Beta-Cell Mass in Obesity and Type 2 Diabetes, and Its Relation to Pancreas Fat: A Mini-Review



Abstract
1. Introduction
Search Strategy
2. Beta Cell Mass in Obesity and Type 2 Diabetes
2.1. Change of Beta Cell Mass in Subjects with Obesity
2.2. Change of Beta Cell Mass in Prediabetes
2.3. Change of Beta Cell Mass in T2DM
2.4. The Mechanism of Beta-Cell Deficit and Change in Alpha-Cell Mass in T2DM
2.5. Decline in Beta-Cell Function with Worsening Glucose Tolerance
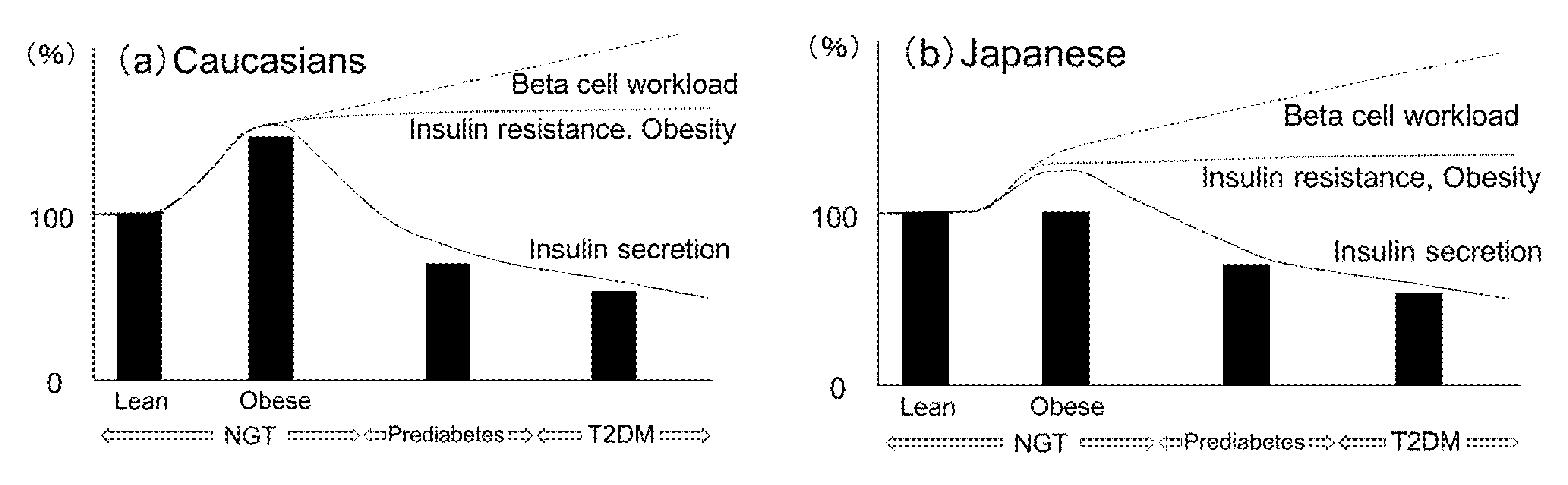
2.6. Ethnic Similarities and Differences in the Change of Beta-Cell Mass
2.7. Change of Pancreas Mass in Obesity and Diabetes
2.8. Association between Pancreas Fat and Glucose Metabolism
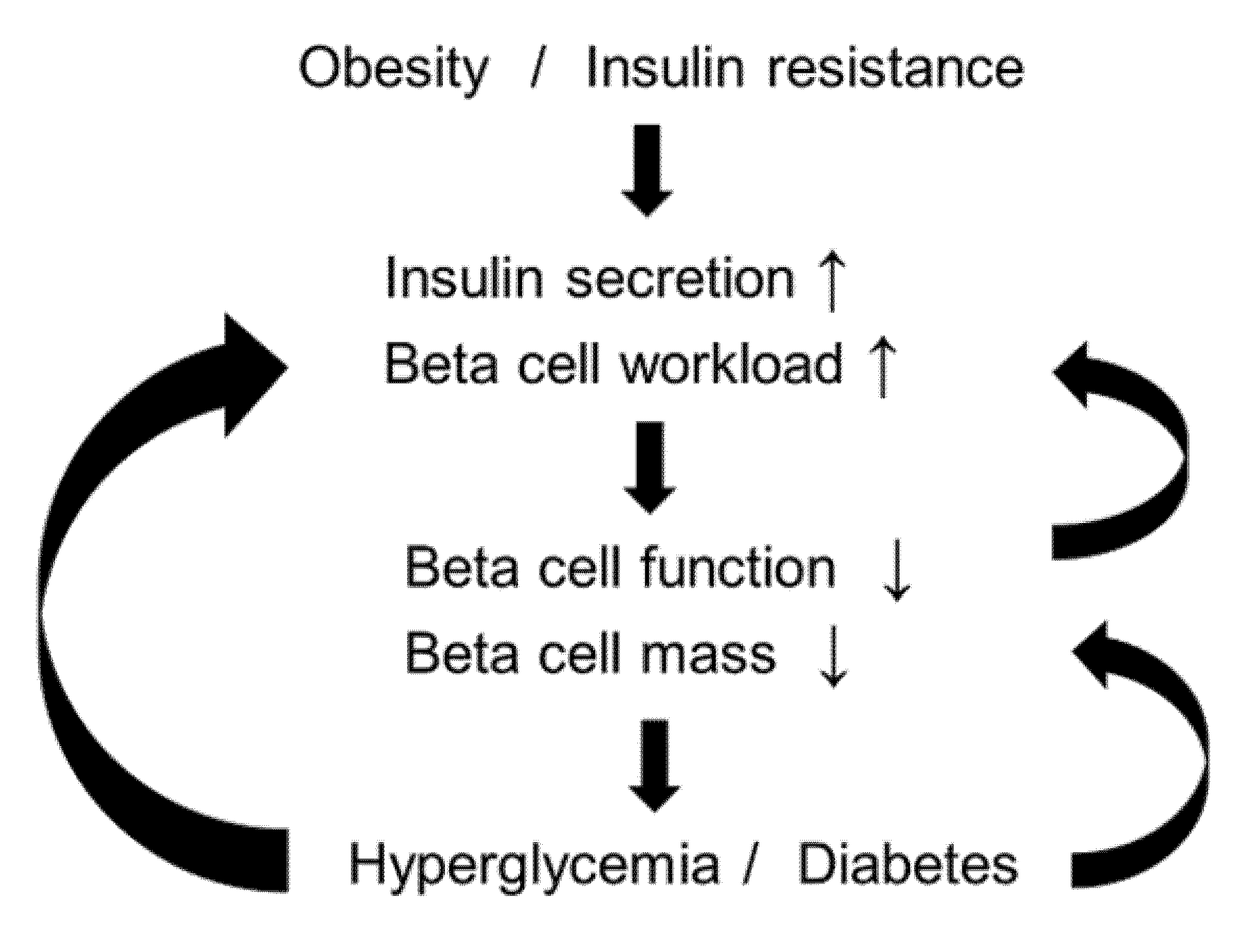
2.9. Beta-Cell Workload Hypothesis
3. Treatment Strategy for T2DM in Relation to the Beta-Cell Workload Hypothesis
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- International Diabetes Federation Diabetes Atlas, 8th ed. Available online: https://www.idf.org/e-library/epidemiology-research/diabetes-atlas/134-idfdiabetesatlas-8th-edition.html (accessed on 12 October 2020).
