Bile Acids and GPBAR-1: Dynamic Interaction Involving Genes, Environment and Gut Microbiome

 ,
,  ,
,  , and
, and 
Abstract
:1. Introduction
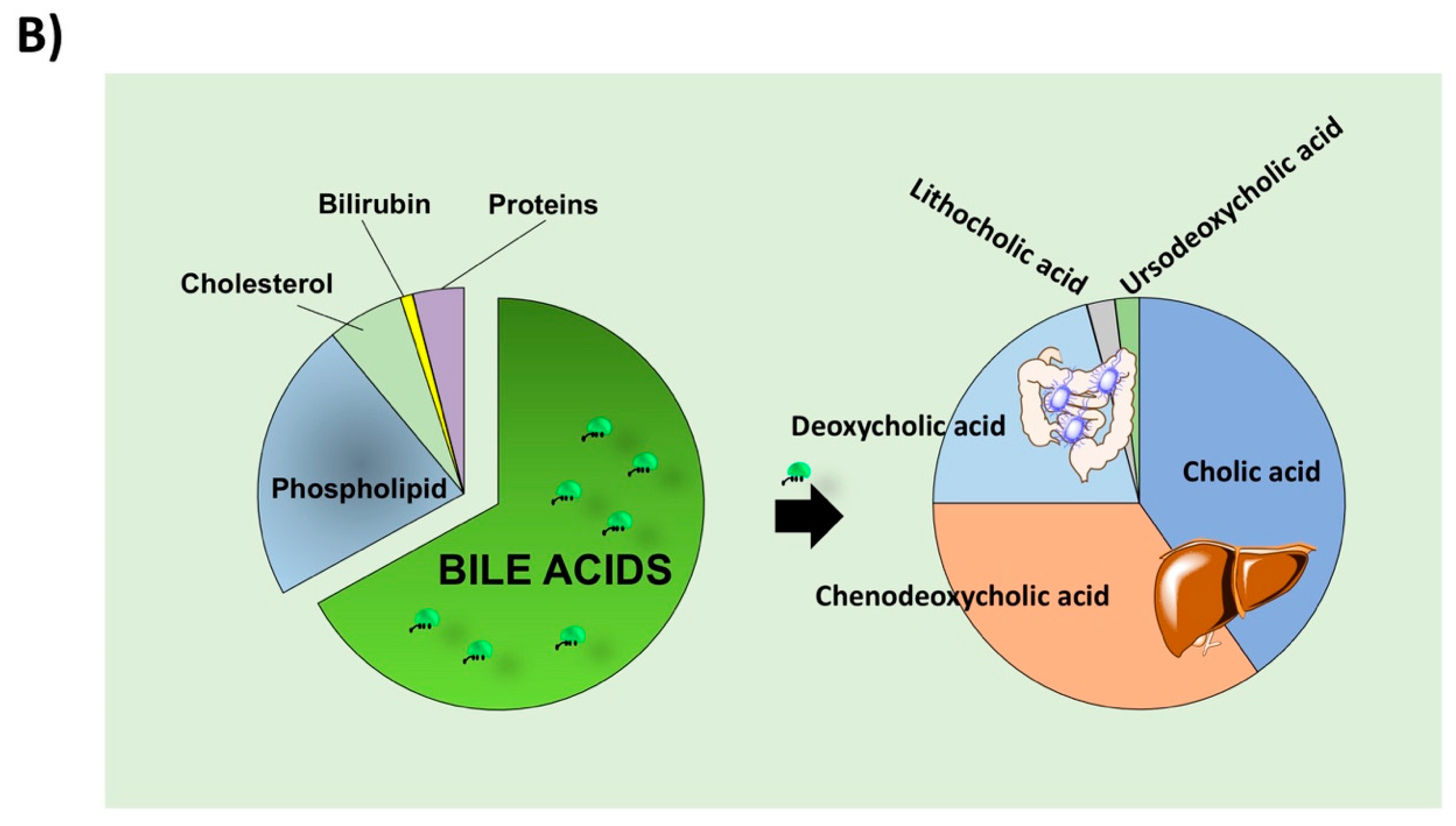
2. BA Synthesis, Secretion, Biotransformation, and Absorption
3. BA as Signalling Molecules
3.1. GPBAR-1
3.1.1. Effect of GPBAR-1 on Bile Composition
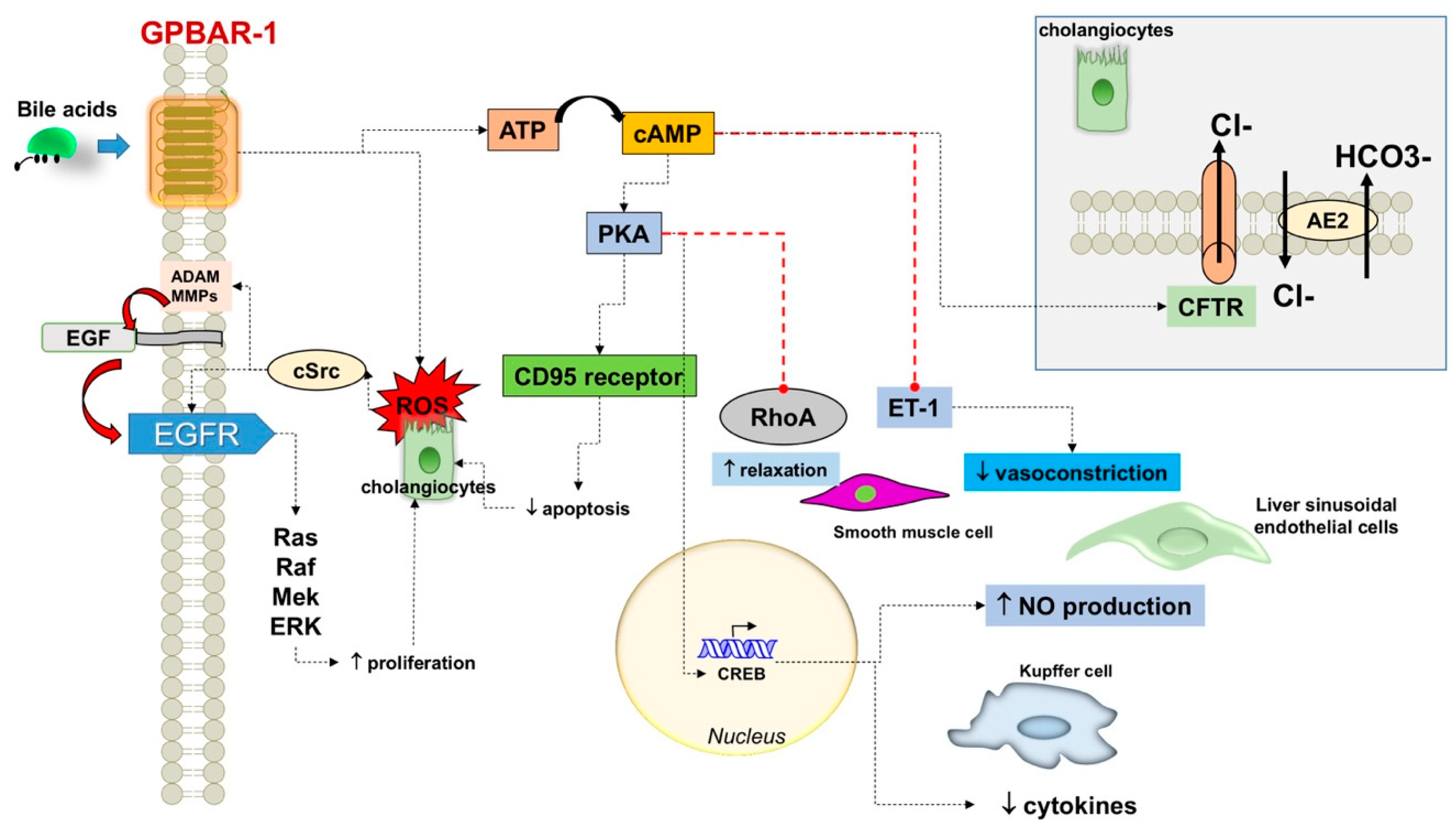
3.1.2. Various Effects on Cell Lines
4. Mechanisms of Damage
4.1. BA Overload
4.2. Biliary Epithelial Barrier
4.3. Inflammation
5. Gene-Environment Interactions Involving GPBAR-1
6. Therapeutic Aspects
6.1. Liver Steatosis
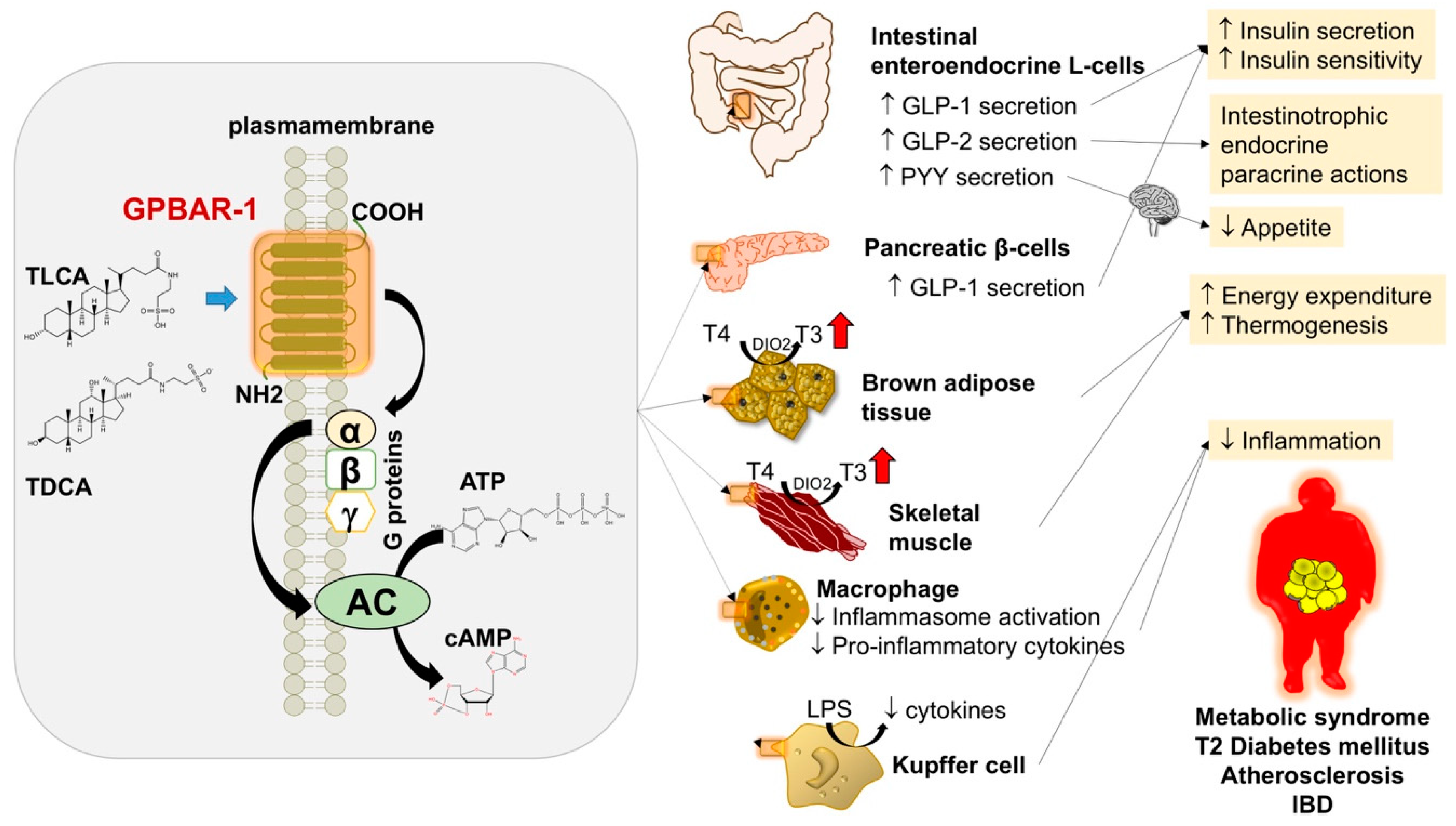
6.2. Obesity and Diabetes
6.3. Atherosclerosis
6.4. Inflammatory Bowel Diseases
6.5. Potential Drawbacks Associated with GPBAR-1 Stimulation
6.6. Summary of Protective Mechanisms and Action Target Involving GPBAR-1.
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Di Ciaula, A.; Garruti, G.; Lunardi Baccetto, R.; Molina-Molina, E.; Bonfrate, L.; Wang, D.Q.; Portincasa, P. Bile acid physiology. Ann. Hepatol. 2017, 16, s4–s14. [Google Scholar] [CrossRef] [PubMed]
- Donkers, J.M.; Roscam Abbing, R.L.P.; van de Graaf, S.F.J. Developments in bile salt based therapies: A critical overview. Biochem. Pharmacol. 2019, 161, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Merlen, G.; Ursic-Bedoya, J.; Jourdainne, V.; Kahale, N.; Glenisson, M.; Doignon, I.; Rainteau, D.; Tordjmann, T. Bile acids and their receptors during liver regeneration: “Dangerous protectors”. Mol. Asp. Med. 2017, 56, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Dawson, P.A.; Karpen, S.J. Intestinal transport and metabolism of bile acids. J. Lipid Res. 2015, 56, 1085–1099. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.Q.H.; Neuschwander-Tetri, B.A.; Portincasa, P. The Biliary System, 2nd ed.; Morgan & Claypool Life Sciences: San Rafael, CA, USA, 2017; Volume 8, p. 178. [Google Scholar]
- Sahin, M.; Kayadibi, H. Importance of the bile acid receptors in different metabolisms. Integr. Obes. Diabetes 2017. [Google Scholar] [CrossRef] [Green Version]
