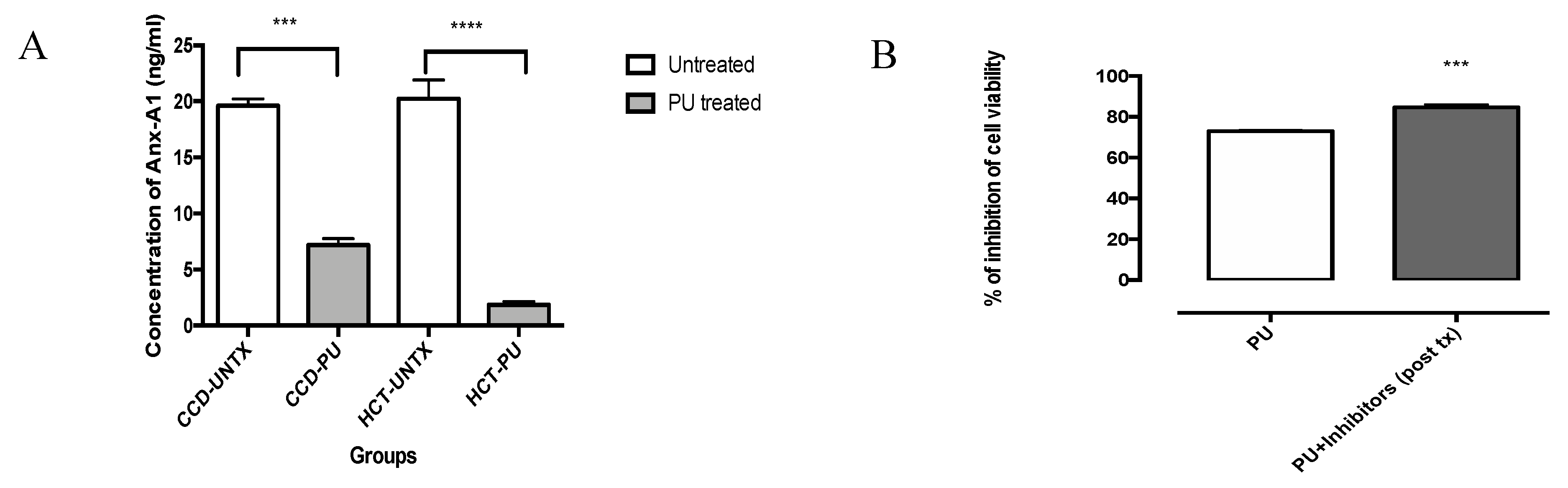
3.3. PU Downregulates Anx-A1 in HCT 116
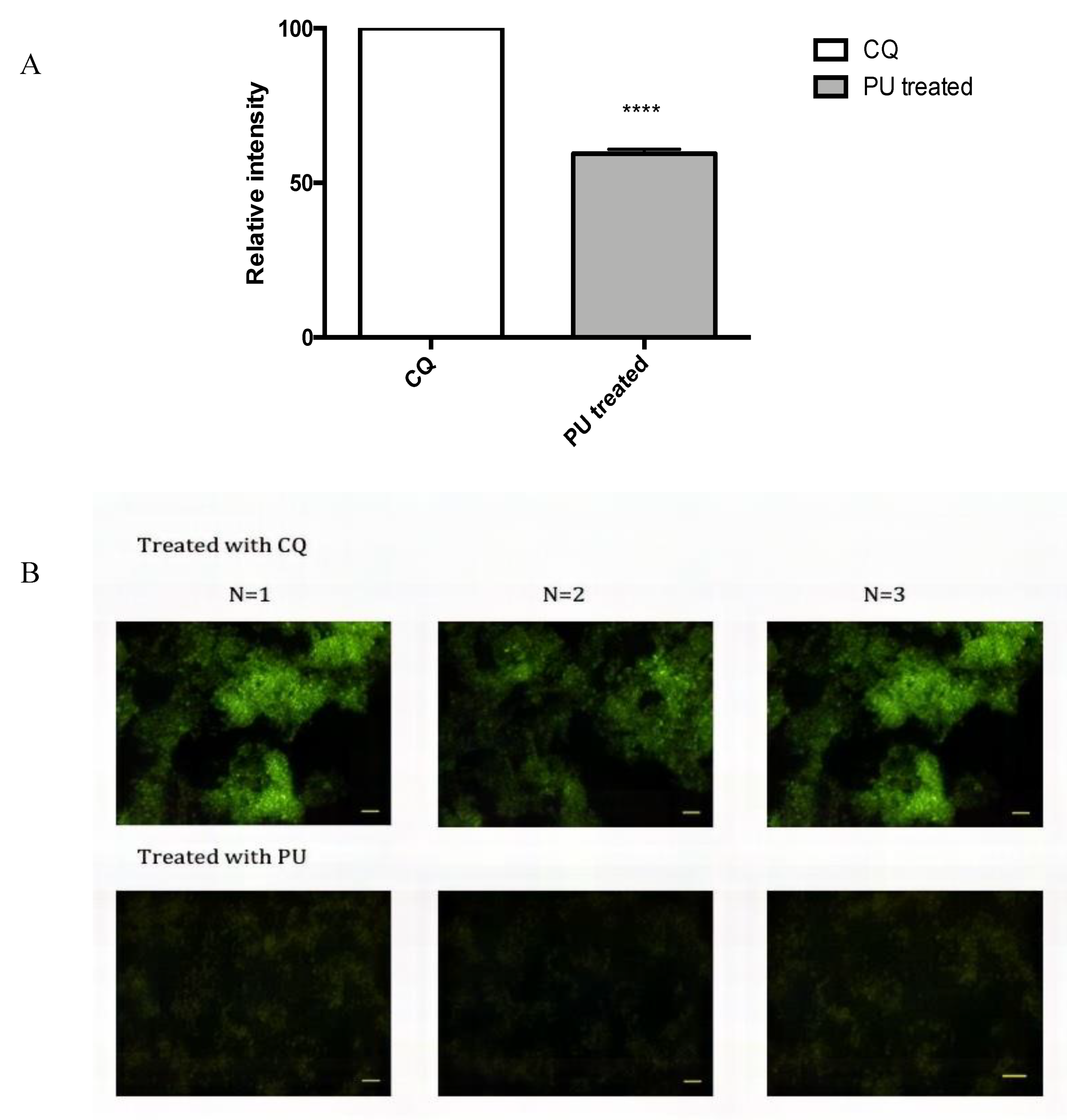
3.4. PU Exhibits Autophagic Properties on HCT 116
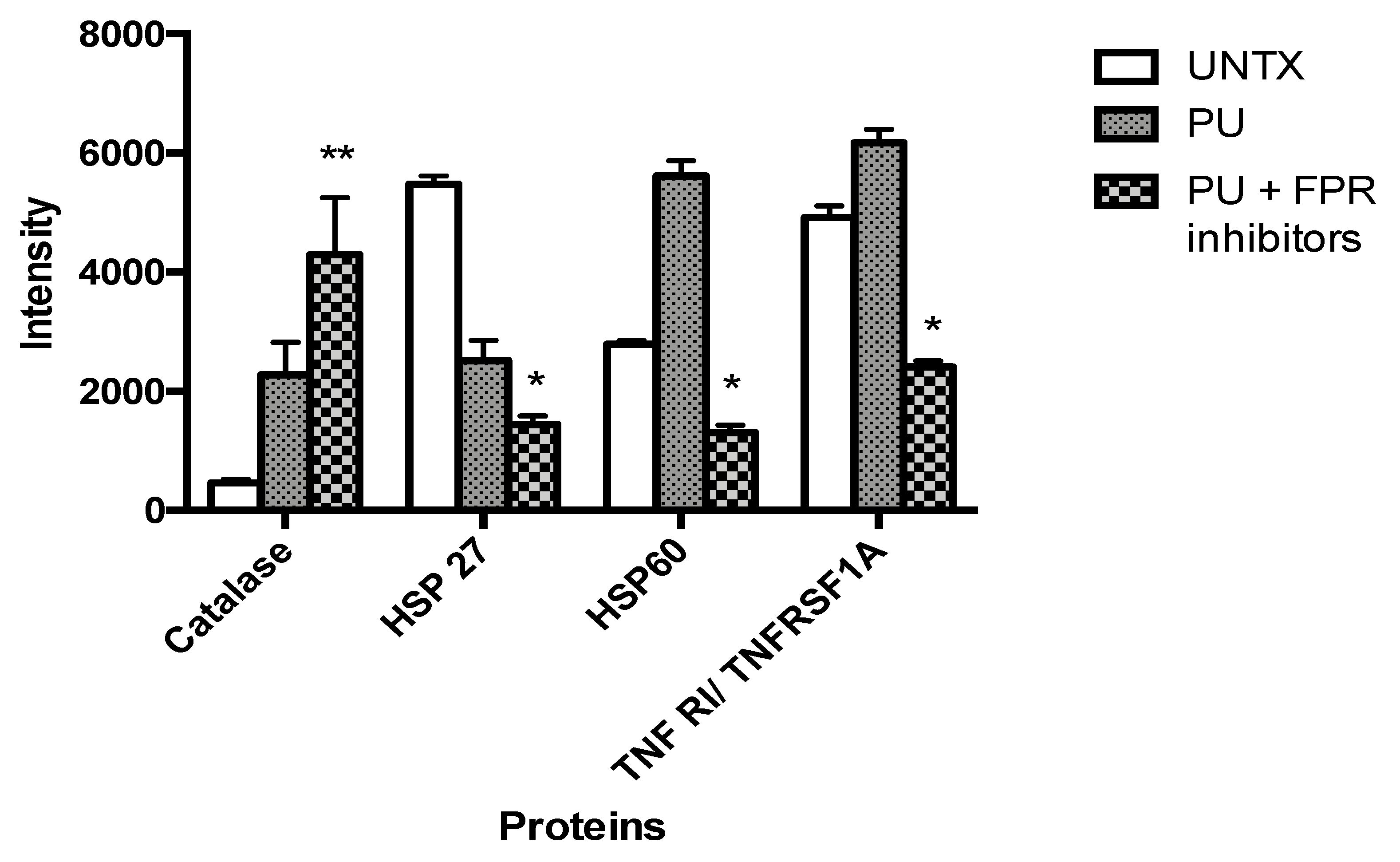
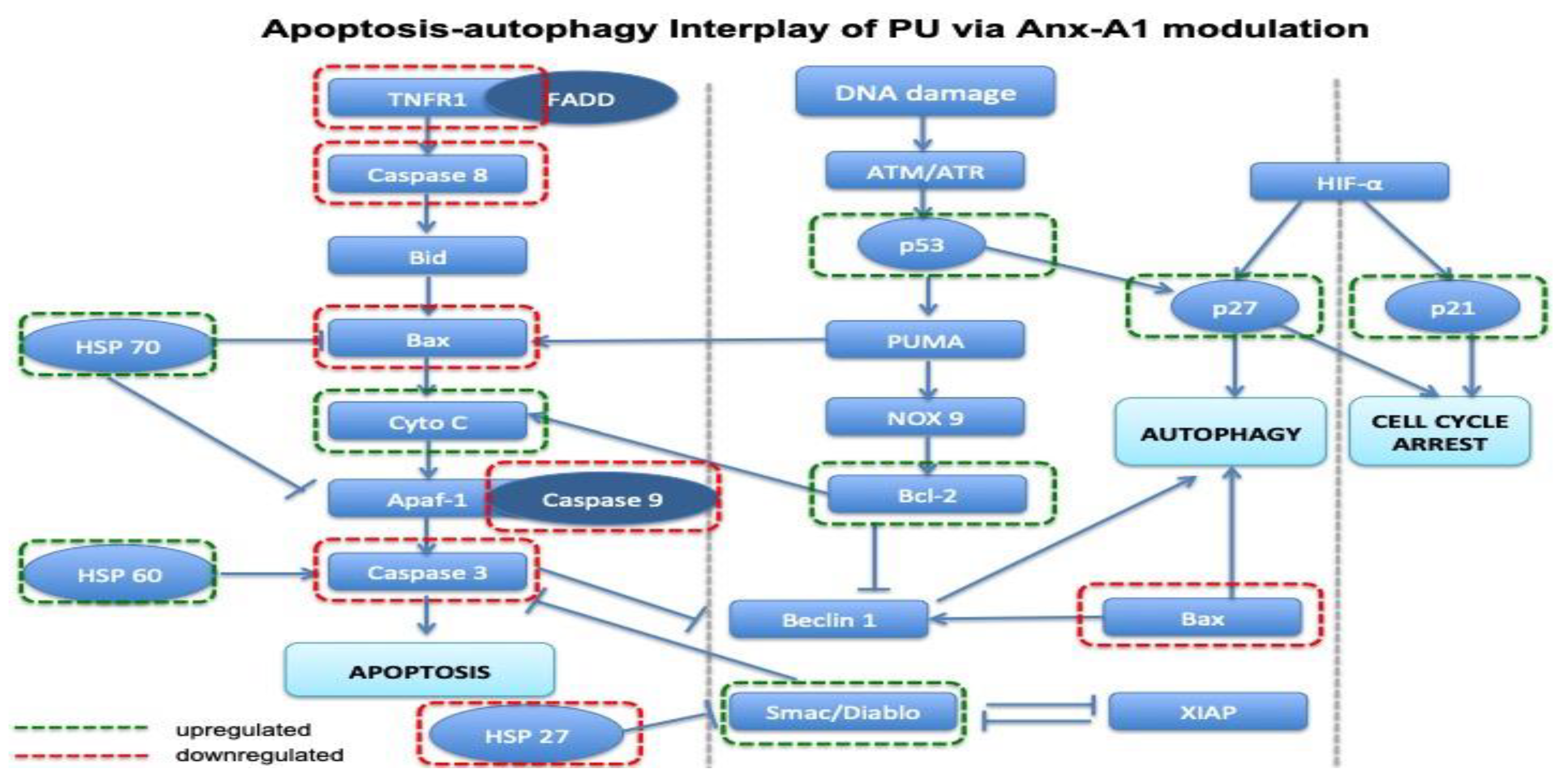
3.5. PU Regulates Apoptosis-Autophagy Interplay in HCT 116 Cells
4. Discussion
4.1. Anti-Proliferative and Apoptotic Effects of PU
4.2. Modulation of Anx-A1 by PU
4.3. Autophagy-Apoptosis Switch and Anx-A1
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, 359–386. [Google Scholar] [CrossRef] [PubMed]
- UK World Cancer Research Fund. Colorectal Cancer; World Cancer Research Fund: London, UK, 2018. [Google Scholar]
- Yao, X.; Cheng, X.; Zhang, L.; Yu, H.; Bao, J.; Guan, H.; Lu, R. Punicalagin from pomegranate promotes human papillary thyroid carcinoma BCPAP cell death by triggering ATM-mediated DNA damage response. Nutr. Res. 2017, 47, 63–71. [Google Scholar] [CrossRef]
- Seeram, N.P.; Adams, L.S.; Henning, S.M.; Niu, Y.; Zhang, Y.; Nair, M.G.; Heber, D. In vitro antiproliferative, apoptotic and antioxidant activities of punicalagin, ellagic acid and a total pomegranate tannin extract are enhanced in combination with other polyphenols as found in pomegranate juice. J. Nutr. Biochem. 2005, 16, 360–367. [Google Scholar] [CrossRef] [PubMed]
- El-Missiry, M.A.; Amer, M.A.; Hemieda, F.A.E.; Othman, A.I.; Sakr, D.A.; Abdulhadi, H.L. Cardioameliorative effect of punicalagin against streptozotocin-induced apoptosis, redox imbalance, metabolic changes and inflammation. Egypt. J. Basic Appl. Sci. 2015, 2, 247–260. [Google Scholar] [CrossRef]