- Khaw, K.T.; Wareham, N.; Bingham, S.; Luben, R.; Welch, A.; Day, N. Association of hemoglobin A1c with cardiovascular disease and mortality in adults: The European prospective investigation into cancer in Norfolk. Ann. Intern. Med. 2004, 141, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Klein, R. Hyperglycemia and microvascular and macrovascular disease in diabetes. Diabetes Care 1995, 18, 258–268. [Google Scholar] [CrossRef] [PubMed]
- Haffner, S.M.; Lehto, S.; Rönnemaa, T.; Pyörälä, K.; Laakso, M. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N. Engl. J. Med. 1998, 339, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Abdullah, A.; Peeters, A.; de Courten, M.; Stoelwinder, J. The magnitude of association between overweight and obesity and the risk of diabetes: A meta-analysis of prospective cohort studies. Diabetes Res. Clin. Pract. 2010, 89, 309–319. [Google Scholar] [CrossRef]
- Lee, Y.; Lingvay, I.; Szczepaniak, L.S.; Ravazzola, M.; Orci, L.; Unger, R.H. Pancreatic steatosis: Harbinger of type 2 diabetes in obese rodents. Int. J. Obes. 2010, 34, 396–400. [Google Scholar] [CrossRef]
- Lee, Y.; Hirose, H.; Ohneda, M.; Johnson, J.H.; McGarry, J.D.; Unger, R.H. Beta-cell lipotoxicity in the pathogenesis of non-insulin-dependent diabetes mellitus of obese rats: Impairment in adipocyte-beta-cell relationships. Proc. Natl. Acad. Sci. USA 1994, 91, 10878–10882. [Google Scholar] [CrossRef]
- Sakuraba, H.; Mizukami, H.; Yagihashi, N.; Wada, R.; Hanyu, C.; Yagihashi, S. Reduced beta-cell mass and expression of oxidative stress-related DNA damage in the islet of Japanese Type II diabetic patients. Diabetologia 2002, 45, 85–96. [Google Scholar] [CrossRef]
- Yoon, K.H.; Ko, S.H.; Cho, J.H.; Lee, J.M.; Ahn, Y.B.; Song, K.H.; Yoo, S.J.; Kang, M.I.; Cha, B.Y.; Lee, K.W.; et al. Selective beta-cell loss and alpha-cell expansion in patients with type 2 diabetes mellitus in Korea. J. Clin. Endocrinol. Metab. 2003, 88, 2300–2308. [Google Scholar] [CrossRef]
- Butler, A.E.; Janson, J.; Bonner-Weir, S.; Ritzel, R.; Rizza, R.A.; Butler, P.C. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 2003, 52, 102–110. [Google Scholar] [CrossRef]
- Rahier, J.; Guiot, Y.; Goebbels, R.M.; Sempoux, C.; Henquin, J.C. Pancreatic beta-cell mass in European subjects with type 2 diabetes. Diabetes Obes. Metab. 2008, 10 (Suppl. S4), 32–42. [Google Scholar] [CrossRef]
- Meier, J.J.; Menge, B.A.; Breuer, T.G.; Muller, C.A.; Tannapfel, A.; Uhl, W.; Schmidt, W.E.; Schrader, H. Functional assessment of pancreatic beta-cell area in humans. Diabetes 2009, 58, 1595–1603. [Google Scholar] [CrossRef] [PubMed]
- Hanley, S.C.; Austin, E.; Assouline-Thomas, B.; Kapeluto, J.; Blaichman, J.; Moosavi, M.; Petropavlovskaia, M.; Rosenberg, L. {β}-Cell mass dynamics and islet cell plasticity in human type 2 diabetes. Endocrinology 2010, 151, 1462–1472. [Google Scholar] [CrossRef] [PubMed]
- Henquin, J.C.; Rahier, J. Pancreatic alpha cell mass in European subjects with type 2 diabetes. Diabetologia 2011, 54, 1720–1725. [Google Scholar] [CrossRef]
- Meier, J.J.; Breuer, T.G.; Bonadonna, R.C.; Tannapfel, A.; Uhl, W.; Schmidt, W.E.; Schrader, H.; Menge, B.A. Pancreatic diabetes manifests when beta cell area declines by approximately 65% in humans. Diabetologia 2012, 55, 1346–1354. [Google Scholar] [CrossRef] [PubMed]
- Yoneda, S.; Uno, S.; Iwahashi, H.; Fujita, Y.; Yoshikawa, A.; Kozawa, J.; Okita, K.; Takiuchi, D.; Eguchi, H.; Nagano, H.; et al. Predominance of beta-cell neogenesis rather than replication in humans with an impaired glucose tolerance and newly diagnosed diabetes. J. Clin. Endocrinol. Metab. 2013, 98, 2053–2061. [Google Scholar] [CrossRef]
- Saisho, Y.; Butler, A.E.; Manesso, E.; Elashoff, D.; Rizza, R.A.; Butler, P.C. beta-cell mass and turnover in humans: Effects of obesity and aging. Diabetes Care 2013, 36, 111–117. [Google Scholar] [CrossRef]
- Kou, K.; Saisho, Y.; Satoh, S.; Yamada, T.; Itoh, H. Change in beta-cell mass in Japanese nondiabetic obese individuals. J. Clin. Endocrinol. Metab. 2013, 98, 3724–3730. [Google Scholar] [CrossRef]
- Mezza, T.; Muscogiuri, G.; Sorice, G.P.; Clemente, G.; Hu, J.; Pontecorvi, A.; Holst, J.J.; Giaccari, A.; Kulkarni, R.N. Insulin resistance alters islet morphology in nondiabetic humans. Diabetes 2014, 63, 994–1007. [Google Scholar] [CrossRef]
- Mizukami, H.; Takahashi, K.; Inaba, W.; Tsuboi, K.; Osonoi, S.; Yoshida, T.; Yagihashi, S. Involvement of oxidative stress-induced DNA damage, endoplasmic reticulum stress, and autophagy deficits in the decline of beta-cell mass in Japanese type 2 diabetic patients. Diabetes Care 2014, 37, 1966–1974. [Google Scholar] [CrossRef]
- Fujita, Y.; Kozawa, J.; Iwahashi, H.; Yoneda, S.; Uno, S.; Yoshikawa, A.; Okita, K.; Eguchi, H.; Nagano, H.; Imagawa, A.; et al. Increment of serum C-peptide measured by glucagon test closely correlates with human relative beta-cell area. Endocr. J. 2015, 62, 329–337. [Google Scholar] [CrossRef]
- Sato, S.; Saisho, Y.; Inaishi, J.; Kou, K.; Murakami, R.; Yamada, T.; Itoh, H. Effects of Glucocorticoid Treatment on beta- and alpha-Cell Mass in Japanese Adults With and Without Diabetes. Diabetes 2015, 64, 2915–2927. [Google Scholar] [CrossRef] [PubMed]