- Bloch, K.; Berg, B.N.; Rittenberg, D. The biological conversion of cholesterol to cholic acid. J. Biol. Chem 1943, 149, 3. [Google Scholar]
- Chiang, J.Y. Regulation of bile acid synthesis. Front. Biosci. A J. Virtual Libr. 1998, 3, 176–193. [Google Scholar] [CrossRef]
- Li, T.; Chiang, J.Y. Bile acid signaling in metabolic disease and drug therapy. Pharm. Rev. 2014, 66, 948–983. [Google Scholar] [CrossRef] [Green Version]
- Russell, D.W. The enzymes, regulation, and genetics of bile acid synthesis. Annu. Rev. Biochem. 2003, 72, 137–174. [Google Scholar] [CrossRef] [Green Version]
- Dowling, R.H. The enterohepatic circulation of bile acids as they relate to lipid disorders. J. Clin. Pathol. Suppl. 1973, 5, 59. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.; Zhao, R.; Zhou, X.; Liang, X.; Campbell, D.J.; Zhang, X.; Zhang, L.; Shi, R.; Wang, G.; Pandak, W.M.; et al. Conjugated bile acids promote cholangiocarcinoma cell invasive growth through activation of sphingosine 1-phosphate receptor 2. Hepatology 2014, 60, 908–918. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, T.; Miyamoto, Y.; Nakamura, T.; Tamai, Y.; Okada, H.; Sugiyama, E.; Nakamura, T.; Itadani, H.; Tanaka, K. Identification of membrane-type receptor for bile acids (M-BAR). Biochem. Biophys. Res. Commun. 2002, 298, 714–719. [Google Scholar] [CrossRef]
- Guo, G.L.; Lambert, G.; Negishi, M.; Ward, J.M.; Brewer, H.B., Jr.; Kliewer, S.A.; Gonzalez, F.J.; Sinal, C.J. Complementary roles of farnesoid X receptor, pregnane X receptor, and constitutive androstane receptor in protection against bile acid toxicity. J. Biol. Chem. 2003, 278, 45062–45071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ory, D.S. Nuclear receptor signaling in the control of cholesterol homeostasis: Have the orphans found a home? Circ. Res. 2004, 95, 660–670. [Google Scholar] [CrossRef] [Green Version]
- Inagaki, T.; Choi, M.; Moschetta, A.; Peng, L.; Cummins, C.L.; McDonald, J.G.; Luo, G.; Jones, S.A.; Goodwin, B.; Richardson, J.A.; et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. 2005, 2, 217–225. [Google Scholar] [CrossRef] [Green Version]