- Yaidikar, L.; Thakur, S. Punicalagin attenuated cerebral ischemia-reperfusion insult via inhibition of proinflammatory cytokines, up-regulation of Bcl-2, down-regulation of Bax, and caspase-3. Mol. Cell. Biochem. 2015, 402, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Sarrias, A.; Gimenez-Bastida, J.A.; Nunez-Sanchez, M.A.; Larrosa, M.; Garcia-Conesa, M.T.; Tomas-Barberan, F.A.; Espin, J.C. Phase-II metabolism limits the antiproliferative activity of urolithins in human colon cancer cells. Eur. J. Nutr. 2014, 53, 853–864. [Google Scholar] [CrossRef]
- Sato, Y.U.; Kumamoto, K.; Saito, K.; Okayama, H.; Hayase, S.; Kofunato, Y.; Miyamoto, K.; Nakamura, I.; Ohki, S.; Koyama, Y.; et al. Up-regulated Annexin A1 expression in gastrointestinal cancer is associated with cancer invasion and lymph node metastasis. Exp. Ther. Med. 2011, 2, 239–243. [Google Scholar] [CrossRef]
- Kozovska, Z.; Gabrisova, V.; Kucerova, L. Colon cancer: Cancer stem cells markers, drug resistance and treatment. Biomed. Pharmacother. 2014, 68, 911–916. [Google Scholar] [CrossRef]
- Cheng, X.; Gao, Y.; Yao, X.; Yu, H.; Bao, J.; Guan, H.; Sun, Y.; Zhang, L. Punicalagin induces apoptosis-independent autophagic cell death in human papillary thyroid carcinoma BCPAP cells. RSC Adv. 2016, 6, 68485–68493. [Google Scholar] [CrossRef]