- Inaishi, J.; Saisho, Y.; Sato, S.; Kou, K.; Murakami, R.; Watanabe, Y.; Kitago, M.; Kitagawa, Y.; Yamada, T.; Itoh, H. Effects of Obesity and Diabetes on alpha- and beta-Cell Mass in Surgically Resected Human Pancreas. J. Clin. Endocrinol. Metab. 2016, 101, 2874–2882. [Google Scholar] [CrossRef] [PubMed]
- Xin, A.; Mizukami, H.; Inaba, W.; Yoshida, T.; Takeuchi, Y.K.; Yagihashi, S. Pancreas Atrophy and Islet Amyloid Deposition in Patients with Elderly-Onset Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2017, 102, 3162–3171. [Google Scholar] [CrossRef] [PubMed]
- Inaishi, J.; Saisho, Y.; Hirakawa, Y.; Yoshida, D.; Hata, J.; Mukai, N.; Watanabe, Y.; Oda, Y.; Itoh, H.; Ninomiya, T. Association of glucose tolerance status with pancreatic β- and α-cell mass in community-based autopsy samples of Japanese individuals: The Hisayama Study. J. Diabetes Investig. 2020, 11, 1197–1206. [Google Scholar] [CrossRef]
- Sasaki, H.; Saisho, Y.; Inaishi, J.; Watanabe, Y.; Tsuchiya, T.; Makio, M.; Sato, M.; Kitago, M.; Yamada, T.; Itoh, H. Associations of birthweight and history of childhood obesity with beta cell mass in Japanese adults. Diabetologia 2020, 63, 1199–1210. [Google Scholar] [CrossRef]
- Polonsky, K.S.; Given, B.D.; Van Cauter, E. Twenty-four-hour profiles and pulsatile patterns of insulin secretion in normal and obese subjects. J. Clin. Investig. 1988, 81, 442–448. [Google Scholar] [CrossRef]
- Tschen, S.I.; Dhawan, S.; Gurlo, T.; Bhushan, A. Age-dependent decline in beta-cell proliferation restricts the capacity of beta-cell regeneration in mice. Diabetes 2009, 58, 1312–1320. [Google Scholar] [CrossRef]
- Meier, J.J.; Butler, A.E.; Saisho, Y.; Monchamp, T.; Galasso, R.; Bhushan, A.; Rizza, R.A.; Butler, P.C. Beta-cell replication is the primary mechanism subserving the postnatal expansion of beta-cell mass in humans. Diabetes 2008, 57, 1584–1594. [Google Scholar] [CrossRef]
- Gregg, B.E.; Moore, P.C.; Demozay, D.; Hall, B.A.; Li, M.; Husain, A.; Wright, A.J.; Atkinson, M.A.; Rhodes, C.J. Formation of a human β-cell population within pancreatic islets is set early in life. J. Clin. Endocrinol. Metab. 2012, 97, 3197–3206. [Google Scholar] [CrossRef]
- Atkinson, M.A.; Eisenbarth, G.S.; Michels, A.W. Type 1 diabetes. Lancet 2014, 383, 69–82. [Google Scholar] [CrossRef]
- Löhr, M.; Klöppel, G. Residual insulin positivity and pancreatic atrophy in relation to duration of chronic type 1 (insulin-dependent) diabetes mellitus and microangiopathy. Diabetologia 1987, 30, 757–762. [Google Scholar] [CrossRef] [PubMed]
- Meier, J.J.; Bhushan, A.; Butler, A.E.; Rizza, R.A.; Butler, P.C. Sustained beta cell apoptosis in patients with long-standing type 1 diabetes: Indirect evidence for islet regeneration? Diabetologia 2005, 48, 2221–2228. [Google Scholar] [CrossRef] [PubMed]
- Dor, Y.; Brown, J.; Martinez, O.I.; Melton, D.A. Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature 2004, 429, 41–46. [Google Scholar] [CrossRef]
- Xiao, X.; Chen, Z.; Shiota, C.; Prasadan, K.; Guo, P.; El-Gohary, Y.; Paredes, J.; Welsh, C.; Wiersch, J.; Gittes, G.K. No evidence for β cell neogenesis in murine adult pancreas. J. Clin. Investig. 2013, 123, 2207–2217. [Google Scholar] [CrossRef] [PubMed]
- Poitout, V.; Robertson, R.P. Glucolipotoxicity: Fuel excess and beta-cell dysfunction. Endocr. Rev. 2008, 29, 351–366. [Google Scholar] [CrossRef] [PubMed]
- Haataja, L.; Gurlo, T.; Huang, C.J.; Butler, P.C. Islet amyloid in type 2 diabetes, and the toxic oligomer hypothesis. Endocr. Rev. 2008, 29, 303–316. [Google Scholar] [CrossRef] [PubMed]
- Robertson, R.P. Antioxidant drugs for treating beta-cell oxidative stress in type 2 diabetes: Glucose-centric versus insulin-centric therapy. Discov. Med. 2010, 9, 132–137. [Google Scholar]
- Eizirik, D.L.; Cardozo, A.K.; Cnop, M. The role for endoplasmic reticulum stress in diabetes mellitus. Endocr. Rev. 2008, 29, 42–61. [Google Scholar] [CrossRef]
- Dinarello, C.A.; Donath, M.Y.; Mandrup-Poulsen, T. Role of IL-1beta in type 2 diabetes. Curr. Opin. Endocrinol. Diabetes Obes. 2010, 17, 314–321. [Google Scholar] [CrossRef]
- Masini, M.; Bugliani, M.; Lupi, R.; del Guerra, S.; Boggi, U.; Filipponi, F.; Marselli, L.; Masiello, P.; Marchetti, P. Autophagy in human type 2 diabetes pancreatic beta cells. Diabetologia 2009, 52, 1083–1086. [Google Scholar] [CrossRef]
- Kusminski, C.M.; Shetty, S.; Orci, L.; Unger, R.H.; Scherer, P.E. Diabetes and apoptosis: Lipotoxicity. Apoptosis 2009, 14, 1484–1495. [Google Scholar] [CrossRef] [PubMed]
- Alejandro, E.U.; Gregg, B.; Blandino-Rosano, M.; Cras-Méneur, C.; Bernal-Mizrachi, E. Natural history of β-cell adaptation and failure in type 2 diabetes. Mol. Asp. Med. 2015, 42, 19–41. [Google Scholar] [CrossRef] [PubMed]
- Gerber, P.A.; Rutter, G.A. The Role of Oxidative Stress and Hypoxia in Pancreatic Beta-Cell Dysfunction in Diabetes Mellitus. Antioxid. Redox Signal. 2017, 26, 501–518. [Google Scholar] [CrossRef] [PubMed]
- Al-Mrabeh, A. Pathogenesis and remission of type 2 diabetes: What has the twin cycle hypothesis taught us? Cardiovasc. Endocrinol. Metab. 2020, 9, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Talchai, C.; Xuan, S.; Lin, H.V.; Sussel, L.; Accili, D. Pancreatic beta cell dedifferentiation as a mechanism of diabetic beta cell failure. Cell 2012, 150, 1223–1234. [Google Scholar] [CrossRef]