- Garruti, G.; Wang, H.H.; Bonfrate, L.; de Bari, O.; Wang, D.Q.; Portincasa, P. A pleiotropic role for the orphan nuclear receptor small heterodimer partner in lipid homeostasis and metabolic pathways. J. Lipids 2012, 2012, 304292. [Google Scholar] [CrossRef] [Green Version]
- Ponz De Leon, M.; Murphy, G.M.; Dowling, R.H. Physiological factors influencing serum bile acid levels. Gut 1978, 19, 32–39. [Google Scholar] [CrossRef] [Green Version]
- Schalm, S.W.; LaRusso, N.F.; Hofmann, A.F.; Hoffman, N.E.; van Berge-Henegouwen, G.P.; Korman, M.G. Diurnal serum levels of primary conjugated bile acids. Assessment by specific radioimmunoassays for conjugates of cholic and chenodeoxycholic acid. Gut 1978, 19, 1006–1014. [Google Scholar] [CrossRef] [Green Version]
- LaRusso, N.F.; Korman, M.G.; Hoffman, N.E.; Hofmann, A.F. Dynamics of the enterohepatic circulation of bile acids. Postprandial serum concentrations of conjugates of cholic acid in health, cholecystectomized patients, and patients with bile acid malabsorption. New Engl. J. Med. 1974, 291, 689–692. [Google Scholar] [CrossRef]
- Trauner, M.; Boyer, J.L. Bile salt transporters: Molecular characterization, function, and regulation. Physiol. Rev. 2003, 83, 633–671. [Google Scholar] [CrossRef] [Green Version]
- Brighton, C.A.; Rievaj, J.; Kuhre, R.E.; Glass, L.L.; Schoonjans, K.; Holst, J.J.; Gribble, F.M.; Reimann, F. Bile acids trigger GLP-1 release predominantly by accessing basolaterally located G protein-coupled bile acid receptors. Endocrinology 2015, 156, 3961–3970. [Google Scholar] [CrossRef] [Green Version]
- Dawson, P.A.; Lan, T.; Rao, A. Bile acid transporters. J. Lipid Res. 2009, 50, 2340–2357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kemper, J.K. Regulation of FXR transcriptional activity in health and disease: Emerging roles of FXR cofactors and post-translational modifications. Biochim. Biophys. Acta 2011, 1812, 842–850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodwin, B.; Jones, S.A.; Price, R.R.; Watson, M.A.; McKee, D.D.; Moore, L.B.; Galardi, C.; Wilson, J.G.; Lewis, M.C.; Roth, M.E.; et al. A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol. Cell 2000, 6, 517–526. [Google Scholar] [CrossRef]
