Commensal Bacterium Rothia aeria Degrades and Detoxifies Gluten via a Highly Effective Subtilisin Enzyme

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. In Vivo Digestion Experiment
2.2. In Vitro Gliadin Digestion by Live and Dead Rothia Bacteria
2.3. Isolation and Identification of the Gluten-Degrading Enzyme from R. aeria
3. Results
3.1. In Vivo Digestion by life Rothia Bacteria
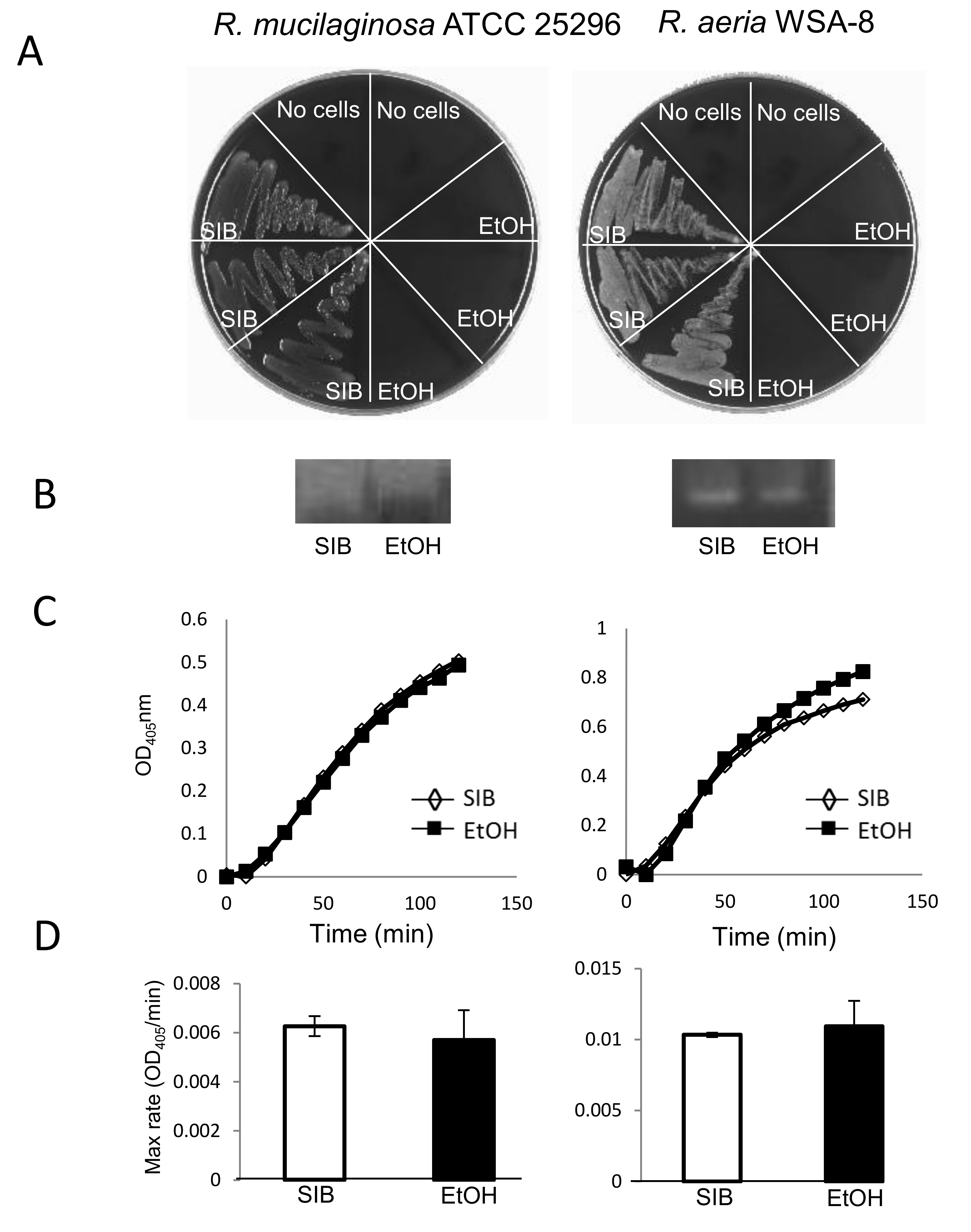
3.2. In Vitro Gluten Digestion by Dead Rothia Bacteria
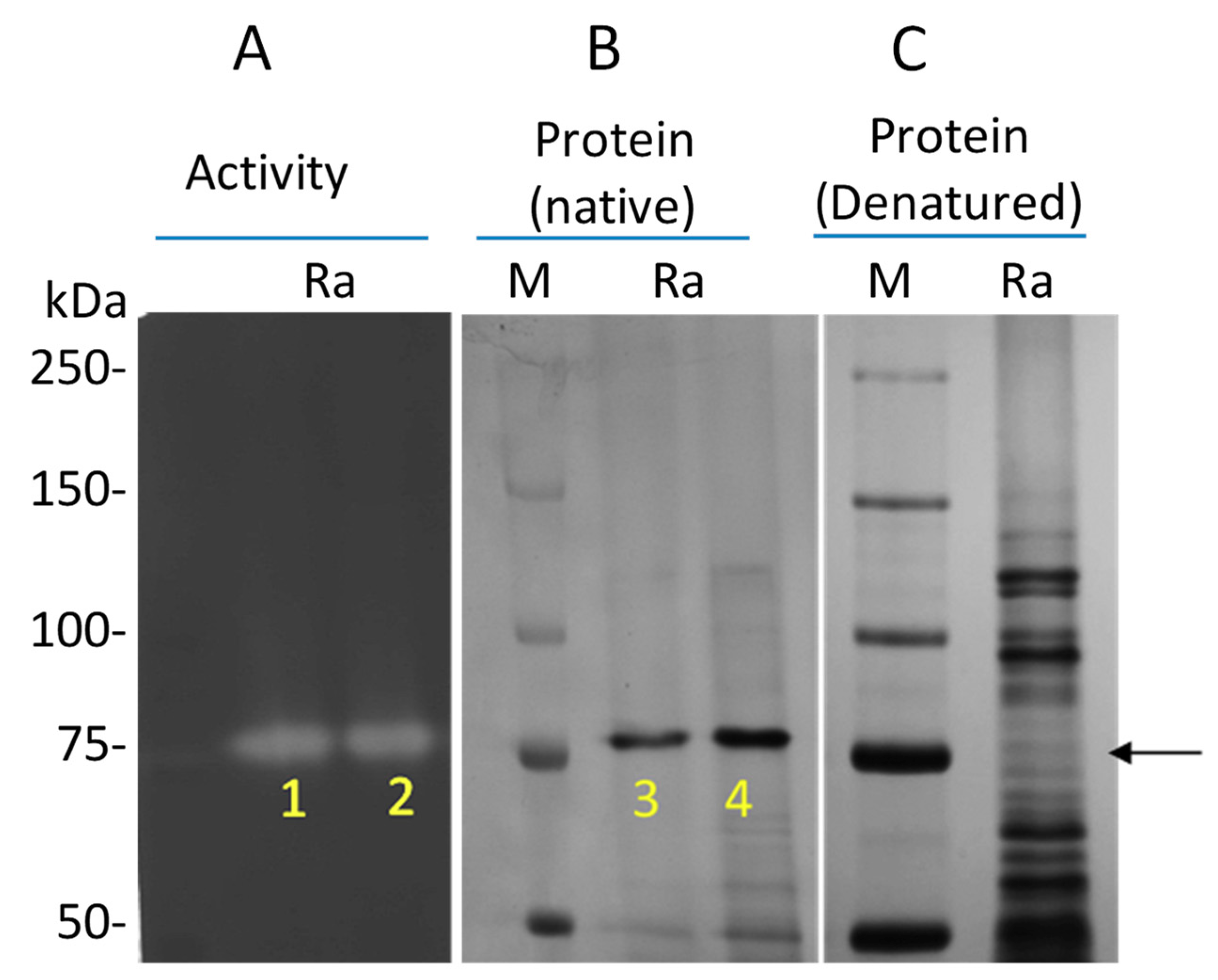
3.3. Isolation and Identification of the Gluten-Degrading Enzyme from R. aeria
4. Discussions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schuppan, D.; Junker, Y.; Barisani, D. Celiac disease: From pathogenesis to novel therapies. Gastroenterology 2009, 137, 1912–1933. [Google Scholar] [CrossRef]
- Fasano, A.; Catassi, C. Clinical practice. Celiac disease. N. Engl. J. Med. 2012, 367, 2419–2426. [Google Scholar] [CrossRef] [PubMed]
- Helmerhorst, E.J.; Zamakhchari, M.; Schuppan, D.; Oppenheim, F.G. Discovery of a novel and rich source of gluten-degrading microbial enzymes in the oral cavity. PLoS ONE 2010, 5, e13264. [Google Scholar] [CrossRef] [Green Version]
- Gass, J.; Khosla, C. Prolyl endopeptidases. Cell Mol. Life Sci 2007, 64, 345–355. [Google Scholar] [CrossRef] [PubMed]
- Hausch, F.; Shan, L.; Santiago, N.A.; Gray, G.M.; Khosla, C. Intestinal digestive resistance of immunodominant gliadin peptides. Am. J. Physiol. Gastrointest. Liver Physiol. 2002, 283, G996–G1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitea, C.; Havenaar, R.; Drijfhout, J.W.; Edens, L.; Dekking, L.; Koning, F. Efficient degradation of gluten by a prolyl endoprotease in a gastrointestinal model: Implications for coeliac disease. Gut 2008, 57, 25–32. [Google Scholar] [CrossRef]
- Vader, L.W.; Stepniak, D.T.; Bunnik, E.M.; Kooy, Y.M.; de Haan, W.; Drijfhout, J.W.; Van Veelen, P.A.; Koning, F. Characterization of cereal toxicity for celiac disease patients based on protein homology in grains. Gastroenterology 2003, 125, 1105–1113. [Google Scholar] [CrossRef]