- Wang, S.G.; Huang, M.H.; Li, J.H.; Lai, F.I.; Lee, H.M.; Hsu, Y.N. Punicalagin induces apoptotic and autophagic cell death in human U87MG glioma cells. Acta Pharmacol. Sin. 2013, 34, 1411–1419. [Google Scholar] [CrossRef]
- Larrosa, M.; Tomás-Barberán, F.A.; Espín, J.C. The dietary hydrolysable tannin punicalagin releases ellagic acid that induces apoptosis in human colon adenocarcinoma Caco-2 cells by using the mitochondrial pathway. J. Nutr. Biochem. 2006, 17, 611–625. [Google Scholar] [CrossRef] [PubMed]
- Adaramoye, O.; Erguen, B.; Nitzsche, B.; Hopfner, M.; Jung, K.; Rabien, A. Punicalagin, a polyphenol from pomegranate fruit, induces growth inhibition and apoptosis in human PC-3 and LNCaP cells. Chem.-Biol. Interact. 2017, 274, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Fouad, Y.A.; Aanei, C. Revisiting the hallmarks of cancer. Am. J. Cancer Res. 2017, 7, 1016–1036. [Google Scholar] [PubMed]
- Grootaert, C.; Kamiloglu, S.; Capanoglu, E.; Van Camp, J. Cell Systems to Investigate the Impact of Polyphenols on Cardiovascular Health. Nutrients 2015, 7, 9229–9255. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Zorova, L.D.; Popkov, V.A.; Plotnikov, E.Y.; Silachev, D.N.; Pevzner, I.B.; Jankauskas, S.S.; Babenko, V.A.; Zorov, S.D.; Balakireva, A.V.; Juhaszova, M.; et al. Mitochondrial membrane potential. Anal. Biochem. 2018, 552, 50–59. [Google Scholar] [CrossRef]