- Lu, T.T.; Makishima, M.; Repa, J.J.; Schoonjans, K.; Kerr, T.A.; Auwerx, J.; Mangelsdorf, D.J. Molecular basis for feedback regulation of bile acid synthesis by nuclear receptors. Mol. Cell 2000, 6, 507–515. [Google Scholar] [CrossRef]
- Nishimaki-Mogami, T.; Une, M.; Fujino, T.; Sato, Y.; Tamehiro, N.; Kawahara, Y.; Shudo, K.; Inoue, K. Identification of intermediates in the bile acid synthetic pathway as ligands for the farnesoid X receptor. J. Lipid Res. 2004, 45, 1538–1545. [Google Scholar] [CrossRef] [Green Version]
- Wagner, M.; Zollner, G.; Trauner, M. New molecular insights into the mechanisms of cholestasis. J. Hepatol. 2009, 51, 565–580. [Google Scholar] [CrossRef] [Green Version]
- Chiang, J.Y. Bile acids: Regulation of synthesis. J. Lipid Res. 2009, 50, 1955–1966. [Google Scholar] [CrossRef] [Green Version]
- Csanaky, I.L.; Aleksunes, L.M.; Tanaka, Y.; Klaassen, C.D. Role of hepatic transporters in prevention of bile acid toxicity after partial hepatectomy in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 297, 419–433. [Google Scholar] [CrossRef] [Green Version]
- Geier, A.; Wagner, M.; Dietrich, C.G.; Trauner, M. Principles of hepatic organic anion transporter regulation during cholestasis, inflammation and liver regeneration. Biochim. Biophys. Acta 2007, 1773, 283–308. [Google Scholar] [CrossRef] [Green Version]
- Ticho, A.L.; Malhotra, P.; Dudeja, P.K.; Gill, R.K.; Alrefai, W.A. Intestinal absorption of bile acids in health and disease. Compr. Physiol. 2019, 10, 21–56. [Google Scholar] [CrossRef] [PubMed]
- Visschers, R.G.; Luyer, M.D.; Schaap, F.G.; Olde Damink, S.W.; Soeters, P.B. The gut-liver axis. Curr. Opin. Clin. Nutr. Metab. Care 2013, 16, 576–581. [Google Scholar] [CrossRef] [PubMed]