- Dewhirst, F.E.; Chen, T.; Izard, J.; Paster, B.J.; Tanner, A.C.; Yu, W.H.; Lakshmanan, A.; Wade, W.G. The human oral microbiome. J. Bacteriol. 2010, 192, 5002–5017. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Feo, M.; Wei, G.; Blumenkranz, G.; Dewhirst, F.E.; Schuppan, D.; Oppenheim, F.G.; Helmerhorst, E.J. The cultivable human oral gluten-degrading microbiome and its potential implications in coeliac disease and gluten sensitivity. Clin. Microbiol. Infect. 2013, 19, E386–E394. [Google Scholar] [CrossRef] [Green Version]
- Stearns, J.C.; Lynch, M.D.; Senadheera, D.B.; Tenenbaum, H.C.; Goldberg, M.B.; Cvitkovitch, D.G.; Croitoru, K.; Moreno-Hagelsieb, G.; Neufeld, J.D. Bacterial biogeography of the human digestive tract. Sci. Rep. 2011, 1, 170. [Google Scholar] [CrossRef] [Green Version]
- Zamakhchari, M.; Wei, G.; Dewhirst, F.; Lee, J.; Schuppan, D.; Oppenheim, F.G.; Helmerhorst, E.J. Identification of Rothia bacteria as gluten-degrading natural colonizers of the upper gastro-intestinal tract. PLoS ONE 2011, 6, e24455. [Google Scholar] [CrossRef] [Green Version]
- Ou, G.; Hedberg, M.; Horstedt, P.; Baranov, V.; Forsberg, G.; Drobni, M.; Sandstrom, O.; Wai, S.N.; Johansson, I.; Hammarstrom, M.L.; et al. Proximal small intestinal microbiota and identification of rod-shaped bacteria associated with childhood celiac disease. Am. J. Gastroenterol. 2009, 104, 3058–3067. [Google Scholar] [CrossRef] [PubMed]
- Ramanan, P.; Barreto, J.N.; Osmon, D.R.; Tosh, P.K. Rothia bacteremia: A 10-year experience at Mayo Clinic, Rochester, Minnesota. J. Clin. Microbiol. 2014, 52, 3184–3189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, N.; Wei, G.; Schuppan, D.; Helmerhorst, E.J. Effect of Rothia mucilaginosa enzymes on gliadin (gluten) structure, deamidation, and immunogenic epitopes relevant to celiac disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 307, G769–G776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, G.; Tian, N.; Siezen, R.; Schuppan, D.; Helmerhorst, E.J. Identification of food-grade subtilisins as gluten-degrading enzymes to treat celiac disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 311, G571–G580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helmerhorst, E.J.; Wei, G. Experimental Strategy to Discover Microbes with Gluten-degrading Enzyme Activities. Proc. Spie Int. Soc. Opt. Eng. 2014, 9112. [Google Scholar] [CrossRef] [Green Version]