- Cinti, F.; Bouchi, R.; Kim-Muller, J.Y.; Ohmura, Y.; Sandoval, P.R.; Masini, M.; Marselli, L.; Suleiman, M.; Ratner, L.E.; Marchetti, P.; et al. Evidence of beta-Cell Dedifferentiation in Human Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2016, 101, 1044–1054. [Google Scholar] [CrossRef]
- Butler, A.E.; Dhawan, S.; Hoang, J.; Cory, M.; Zeng, K.; Fritsch, H.; Meier, J.J.; Rizza, R.A.; Butler, P.C. β-Cell Deficit in Obese Type 2 Diabetes, a Minor Role of β-Cell Dedifferentiation and Degranulation. J. Clin. Endocrinol. Metab. 2016, 101, 523–532. [Google Scholar] [CrossRef]
- Defronzo, R.A. Banting Lecture. From the triumvirate to the ominous octet: A new paradigm for the treatment of type 2 diabetes mellitus. Diabetes 2009, 58, 773–795. [Google Scholar] [CrossRef]
- Jensen, C.C.; Cnop, M.; Hull, R.L.; Fujimoto, W.Y.; Kahn, S.E. Beta-cell function is a major contributor to oral glucose tolerance in high-risk relatives of four ethnic groups in the U.S. Diabetes 2002, 51, 2170–2178. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Eldor, R.; Abdul-Ghani, M. Pathophysiologic approach to therapy in patients with newly diagnosed type 2 diabetes. Diabetes Care 2013, 36 (Suppl. S2), S127–S138. [Google Scholar] [CrossRef]
- U.K. Prospective Diabetes Study Group. Overview of 6 years’ therapy of type II diabetes: A progressive disease. U.K. Prospective Diabetes Study 16. Diabetes 1995, 44, 1249–1258. [Google Scholar] [CrossRef]
- Utzschneider, K.M.; Prigeon, R.L.; Faulenbach, M.V.; Tong, J.; Carr, D.B.; Boyko, E.J.; Leonetti, D.L.; McNeely, M.J.; Fujimoto, W.Y.; Kahn, S.E. Oral disposition index predicts the development of future diabetes above and beyond fasting and 2-h glucose levels. Diabetes Care 2009, 32, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Weyer, C.; Bogardus, C.; Mott, D.M.; Pratley, R.E. The natural history of insulin secretory dysfunction and insulin resistance in the pathogenesis of type 2 diabetes mellitus. J. Clin. Investig. 1999, 104, 787–794. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Cohrs, C.M.; Stertmann, J.; Bozsak, R.; Speier, S. Human beta cell mass and function in diabetes: Recent advances in knowledge and technologies to understand disease pathogenesis. Mol. Metab. 2017, 6, 943–957. [Google Scholar] [CrossRef]
- Laedtke, T.; Kjems, L.; Pørksen, N.; Schmitz, O.; Veldhuis, J.; Kao, P.C.; Butler, P.C. Overnight inhibition of insulin secretion restores pulsatility and proinsulin/insulin ratio in type 2 diabetes. Am. J. Physiol. Endocrinol. Metab. 2000, 279, E520–E528. [Google Scholar] [CrossRef]
- Kahn, S.E.; Haffner, S.M.; Heise, M.A.; Herman, W.H.; Holman, R.R.; Jones, N.P.; Kravitz, B.G.; Lachin, J.M.; O’Neill, M.C.; Zinman, B.; et al. Glycemic durability of rosiglitazone, metformin, or glyburide monotherapy. N. Engl. J. Med. 2006, 355, 2427–2443. [Google Scholar] [CrossRef]
- Saisho, Y.; Tanaka, K.; Abe, T.; Shimada, A.; Kawai, T.; Itoh, H. Effect of obesity on declining beta cell function after diagnosis of type 2 diabetes: A possible link suggested by cross-sectional analysis. Endocr. J. 2012, 59, 187–195. [Google Scholar] [CrossRef]
- Funakoshi, S.; Fujimoto, S.; Hamasaki, A.; Fujiwara, H.; Fujita, Y.; Ikeda, K.; Hamamoto, Y.; Hosokawa, M.; Seino, Y.; Inagaki, N. Analysis of factors influencing pancreatic beta-cell function in Japanese patients with type 2 diabetes: Association with body mass index and duration of diabetic exposure. Diabetes Res. Clin. Pract. 2008, 82, 353–358. [Google Scholar] [CrossRef]
- Kodama, K.; Tojjar, D.; Yamada, S.; Toda, K.; Patel, C.J.; Butte, A.J. Ethnic differences in the relationship between insulin sensitivity and insulin response: A systematic review and meta-analysis. Diabetes Care 2013, 36, 1789–1796. [Google Scholar] [CrossRef]
- Hsia, D.S.; Larrivee, S.; Cefalu, W.T.; Johnson, W.D. Impact of Lowering BMI Cut Points as Recommended in the Revised American Diabetes Association’s Standards of Medical Care in Diabetes-2015 on Diabetes Screening in Asian Americans. Diabetes Care 2015, 38, 2166–2168. [Google Scholar] [CrossRef]
- Imamura, M.; Takahashi, A.; Yamauchi, T.; Hara, K.; Yasuda, K.; Grarup, N.; Zhao, W.; Wang, X.; Huerta-Chagoya, A.; Hu, C.; et al. Genome-wide association studies in the Japanese population identify seven novel loci for type 2 diabetes. Nat. Commun. 2016, 7, 10531. [Google Scholar] [CrossRef] [PubMed]
- Inaishi, J.; Hirakawa, Y.; Horikoshi, M.; Akiyama, M.; Higashioka, M.; Yoshinari, M.; Hata, J.; Mukai, N.; Kamatani, Y.; Momozawa, Y.; et al. Association Between Genetic Risk and Development of Type 2 Diabetes in a General Japanese Population: The Hisayama Study. J. Clin. Endocrinol. Metab. 2019, 104, 3213–3222. [Google Scholar] [CrossRef] [PubMed]
- Pal, A.; McCarthy, M.I. The genetics of type 2 diabetes and its clinical relevance. Clin. Genet. 2013, 83, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Saisho, Y.; Butler, A.E.; Meier, J.J.; Monchamp, T.; Allen-Auerbach, M.; Rizza, R.A.; Butler, P.C. Pancreas volumes in humans from birth to age one hundred taking into account sex, obesity, and presence of type-2 diabetes. Clin. Anat 2007, 20, 933–942. [Google Scholar] [CrossRef]
- Kou, K.; Saisho, Y.; Jinzaki, M.; Itoh, H. Relationship between body mass index and pancreas volume in Japanese people. J. Pancreas 2014, 15, 626–627. [Google Scholar] [CrossRef]