- Onozawa, H.; Saito, M.; Saito, K.; Kanke, Y.; Watanabe, Y.; Hayase, S.; Sakamoto, W.; Ishigame, T.; Momma, T.; Ohki, S.; et al. Annexin A1 is involved in resistance to 5-FU in colon cancer cells. Oncol. Rep. 2017, 37, 235–240. [Google Scholar] [CrossRef]
- Vecchi, L.; Alves Pereira Zoia, M.; Goss Santos, T.; de Oliveira Beserra, A.; Colaco Ramos, C.M.; Franca Matias Colombo, B.; Paiva Maia, Y.C.; Piana de Andrade, V.; Teixeira Soares Mota, S.; Goncalves de Araujo, T.; et al. Inhibition of the AnxA1/FPR1 autocrine axis reduces MDA-MB-231 breast cancer cell growth and aggressiveness in vitro and in vivo. Biochim. Biophys. Acta. Mol. Cell Res. 2018, 1865, 1368–1382. [Google Scholar] [CrossRef] [PubMed]
- Parisi, J.D.S.; Correa, M.P.; Gil, C.D. Lack of Endogenous Annexin A1 Increases Mast Cell Activation and Exacerbates Experimental Atopic Dermatitis. Cells 2019, 8, 51. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, B.; Longtine, M.S.; Nelson, D.M. Punicalagin promotes autophagy to protect primary human syncytiotrophoblasts from apoptosis. Reproduction 2016, 151, 97–104. [Google Scholar] [CrossRef]
- Santiago-O’Farrill, J.M.; Kleinerman, E.S.; Hollomon, M.G.; Livingston, A.; Wang, W.L.; Tsai, J.W.; Gordon, N.B. Phosphorylated heat shock protein 27 as a potential biomarker to predict the role of chemotherapy-induced autophagy in osteosarcoma response to therapy. Oncotarget 2018, 9, 1602–1616. [Google Scholar] [CrossRef]