- Wei, G.; Tian, N.; Valery, A.C.; Zhong, Y.; Schuppan, D.; Helmerhorst, E.J. Identification of Pseudolysin (lasB) as an Aciduric Gluten-Degrading Enzyme with High Therapeutic Potential for Celiac Disease. Am. J. Gastroenterol. 2015, 110, 899–908. [Google Scholar] [CrossRef] [Green Version]
- Osman, A.A.; Uhlig, H.H.; Valdes, I.; Amin, M.; Mendez, E.; Mothes, T. A monoclonal antibody that recognizes a potential coeliac-toxic repetitive pentapeptide epitope in gliadins. Eur. J. Gastroenterol. Hepatol. 2001, 13, 1189–1193. [Google Scholar] [CrossRef]
- White, L.E.; Bannerman, E.; Gillett, P.M. Coeliac disease and the gluten-free diet: A review of the burdens; factors associated with adherence and impact on health-related quality of life, with specific focus on adolescence. J. Hum. Nutr. Diet. 2016, 29, 593–606. [Google Scholar] [CrossRef]
- Catassi, C.; Fabiani, E.; Iacono, G.; D’Agate, C.; Francavilla, R.; Biagi, F.; Volta, U.; Accomando, S.; Picarelli, A.; De Vitis, I.; et al. A prospective, double-blind, placebo-controlled trial to establish a safe gluten threshold for patients with celiac disease. Am. J. Clin. Nutr. 2007, 85, 160–166. [Google Scholar] [CrossRef]
- Shan, L.; Marti, T.; Sollid, L.M.; Gray, G.M.; Khosla, C. Comparative biochemical analysis of three bacterial prolyl endopeptidases: Implications for coeliac sprue. Biochem. J. 2004, 383, 311–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bethune, M.T.; Khosla, C. Oral enzyme therapy for celiac sprue. Methods Enzym. 2012, 502, 241–271. [Google Scholar] [CrossRef] [Green Version]
- Khosla, C. Celiac Disease: Lessons for and from Chemical Biology. ACS Chem. Biol. 2017, 12, 1455–1459. [Google Scholar] [CrossRef] [PubMed]