- Van Raalte, D.H.; van der Zijl, N.J.; Diamant, M. Pancreatic steatosis in humans: Cause or marker of lipotoxicity? Curr. Opin. Clin. Nutr. Metab. Care 2010, 13, 478–485. [Google Scholar] [CrossRef]
- Sattar, N.; Gill, J.M. Type 2 diabetes as a disease of ectopic fat? BMC Med. 2014, 12, 123. [Google Scholar] [CrossRef]
- Pencina, M.J.; D’Agostino, R.B., Sr.; D’Agostino, R.B., Jr.; Vasan, R.S. Evaluating the added predictive ability of a new marker: From area under the ROC curve to reclassification and beyond. Stat. Med. 2008, 27, 157–172, discussion 207–112. [Google Scholar] [CrossRef]
- Roden, M.; Price, T.B.; Perseghin, G.; Petersen, K.F.; Rothman, D.L.; Cline, G.W.; Shulman, G.I. Mechanism of free fatty acid-induced insulin resistance in humans. J. Clin. Investig. 1996, 97, 2859–2865. [Google Scholar] [CrossRef]
- Cnop, M. Fatty acids and glucolipotoxicity in the pathogenesis of Type 2 diabetes. Biochem. Soc. Trans. 2008, 36, 348–352. [Google Scholar] [CrossRef]
- Taylor, R. Type 2 diabetes: Etiology and reversibility. Diabetes Care 2013, 36, 1047–1055. [Google Scholar] [CrossRef] [PubMed]
- Adiels, M.; Taskinen, M.R.; Packard, C.; Caslake, M.J.; Soro-Paavonen, A.; Westerbacka, J.; Vehkavaara, S.; Häkkinen, A.; Olofsson, S.O.; Yki-Järvinen, H.; et al. Overproduction of large VLDL particles is driven by increased liver fat content in man. Diabetologia 2006, 49, 755–765. [Google Scholar] [CrossRef] [PubMed]
- Saisho, Y. Pancreas Volume and Fat Deposition in Diabetes and Normal Physiology: Consideration of the Interplay Between Endocrine and Exocrine Pancreas. Rev. Diabet. Stud. 2016, 13, 132–147. [Google Scholar] [CrossRef] [PubMed]
- Weir, G.C. Glucolipotoxicity, β-Cells, and Diabetes: The Emperor Has No Clothes. Diabetes 2020, 69, 273–278. [Google Scholar] [CrossRef]
- Campbell-Thompson, M.L.; Kaddis, J.S.; Wasserfall, C.; Haller, M.J.; Pugliese, A.; Schatz, D.A.; Shuster, J.J.; Atkinson, M.A. The influence of type 1 diabetes on pancreatic weight. Diabetologia 2016, 59, 217–221. [Google Scholar] [CrossRef]
- Goda, K.; Sasaki, E.; Nagata, K.; Fukai, M.; Ohsawa, N.; Hahafusa, T. Pancreatic volume in type 1 and type 2 diabetes mellitus. Acta Diabetol. 2001, 38, 145–149. [Google Scholar] [CrossRef]
- Williams, A.J.; Thrower, S.L.; Sequeiros, I.M.; Ward, A.; Bickerton, A.S.; Triay, J.M.; Callaway, M.P.; Dayan, C.M. Pancreatic volume is reduced in adult patients with recently diagnosed type 1 diabetes. J. Clin. Endocrinol. Metab. 2012, 97, E2109–E2113. [Google Scholar] [CrossRef]
- Burute, N.; Nisenbaum, R.; Jenkins, D.J.; Mirrahimi, A.; Anthwal, S.; Colak, E.; Kirpalani, A. Pancreas volume measurement in patients with Type 2 diabetes using magnetic resonance imaging-based planimetry. Pancreatology 2014, 14, 268–274. [Google Scholar] [CrossRef]
- Al-Mrabeh, A.; Hollingsworth, K.G.; Steven, S.; Taylor, R. Morphology of the pancreas in type 2 diabetes: Effect of weight loss with or without normalisation of insulin secretory capacity. Diabetologia 2016, 59, 1753–1759. [Google Scholar] [CrossRef]
- Al-Mrabeh, A.; Hollingsworth, K.G.; Shaw, J.A.M.; McConnachie, A.; Sattar, N.; Lean, M.E.J.; Taylor, R. 2-year remission of type 2 diabetes and pancreas morphology: A post-hoc analysis of the DiRECT open-label, cluster-randomised trial. Lancet Diabetes Endocrinol. 2020, 8, 939–948. [Google Scholar] [CrossRef]
- Hardt, P.D.; Krauss, A.; Bretz, L.; Porsch-Ozcürümez, M.; Schnell-Kretschmer, H.; Mäser, E.; Bretzel, R.G.; Zekhorn, T.; Klör, H.U. Pancreatic exocrine function in patients with type 1 and type 2 diabetes mellitus. Acta Diabetol. 2000, 37, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Larger, E.; Philippe, M.F.; Barbot-Trystram, L.; Radu, A.; Rotariu, M.; Nobécourt, E.; Boitard, C. Pancreatic exocrine function in patients with diabetes. Diabet. Med. 2012, 29, 1047–1054. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; D’Hoker, J.; Stangé, G.; Bonné, S.; De Leu, N.; Xiao, X.; Van de Casteele, M.; Mellitzer, G.; Ling, Z.; Pipeleers, D.; et al. Beta cells can be generated from endogenous progenitors in injured adult mouse pancreas. Cell 2008, 132, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Inada, A.; Nienaber, C.; Katsuta, H.; Fujitani, Y.; Levine, J.; Morita, R.; Sharma, A.; Bonner-Weir, S. Carbonic anhydrase II-positive pancreatic cells are progenitors for both endocrine and exocrine pancreas after birth. Proc. Natl. Acad. Sci. USA 2008, 105, 19915–19919. [Google Scholar] [CrossRef]
- Minami, K.; Okuno, M.; Miyawaki, K.; Okumachi, A.; Ishizaki, K.; Oyama, K.; Kawaguchi, M.; Ishizuka, N.; Iwanaga, T.; Seino, S. Lineage tracing and characterization of insulin-secreting cells generated from adult pancreatic acinar cells. Proc. Natl. Acad. Sci. USA 2005, 102, 15116–15121. [Google Scholar] [CrossRef]
- Zhou, Q.; Brown, J.; Kanarek, A.; Rajagopal, J.; Melton, D.A. In vivo reprogramming of adult pancreatic exocrine cells to beta-cells. Nature 2008, 455, 627–632. [Google Scholar] [CrossRef]
- Baeyens, L.; Lemper, M.; Leuckx, G.; De Groef, S.; Bonfanti, P.; Stangé, G.; Shemer, R.; Nord, C.; Scheel, D.W.; Pan, F.C.; et al. Transient cytokine treatment induces acinar cell reprogramming and regenerates functional beta cell mass in diabetic mice. Nat. Biotechnol. 2014, 32, 76–83. [Google Scholar] [CrossRef]