- Lian, W.S.; Ko, J.Y.; Chen, Y.S.; Ke, H.J.; Wu, S.L.; Kuo, C.W.; Wang, F.S. Chaperonin 60 sustains osteoblast autophagy and counteracts glucocorticoid aggravation of osteoporosis by chaperoning RPTOR. Cell Death Dis. 2018, 9, 938. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Wan, F.; Dutta, S.; Welsh, S.; Liu, Z.; Freundt, E.; Baehrecke, E.H.; Lenardo, M. Autophagic programmed cell death by selective catalase degradation. Proc. Natl. Acad. Sci. USA 2006, 103, 4952–4957. [Google Scholar] [CrossRef] [PubMed]
- Su, M.; Mei, Y.; Sinha, S. Role of the Crosstalk between Autophagy and Apoptosis in Cancer. J. Oncol. 2013, 2013, 102735. [Google Scholar] [CrossRef]
- El-Khattouti, A.; Selimovic, D.; Haikel, Y.; Hassan, M. Crosstalk between apoptosis and autophagy: Molecular mechanisms and therapeutic strategies in cancer. J. Cell Death 2013, 6, 37–55. [Google Scholar] [CrossRef]
- Muller, M.; Wilder, S.; Bannasch, D.; Israeli, D.; Lehlbach, K.; Li-Weber, M.; Friedman, S.L.; Galle, P.R.; Stremmel, W.; Oren, M.; et al. p53 activates the CD95 (APO-1/Fas) gene in response to DNA damage by anticancer drugs. J. Exp. Med. 1998, 188, 2033–2045. [Google Scholar] [CrossRef]
- Kuribayashi, K.; El-Deiry, W.S. Regulation of programmed cell death by the p53 pathway. Adv. Exp. Med. Biol. 2008, 615, 201–221. [Google Scholar]
- Maiuri, M.C.; Zalckvar, E.; Kimchi, A.; Kroemer, G. Self-eating and self-killing: Crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2007, 8, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Reuter, S.; Eifes, S.; Dicato, M.; Aggarwal, B.B.; Diederich, M. Modulation of anti-apoptotic and survival pathways by curcumin as a strategy to induce apoptosis in cancer cells. Biochem. Pharmacol. 2008, 76, 1340–1351. [Google Scholar] [CrossRef] [PubMed]