- Kir, S.; Kliewer, S.A.; Mangelsdorf, D.J. Roles of FGF19 in liver metabolism. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 139–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goetz, R.; Beenken, A.; Ibrahimi, O.A.; Kalinina, J.; Olsen, S.K.; Eliseenkova, A.V.; Xu, C.; Neubert, T.A.; Zhang, F.; Linhardt, R.J.; et al. Molecular insights into the klotho-dependent, endocrine mode of action of fibroblast growth factor 19 subfamily members. Mol. Cell. Biol. 2007, 27, 3417–3428. [Google Scholar] [CrossRef] [Green Version]
- Owen, B.M.; Mangelsdorf, D.J.; Kliewer, S.A. Tissue-specific actions of the metabolic hormones FGF15/19 and FGF21. Trends Endocrinol. Metab. Tem. 2015, 26, 22–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landrier, J.F.; Eloranta, J.J.; Vavricka, S.R.; Kullak-Ublick, G.A. The nuclear receptor for bile acids, FXR, transactivates human organic solute transporter-alpha and -beta genes. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, 476–485. [Google Scholar] [CrossRef] [Green Version]
- Plass, J.R.; Mol, O.; Heegsma, J.; Geuken, M.; Faber, K.N.; Jansen, P.L.; Muller, M. Farnesoid X receptor and bile salts are involved in transcriptional regulation of the gene encoding the human bile salt export pump. Hepatology 2002, 35, 589–596. [Google Scholar] [CrossRef]
- Denson, L.A.; Sturm, E.; Echevarria, W.; Zimmerman, T.L.; Makishima, M.; Mangelsdorf, D.J.; Karpen, S.J. The orphan nuclear receptor, shp, mediates bile acid-induced inhibition of the rat bile acid transporter, ntcp. Gastroenterology 2001, 121, 140–147. [Google Scholar] [CrossRef]
- Inagaki, T.; Moschetta, A.; Lee, Y.K.; Peng, L.; Zhao, G.; Downes, M.; Yu, R.T.; Shelton, J.M.; Richardson, J.A.; Repa, J.J.; et al. Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc. Natl. Acad. Sci. USA 2006, 103, 3920–3925. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Jiang, C.; Krausz, K.W.; Li, Y.; Albert, I.; Hao, H.; Fabre, K.M.; Mitchell, J.B.; Patterson, A.D.; Gonzalez, F.J. Microbiome remodelling leads to inhibition of intestinal farnesoid X receptor signalling and decreased obesity. Nat. Commun 2013, 4, 2384. [Google Scholar] [CrossRef]