- Philippe, M.F.; Benabadji, S.; Barbot-Trystram, L.; Vadrot, D.; Boitard, C.; Larger, E. Pancreatic volume and endocrine and exocrine functions in patients with diabetes. Pancreas 2011, 40, 359–363. [Google Scholar] [CrossRef]
- Murakami, R.; Saisho, Y.; Watanabe, Y.; Inaishi, J.; Tsuchiya, T.; Kou, K.; Sato, S.; Kitago, M.; Kitagawa, Y.; Yamada, T.; et al. Pancreas Fat and β Cell Mass in Humans With and Without Diabetes: An Analysis in the Japanese Population. J. Clin. Endocrinol. Metab. 2017, 102, 3251–3260. [Google Scholar] [CrossRef]
- Horii, T.; Fujita, Y.; Ishibashi, C.; Fukui, K.; Eguchi, H.; Kozawa, J.; Shimomura, I. Islet inflammation is associated with pancreatic fatty infiltration and hyperglycemia in type 2 diabetes. BMJ Open Diabetes Res. Care 2020, 8, e001508. [Google Scholar] [CrossRef]
- Van der Zijl, N.J.; Goossens, G.H.; Moors, C.C.; van Raalte, D.H.; Muskiet, M.H.; Pouwels, P.J.; Blaak, E.E.; Diamant, M. Ectopic fat storage in the pancreas, liver, and abdominal fat depots: Impact on β-cell function in individuals with impaired glucose metabolism. J. Clin. Endocrinol. Metab. 2011, 96, 459–467. [Google Scholar] [CrossRef] [PubMed]
- Gaborit, B.; Abdesselam, I.; Kober, F.; Jacquier, A.; Ronsin, O.; Emungania, O.; Lesavre, N.; Alessi, M.C.; Martin, J.C.; Bernard, M.; et al. Ectopic fat storage in the pancreas using 1H-MRS: Importance of diabetic status and modulation with bariatric surgery-induced weight loss. Int. J. Obes. 2015, 39, 480–487. [Google Scholar] [CrossRef] [PubMed]
- Steven, S.; Hollingsworth, K.G.; Small, P.K.; Woodcock, S.A.; Pucci, A.; Aribisala, B.; Al-Mrabeh, A.; Daly, A.K.; Batterham, R.L.; Taylor, R. Weight Loss Decreases Excess Pancreatic Triacylglycerol Specifically in Type 2 Diabetes. Diabetes Care 2016, 39, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Tushuizen, M.E.; Bunck, M.C.; Pouwels, P.J.; Bontemps, S.; van Waesberghe, J.H.; Schindhelm, R.K.; Mari, A.; Heine, R.J.; Diamant, M. Pancreatic fat content and beta-cell function in men with and without type 2 diabetes. Diabetes Care 2007, 30, 2916–2921. [Google Scholar] [CrossRef] [PubMed]
- Heni, M.; Machann, J.; Staiger, H.; Schwenzer, N.F.; Peter, A.; Schick, F.; Claussen, C.D.; Stefan, N.; Häring, H.U.; Fritsche, A. Pancreatic fat is negatively associated with insulin secretion in individuals with impaired fasting glucose and/or impaired glucose tolerance: A nuclear magnetic resonance study. Diabetes Metab. Res. Rev. 2010, 26, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Yokota, K.; Fukushima, M.; Takahashi, Y.; Igaki, N.; Seino, S. Insulin secretion and computed tomography values of the pancreas in the early stage of the development of diabetes. J. Diabetes Investig. 2012, 3, 371–376. [Google Scholar] [CrossRef]
- Begovatz, P.; Koliaki, C.; Weber, K.; Strassburger, K.; Nowotny, B.; Nowotny, P.; Müssig, K.; Bunke, J.; Pacini, G.; Szendrödi, J.; et al. Pancreatic adipose tissue infiltration, parenchymal steatosis and beta cell function in humans. Diabetologia 2015, 58, 1646–1655. [Google Scholar] [CrossRef]
- Wagner, R.; Jaghutriz, B.A.; Gerst, F.; Barroso Oquendo, M.; Machann, J.; Schick, F.; Löffler, M.W.; Nadalin, S.; Fend, F.; Königsrainer, A.; et al. Pancreatic Steatosis Associates With Impaired Insulin Secretion in Genetically Predisposed Individuals. J. Clin. Endocrinol. Metab. 2020, 105, dgaa435. [Google Scholar] [CrossRef]
- Yamazaki, H.; Tauchi, S.; Wang, J.; Dohke, M.; Hanawa, N.; Kodama, Y.; Katanuma, A.; Saisho, Y.; Kamitani, T.; Fukuhara, S.; et al. Longitudinal association of fatty pancreas with the incidence of type-2 diabetes in lean individuals: A 6-year computed tomography-based cohort study. J. Gastroenterol. 2020, 55, 712–721. [Google Scholar] [CrossRef]
- Saisho, Y. Changing the Concept of Type 2 Diabetes: Beta Cell Workload Hypothesis Revisited. Endocr. Metab. Immune Disord. Drug Targets 2019, 19, 121–127. [Google Scholar] [CrossRef]
- Saisho, Y. β-cell dysfunction: Its critical role in prevention and management of type 2 diabetes. World J. Diabetes 2015, 6, 109–124. [Google Scholar] [CrossRef] [PubMed]
- Tuomilehto, J.; Lindström, J.; Eriksson, J.G.; Valle, T.T.; Hämäläinen, H.; Ilanne-Parikka, P.; Keinänen-Kiukaanniemi, S.; Laakso, M.; Louheranta, A.; Rastas, M.; et al. Prevention of type 2 diabetes mellitus by changes in lifestyle among subjects with impaired glucose tolerance. N. Engl. J. Med. 2001, 344, 1343–1350. [Google Scholar] [CrossRef] [PubMed]
- Knowler, W.C.; Barrett-Connor, E.; Fowler, S.E.; Hamman, R.F.; Lachin, J.M.; Walker, E.A.; Nathan, D.M. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N. Engl. J. Med. 2002, 346, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Knowler, W.C.; Fowler, S.E.; Hamman, R.F.; Christophi, C.A.; Hoffman, H.J.; Brenneman, A.T.; Brown-Friday, J.O.; Goldberg, R.; Venditti, E.; Nathan, D.M. 10-year follow-up of diabetes incidence and weight loss in the Diabetes Prevention Program Outcomes Study. Lancet 2009, 374, 1677–1686. [Google Scholar] [CrossRef]