- Moroni, M.C.; Hickman, E.S.; Lazzerini Denchi, E.; Caprara, G.; Colli, E.; Cecconi, F.; Muller, H.; Helin, K. Apaf-1 is a transcriptional target for E2F and p53. Nat. Cell Biol. 2001, 3, 552–558. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z. p53 regulation of the IGF-1/AKT/mTOR pathways and the endosomal compartment. Cold Spring Harb. Perspect. Biol. 2010, 2, a001057. [Google Scholar] [CrossRef] [PubMed]
- Crighton, D.; Wilkinson, S.; O’Prey, J.; Syed, N.; Smith, P.; Harrison, P.R.; Gasco, M.; Garrone, O.; Crook, T.; Ryan, K.M. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 2006, 126, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, K.; Daido, S.; Yamamoto, A.; Kobayashi, R.; Yokoyama, T.; Aoki, H.; Iwado, E.; Shinojima, N.; Kondo, Y.; Kondo, S. Pivotal role of the cyclin-dependent kinase inhibitor p21WAF1/CIP1 in apoptosis and autophagy. J. Biol. Chem. 2008, 283, 388–397. [Google Scholar] [CrossRef] [PubMed]
- White, J.P.; Billin, A.N.; Campbell, M.E.; Russell, A.J.; Huffman, K.M.; Kraus, W.E. The AMPK/p27 (Kip1) Axis Regulates Autophagy/Apoptosis Decisions in Aged Skeletal Muscle Stem Cells. Stem Cell Rep. 2018, 11, 425–439. [Google Scholar] [CrossRef] [PubMed]