- Ehren, J.; Moron, B.; Martin, E.; Bethune, M.T.; Gray, G.M.; Khosla, C. A food-grade enzyme preparation with modest gluten detoxification properties. PLoS ONE 2009, 4, e6313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gass, J.; Bethune, M.T.; Siegel, M.; Spencer, A.; Khosla, C. Combination enzyme therapy for gastric digestion of dietary gluten in patients with celiac sprue. Gastroenterology 2007, 133, 472–480. [Google Scholar] [CrossRef] [Green Version]
- Wei, G.; Helmerhorst, E.J.; Darwish, G.; Blumenkranz, G.; Schuppan, D. Gluten Degrading Enzymes for Treatment of Celiac Disease. Nutrients 2020, 12, 2095. [Google Scholar] [CrossRef]
- Krishnareddy, S.; Stier, K.; Recanati, M.; Lebwohl, B.; Green, P.H. Commercially available glutenases: A potential hazard in coeliac disease. Ther. Adv. Gastroenterol. 2017, 10, 473–481. [Google Scholar] [CrossRef]
- Jose, N.M.; Bunt, C.R.; Hussain, M.A. Comparison of Microbiological and Probiotic Characteristics of Lactobacilli Isolates from Dairy Food Products and Animal Rumen Contents. Microorganisms 2015, 3, 198–212. [Google Scholar] [CrossRef] [Green Version]
- Padmanabhan, P.; Grosse, J.; Asad, A.B.; Radda, G.K.; Golay, X. Gastrointestinal transit measurements in mice with 99mTc-DTPA-labeled activated charcoal using NanoSPECT-CT. EJNMMI Res. 2013, 3, 60. [Google Scholar] [CrossRef] [Green Version]
- Rizzello, C.G.; De Angelis, M.; Di Cagno, R.; Camarca, A.; Silano, M.; Losito, I.; De Vincenzi, M.; De Bari, M.D.; Palmisano, F.; Maurano, F.; et al. Highly efficient gluten degradation by lactobacilli and fungal proteases during food processing: New perspectives for celiac disease. Appl. Environ. Microbiol. 2007, 73, 4499–4507. [Google Scholar] [CrossRef] [Green Version]
- Klausen, B. Microbiological and immunological aspects of experimental periodontal disease in rats: A review article. J. Periodontol. 1991, 62, 59–73. [Google Scholar] [CrossRef] [PubMed]
- Tanner, A. Microbial etiology of periodontal diseases. Where are we? Where are we going? Curr. Opin. Dent. 1992, 2, 12–24. [Google Scholar] [PubMed]
- Watanabe, K. Prepubertal periodontitis: A review of diagnostic criteria, pathogenesis, and differential diagnosis. J. Periodontal. Res. 1990, 25, 31–48. [Google Scholar] [CrossRef] [PubMed]