- Chiasson, J.L.; Josse, R.G.; Gomis, R.; Hanefeld, M.; Karasik, A.; Laakso, M. Acarbose for prevention of type 2 diabetes mellitus: The STOP-NIDDM randomised trial. Lancet 2002, 359, 2072–2077. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Abdul-Ghani, M.A. Preservation of beta-cell function: The key to diabetes prevention. J. Clin. Endocrinol. Metab. 2011, 96, 2354–2366. [Google Scholar] [CrossRef]
- Gerstein, H.C.; Yusuf, S.; Bosch, J.; Pogue, J.; Sheridan, P.; Dinccag, N.; Hanefeld, M.; Hoogwerf, B.; Laakso, M.; Mohan, V.; et al. Effect of rosiglitazone on the frequency of diabetes in patients with impaired glucose tolerance or impaired fasting glucose: A randomised controlled trial. Lancet 2006, 368, 1096–1105. [Google Scholar] [CrossRef]
- Holman, R.R.; Haffner, S.M.; McMurray, J.J.; Bethel, M.A.; Holzhauer, B.; Hua, T.A.; Belenkov, Y.; Boolell, M.; Buse, J.B.; Buckley, B.M.; et al. Effect of nateglinide on the incidence of diabetes and cardiovascular events. N. Engl. J. Med. 2010, 362, 1463–1476. [Google Scholar] [CrossRef]
- Gregg, E.W.; Chen, H.; Wagenknecht, L.E.; Clark, J.M.; Delahanty, L.M.; Bantle, J.; Pownall, H.J.; Johnson, K.C.; Safford, M.M.; Kitabchi, A.E.; et al. Association of an intensive lifestyle intervention with remission of type 2 diabetes. JAMA 2012, 308, 2489–2496. [Google Scholar] [CrossRef]
- Lean, M.E.; Leslie, W.S.; Barnes, A.C.; Brosnahan, N.; Thom, G.; McCombie, L.; Peters, C.; Zhyzhneuskaya, S.; Al-Mrabeh, A.; Hollingsworth, K.G.; et al. Primary care-led weight management for remission of type 2 diabetes (DiRECT): An open-label, cluster-randomised trial. Lancet 2018, 391, 541–551. [Google Scholar] [CrossRef]
- Taylor, R.; Al-Mrabeh, A.; Zhyzhneuskaya, S.; Peters, C.; Barnes, A.C.; Aribisala, B.S.; Hollingsworth, K.G.; Mathers, J.C.; Sattar, N.; Lean, M.E.J. Remission of Human Type 2 Diabetes Requires Decrease in Liver and Pancreas Fat Content but Is Dependent upon Capacity for β Cell Recovery. Cell Metab. 2018, 28, 667. [Google Scholar] [CrossRef]
- Drucker, D.J.; Nauck, M.A. The incretin system: Glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet 2006, 368, 1696–1705. [Google Scholar] [CrossRef]
- Leibowitz, G.; Cahn, A.; Bhatt, D.L.; Hirshberg, B.; Mosenzon, O.; Wei, C.; Jermendy, G.; Sheu, W.H.; Sendon, J.L.; Im, K.; et al. Impact of treatment with saxagliptin on glycaemic stability and β-cell function in the SAVOR-TIMI 53 study. Diabetes Obes. Metab. 2015, 17, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Del Prato, S.; Camisasca, R.; Wilson, C.; Fleck, P. Durability of the efficacy and safety of alogliptin compared with glipizide in type 2 diabetes mellitus: A 2-year study. Diabetes Obes. Metab. 2014, 16, 1239–1246. [Google Scholar] [CrossRef] [PubMed]
- Marso, S.P.; Daniels, G.H.; Brown-Frandsen, K.; Kristensen, P.; Mann, J.F.; Nauck, M.A.; Nissen, S.E.; Pocock, S.; Poulter, N.R.; Ravn, L.S.; et al. Liraglutide and Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2016, 375, 311–322. [Google Scholar] [CrossRef]
- Marso, S.P.; Bain, S.C.; Consoli, A.; Eliaschewitz, F.G.; Jódar, E.; Leiter, L.A.; Lingvay, I.; Rosenstock, J.; Seufert, J.; Warren, M.L.; et al. Semaglutide and Cardiovascular Outcomes in Patients with Type 2 Diabetes. N. Engl. J. Med. 2016, 375, 1834–1844. [Google Scholar] [CrossRef]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef]
- Neal, B.; Perkovic, V.; Mahaffey, K.W.; de Zeeuw, D.; Fulcher, G.; Erondu, N.; Shaw, W.; Law, G.; Desai, M.; Matthews, D.R. Canagliflozin and Cardiovascular and Renal Events in Type 2 Diabetes. N. Engl. J. Med. 2017, 377, 644–657. [Google Scholar] [CrossRef]
- Pharmacologic Approaches to Glycemic Treatment: Standards of Medical Care in Diabetes-2020. Diabetes Care 2020, 43, S98–S110. [CrossRef]
- Ghosh-Swaby, O.R.; Goodman, S.G.; Leiter, L.A.; Cheng, A.; Connelly, K.A.; Fitchett, D.; Jüni, P.; Farkouh, M.E.; Udell, J.A. Glucose-lowering drugs or strategies, atherosclerotic cardiovascular events, and heart failure in people with or at risk of type 2 diabetes: An updated systematic review and meta-analysis of randomised cardiovascular outcome trials. Lancet Diabetes Endocrinol. 2020, 8, 418–435. [Google Scholar] [CrossRef]
- Weng, J.; Li, Y.; Xu, W.; Shi, L.; Zhang, Q.; Zhu, D.; Hu, Y.; Zhou, Z.; Yan, X.; Tian, H.; et al. Effect of intensive insulin therapy on beta-cell function and glycaemic control in patients with newly diagnosed type 2 diabetes: A multicentre randomised parallel-group trial. Lancet 2008, 371, 1753–1760. [Google Scholar] [CrossRef]
- Matthews, D.R.; Paldánius, P.M.; Proot, P.; Chiang, Y.; Stumvoll, M.; Del Prato, S. Glycaemic durability of an early combination therapy with vildagliptin and metformin versus sequential metformin monotherapy in newly diagnosed type 2 diabetes (VERIFY): A 5-year, multicentre, randomised, double-blind trial. Lancet 2019, 394, 1519–1529. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Study Year, Reference | Approach for Sample Collection | Country Where Performed (Ethnicity) | Metabolic Status/ Number of Samples (Male) | Mean Age (Year) | Mean BMI (kg/m2) | Change in Beta-Cell Mass (BCM) or Beta-Cell Area (BCA) | |
|---|---|---|---|---|---|---|---|
| Obesity | Diabetes | ||||||
| Sakuraba et al., 2002, [8] | autopsy | Japan (n.a.) | NDM 15 (10) | 52 | 21.3 | n.a. | BCM: 1.14 g |
| DM 14 (9) | 61 | 20.7 | BCM: 0.82 g * 30% decreased vs. NDM | ||||