- Parseus, A.; Sommer, N.; Sommer, F.; Caesar, R.; Molinaro, A.; Stahlman, M.; Greiner, T.U.; Perkins, R.; Backhed, F. Microbiota-induced obesity requires farnesoid X receptor. Gut 2017, 66, 429–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, C.; Gioiello, A.; Noriega, L.; Strehle, A.; Oury, J.; Rizzo, G.; Macchiarulo, A.; Yamamoto, H.; Mataki, C.; Pruzanski, M.; et al. TGR5-mediated bile acid sensing controls glucose homeostasis. Cell Metab. 2009, 10, 167–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katsuma, S.; Hirasawa, A.; Tsujimoto, G. Bile acids promote glucagon-like peptide-1 secretion through TGR5 in a murine enteroendocrine cell line STC-1. Biochem. Biophys. Res. Commun. 2005, 329, 386–390. [Google Scholar] [CrossRef] [PubMed]
- Keitel, V.; Haussinger, D. Role of TGR5 (GPBAR1) in Liver Disease. Semin. Liver Dis. 2018, 38, 333–339. [Google Scholar] [CrossRef]
- Jourdainne, V.; Pean, N.; Doignon, I.; Humbert, L.; Rainteau, D.; Tordjmann, T. The bile acid receptor TGR5 and liver regeneration. Dig. Dis. 2015, 33, 319–326. [Google Scholar] [CrossRef]
- Pean, N.; Doignon, I.; Garcin, I.; Besnard, A.; Julien, B.; Liu, B.; Branchereau, S.; Spraul, A.; Guettier, C.; Humbert, L.; et al. The receptor TGR5 protects the liver from bile acid overload during liver regeneration in mice. Hepatology 2013, 58, 1451–1460. [Google Scholar] [CrossRef]
- Di Ciaula, A.; Garruti, G.; Wang, D.Q.; Portincasa, P. Cholecystectomy and risk of metabolic syndrome. Eur. J. Intern. Med. 2018, 53, 3–11. [Google Scholar] [CrossRef]
- Garruti, G.; Di Ciaula, A.; Wang, H.H.; Wang, D.Q.; Portincasa, P. Cross-talk between bile acids and gastro-intestinal and thermogenic hormones: Clues from bariatric surgery. Ann. Hepatol. 2017, 16, s68–s82. [Google Scholar] [CrossRef]
- Garruti, G.; Wang, D.Q.; Di Ciaula, A.; Portincasa, P. Cholecystectomy: A way forward and back to metabolic syndrome? Lab. Invest. 2018, 98, 4–6. [Google Scholar] [CrossRef] [Green Version]