- Sassone, L.M.; Fidel, R.; Faveri, M.; Fidel, S.; Figueiredo, L.; Feres, M. Microbiological evaluation of primary endodontic infections in teeth with and without sinus tract. Int. Endod. J. 2008, 41, 508–515. [Google Scholar] [CrossRef]
- Caminero, A.; Galipeau, H.J.; McCarville, J.L.; Johnston, C.W.; Bernier, S.P.; Russell, A.K.; Jury, J.; Herran, A.R.; Casqueiro, J.; Tye-Din, J.A.; et al. Duodenal Bacteria From Patients With Celiac Disease and Healthy Subjects Distinctly Affect Gluten Breakdown and Immunogenicity. Gastroenterology 2016, 151, 670–683. [Google Scholar] [CrossRef] [Green Version]
- Schuppan, D.; Gisbert-Schuppan, K. Wheat Syndromes: How Wheat, Gluten and ATI Cause Inflammation, IBS and Autoimmune Diseases; Springer: Cham, Germany, 2019; 19p. [Google Scholar]
- Wilkens, S. Structure and mechanism of ABC transporters. F1000Prime Rep. 2015, 7, 14. [Google Scholar] [CrossRef]
- Ottesen, M.; Spector, A. A comparison of two proteinases from Bacillus subtilis. C. R. Trav. Lab. Carlsberg 1960, 32, 63–74. [Google Scholar]
- Dressman, J.B. Comparison of canine and human gastrointestinal physiology. Pharm. Res. 1986, 3, 123–131. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, G.; Darwish, G.; Oppenheim, F.G.; Schuppan, D.; Helmerhorst, E.J. Commensal Bacterium Rothia aeria Degrades and Detoxifies Gluten via a Highly Effective Subtilisin Enzyme. Nutrients 2020, 12, 3724. https://doi.org/10.3390/nu12123724
Wei G, Darwish G, Oppenheim FG, Schuppan D, Helmerhorst EJ. Commensal Bacterium Rothia aeria Degrades and Detoxifies Gluten via a Highly Effective Subtilisin Enzyme. Nutrients. 2020; 12(12):3724. https://doi.org/10.3390/nu12123724
Chicago/Turabian StyleWei, Guoxian, Ghassan Darwish, Frank G. Oppenheim, Detlef Schuppan, and Eva J. Helmerhorst. 2020. "Commensal Bacterium Rothia aeria Degrades and Detoxifies Gluten via a Highly Effective Subtilisin Enzyme" Nutrients 12, no. 12: 3724. https://doi.org/10.3390/nu12123724
APA StyleWei, G., Darwish, G., Oppenheim, F. G., Schuppan, D., & Helmerhorst, E. J. (2020). Commensal Bacterium Rothia aeria Degrades and Detoxifies Gluten via a Highly Effective Subtilisin Enzyme. Nutrients, 12(12), 3724. https://doi.org/10.3390/nu12123724