| Yoon et al., 2003, [9] | organ donor | Korea (n.a.) | 9 (6) | 41 | 23.8 | Positive correlation between BCA and BMI | n.a. |
| pancreas surgery | NDM 10 (6) | 57 | 22.2 | No difference | BCA: 1.94% | ||
| T2DM 25 (15) | 60 | 22.2 | Positive correlation between BCA and BMI | BCA: 1.37% * 29% decreased vs. NDM | |||
| Butler et al., 2003, [10] | autopsy | USA (n.a.) | NDM lean 17 (10), BMI < 25 kg/m2 | 78 | <25 kg/m2 | BCA:1.71% | |
| T2DM lean 16 (9), BMI < 25 kg/m2 | 80 | n.a. | 41% decreased vs. NDM Lean | ||||
| NDM Obese 31 (16), BMI > 27 kg/m2 | 67 | >27 kg/m2 | BCA:2.6% | ||||
| IFG Obese 19 (10), BMI > 27 kg/m2 | 63 | n.a. | BCA: 1.56% * 40% decreased vs. NDM Obese | ||||
| T2DM Obese 41 (24), BMI > 27 kg/m2 | 63 | BCA: 0.96% * 63% decreased vs. NDM Obese | |||||
| Rahier et al., 2008, [11] | autopsy | Belgium (European) | BMI < 25 kg/m2, NDM 26 (14) | 63 | 21.9 | 20% increased in NDM | 41% decreased vs. NDM with BMI < 25 kg/m2 |
| BMI < 25 kg/m2, T2DM 15 (5) | 72 | 22.3 | |||||
| BMI 26–40 kg/m2, NDM 25 (21) | 68 | 29.9 | 38% decreased vs. NDM with BMI 26-40 kg/m2 | ||||
| BMI 26–40 kg/m2, T2DM 34 (21) | 68 | 31.7 | |||||
| Meier et al., 2009, [12] | pancreas surgery | Germany (n.a.) | NGT 8 (3) | 56 | 23.6 | n.a. | BCA: 1.22% |
| IGT/IFG 14 (6) | 63 | 23.4 | BCA: 1.14% | ||||
| DM 11 (8) | 56 | 24.1 | BCA: 0.43% * 65% decreased vs. NGT | ||||
| Hanley et al., 2010, [13] | organ donor | Canada (n.a) | NDM Lean 18 (13), BMI < 27 kg/m2 | 59 | 24.2 | BCA:1.15% | |
| T2DM Lean 8 (6), BMI < 27 kg/m2 | 59 | 23.7 | n.a. | BCA: 1.28% | |||
| NDM Obese 23 (10), BMI > 27 kg/m2 | 59 | 31.4 | BCA: 2.20% * 91% increased vs. NDM lean | BCA: 2.20% | |||
| T2DM Obese 11 (5), BMI > 27 kg/m2 | 61 | 32.6 | n.a. | BCA: 1.41% * 36% decreased vs. NDM Obese | |||
| Henquin et al., 2011, [14] | autopsy | Belgium (n.a.) | NDM 52 (35) | 66 | 25.8 | No difference | 36% decreased vs. NDM |
| T2DM 50 (26) | 68 | 30.1 | No difference | ||||
| Meier et al., 2012, [15] | pancreas surgery | Germany (n.a.) | 82 (42) | 60 | 24.4 | n.a. | BCA in IGT 21% decreased vs. NGT |
| Yoneda et al., 2013, [16] | pancreas surgery | Japan (n.a.) | NGT 11 (7) | 67 | 21.1 | n.a. | BCA: 1.60% |
| IGT 11 (3) | 67 | 22.7 | BCA: 0.99% * 38% decreased vs. NGT | ||||
| Newly Diagnosed DM 10 (4) | 66 | 23.4 | BCA: 0.93% * 42% decreased vs. NGT | ||||
| Long-Standing T2DM 10 (6) | 76 | 20.5 | BCA: 0.53% * 67% decreased vs. NGT | ||||
| Saisho et al., 2013, [17] | autopsy | USA (n.a.) | NDM Lean 53 (30), BMI < 25 kg/m2 | 37 | 21.2 | BCM: 0.8 g | n.a. |
| NDM Obese 61 (43), BMI ≥ 27 kg/m2 | 41 | 35.1 | BCM: 1.2 g* 50% increased vs. Lean | ||||
| Kou et al., 2013, [18] | autopsy | Japan (Japanese) | NDM Lean 39 (22), BMI < 25 kg/m2 | 47 | 20.4 | BCM: 0.7 g | n.a. |
| NDM Obese 33 (24), BMI ≥ 25 kg/m2 | 47 | 28.5 | BCM: 0.6 g No difference vs. Lean | ||||
| Mezza et al., 2014, [19] | pancreas surgery | Italy (n.a.) | NDM 18 (9) | 53 | 27.9 | BCA in insulin-resistant # (1.10%) increased vs. insulin-sensitive # (0.58%*) | n.a. |
| Mizukami et al., 2014, [20] | autopsy | Japan (n.a.) | NDM 30 (21) | 65 | 22.4 | No difference | BCM: 1.86 g |
| DM 47 (38) | 68 | 22.7 | BCM: 1.27 g * 32% decreased vs. NDM | ||||
| Fujita et al., 2015, [21] | pancreas surgery | Japan (n.a.) | NGT 13 (8) | 64 | 21.5 | No difference | BCA: 1.072% |
| IGT 9 (4) | 61 | 20.8 | BCA: 0.998% | ||||
| DM 10 (7) | 68 | 22.4 | BCA: 0.762% | ||||
| Sato et al., 2015, [22] | autopsy | Japan (Japanese) | NDM 26 (15) | 63 | 20.8 | n.a. | BCA:1.66% |
| DM 25 (21) | 66 | 21.5 | BCA: 0.92% * 45% decreased vs. NDM | ||||
| Inaishi et al., 2016, [23] | pancreas surgery | Japan (Japanese) | NDM Lean 40 (17), BMI < 25 kg/m2 | 64 | 21.5 | BCA:1.42% | BCA: 1.48% |
| NDM Obese 10 (9), BMI ≥ 25 kg/m2 | 63 | 26.4 | BCA: 1.71% No difference vs. Lean | ||||
| DM 49 (35) | 67 | 21.9 | n.a. | BCA: 0.80% * 46% decreased vs. NDM | |||
| Xin et al., 2017, [24] | autopsy | Japan (n.a.) | NDM 22 (11) | 61 | 21.8 | n.a. | BCA in T2DM 30% decreased vs. NDM |
| T2DM 27 (19) | 63 | 22.8 | |||||
| Inaishi et al., 2020, [25] | autopsy | Japan (Japanese) | NGT 40 (24) | 80 | 20.4 | No difference | BCA: 1.85% |
| Prediabetes 31 (25) | 78 | 22.1 | BCA: 1.59% | ||||
| T2DM 32 (19) | 76 | 23.6 | BCA: 1.17% * 37% decreased vs. NGT | ||||
| Sasaki et al., 2020, [26] | pancreas surgery | Japan (Japanese) | NDM 38 (20) | 61 | 22.3 | No difference | BCA: 1.14% |
| DM 26 (23) | 67 | 25.1 | BCA: 0.75% * 34% decreased vs. NDM | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Inaishi, J.; Saisho, Y. Beta-Cell Mass in Obesity and Type 2 Diabetes, and Its Relation to Pancreas Fat: A Mini-Review. Nutrients 2020, 12, 3846. https://doi.org/10.3390/nu12123846
Inaishi J, Saisho Y. Beta-Cell Mass in Obesity and Type 2 Diabetes, and Its Relation to Pancreas Fat: A Mini-Review. Nutrients. 2020; 12(12):3846. https://doi.org/10.3390/nu12123846
Chicago/Turabian StyleInaishi, Jun, and Yoshifumi Saisho. 2020. "Beta-Cell Mass in Obesity and Type 2 Diabetes, and Its Relation to Pancreas Fat: A Mini-Review" Nutrients 12, no. 12: 3846. https://doi.org/10.3390/nu12123846
APA StyleInaishi, J., & Saisho, Y. (2020). Beta-Cell Mass in Obesity and Type 2 Diabetes, and Its Relation to Pancreas Fat: A Mini-Review. Nutrients, 12(12), 3846. https://doi.org/10.3390/nu12123846