- Cerda, B.; Llorach, R.; Ceron, J.J.; Espin, J.C.; Tomas-Barberan, F.A. Evaluation of the bioavailability and metabolism in the rat of punicalagin, an antioxidant polyphenol from pomegranate juice. Eur. J. Nutr. 2003, 42, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Nazzaro, F.; Orlando, P.; Fratianni, F.; Coppola, R. Microencapsulation in food science and biotechnology. Curr. Opin. Biotechnol. 2012, 23, 182–186. [Google Scholar] [CrossRef]
- Sajid, M.; Cameotra, S.; Khan, M.; Ahmad, I. Nanoparticle-Based Delivery of Phytomedicines; Academic Press: Cambridge, MA, USA, 2019; pp. 597–623. [Google Scholar]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ganesan, T.; Sinniah, A.; Chik, Z.; Alshawsh, M.A. Punicalagin Regulates Apoptosis-Autophagy Switch via Modulation of Annexin A1 in Colorectal Cancer. Nutrients 2020, 12, 2430. https://doi.org/10.3390/nu12082430
Ganesan T, Sinniah A, Chik Z, Alshawsh MA. Punicalagin Regulates Apoptosis-Autophagy Switch via Modulation of Annexin A1 in Colorectal Cancer. Nutrients. 2020; 12(8):2430. https://doi.org/10.3390/nu12082430
Chicago/Turabian StyleGanesan, Thanusha, Ajantha Sinniah, Zamri Chik, and Mohammed Abdullah Alshawsh. 2020. "Punicalagin Regulates Apoptosis-Autophagy Switch via Modulation of Annexin A1 in Colorectal Cancer" Nutrients 12, no. 8: 2430. https://doi.org/10.3390/nu12082430
APA StyleGanesan, T., Sinniah, A., Chik, Z., & Alshawsh, M. A. (2020). Punicalagin Regulates Apoptosis-Autophagy Switch via Modulation of Annexin A1 in Colorectal Cancer. Nutrients, 12(8), 2430. https://doi.org/10.3390/nu12082430