- Keitel, V.; Reinehr, R.; Gatsios, P.; Rupprecht, C.; Gorg, B.; Selbach, O.; Haussinger, D.; Kubitz, R. The G-protein coupled bile salt receptor TGR5 is expressed in liver sinusoidal endothelial cells. Hepatology 2007, 45, 695–704. [Google Scholar] [CrossRef]
- Kida, T.; Omori, K.; Hori, M.; Ozaki, H.; Murata, T. Stimulation of G protein-coupled bile acid receptor enhances vascular endothelial barrier function via activation of protein kinase A and Rac1. J. Pharmacol. Exp. Ther. 2014, 348, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Kida, T.; Tsubosaka, Y.; Hori, M.; Ozaki, H.; Murata, T. Bile acid receptor TGR5 agonism induces NO production and reduces monocyte adhesion in vascular endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1663–1669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renga, B.; Cipriani, S.; Carino, A.; Simonetti, M.; Zampella, A.; Fiorucci, S. Reversal of endothelial dysfunction by GPBAR1 agonism in portal hypertension involves a AKT/FOXOA1 dependent regulation of H2S generation and endothelin-1. PLoS ONE 2015, 10, e0141082. [Google Scholar] [CrossRef] [Green Version]
- Klindt, C.; Reich, M.; Hellwig, B.; Stindt, J.; Rahnenfuhrer, J.; Hengstler, J.G.; Kohrer, K.; Schoonjans, K.; Haussinger, D.; Keitel, V. The G protein-coupled bile acid receptor TGR5 (Gpbar1) modulates endothelin-1 signaling in liver. Cells 2019, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pols, T.W.; Nomura, M.; Harach, T.; Lo Sasso, G.; Oosterveer, M.H.; Thomas, C.; Rizzo, G.; Gioiello, A.; Adorini, L.; Pellicciari, R.; et al. TGR5 activation inhibits atherosclerosis by reducing macrophage inflammation and lipid loading. Cell Metab. 2011, 14, 747–757. [Google Scholar] [CrossRef] [Green Version]
- Yoneno, K.; Hisamatsu, T.; Shimamura, K.; Kamada, N.; Ichikawa, R.; Kitazume, M.T.; Mori, M.; Uo, M.; Namikawa, Y.; Matsuoka, K.; et al. TGR5 signalling inhibits the production of pro-inflammatory cytokines by in vitro differentiated inflammatory and intestinal macrophages in Crohn’s disease. Immunology 2013, 139, 19–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reich, M.; Deutschmann, K.; Sommerfeld, A.; Klindt, C.; Kluge, S.; Kubitz, R.; Ullmer, C.; Knoefel, W.T.; Herebian, D.; Mayatepek, E.; et al. TGR5 is essential for bile acid-dependent cholangiocyte proliferation In Vivo and In Vitro. Gut 2016, 65, 487–501. [Google Scholar] [CrossRef] [PubMed]
- Michalopoulos, G.K. Principles of liver regeneration and growth homeostasis. Compr. Physiol. 2013, 3, 485–513. [Google Scholar] [CrossRef]
- Taub, R. Liver regeneration: From myth to mechanism. Nat. Rev. Mol. Cell Biol. 2004, 5, 836–847. [Google Scholar] [CrossRef]
- Guo, C.; Xie, S.; Chi, Z.; Zhang, J.; Liu, Y.; Zhang, L.; Zheng, M.; Zhang, X.; Xia, D.; Ke, Y. Bile acids control inflammation and metabolic disorder through inhibition of NLRP3 inflammasome. Immunity 2016, 45, 802–816. [Google Scholar] [CrossRef] [Green Version]
- Keitel, V.; Stindt, J.; Haussinger, D. Bile Acid-activated receptors: GPBAR1 (TGR5) and other G protein-coupled receptors. Handb. Exp. Pharmacol. 2019, 256, 19–49. [Google Scholar] [CrossRef] [PubMed]
- Jozefczuk, E.; Guzik, T.J.; Siedlinski, M. Significance of sphingosine-1-phosphate in cardiovascular physiology and pathology. Pharmacol. Res. Off. J. Ital. Pharmacol. Soc. 2020, 156, 104793. [Google Scholar] [CrossRef] [PubMed]
- Karimian, G.; Buist-Homan, M.; Schmidt, M.; Tietge, U.J.; de Boer, J.F.; Klappe, K.; Kok, J.W.; Combettes, L.; Tordjmann, T.; Faber, K.N.; et al. Sphingosine kinase-1 inhibition protects primary rat hepatocytes against bile salt-induced apoptosis. Biochim. Biophys. Acta 2013, 1832, 1922–1929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, T.; Kuriyama, N.; Kato, H.; Matsuda, A.; Mizuno, S.; Usui, M.; Sakurai, H.; Isaji, S. Sinusoidal protection by sphingosine-1-phosphate receptor 1 agonist in liver ischemia-reperfusion injury. J. Surg. Res. 2018, 222, 139–152. [Google Scholar] [CrossRef] [Green Version]
- Sarkisyan, G.; Gay, L.J.; Nguyen, N.; Felding, B.H.; Rosen, H. Host endothelial S1PR1 regulation of vascular permeability modulates tumor growth. Am. J. Physiol. Cell Physiol. 2014, 307, 14–24. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Yue, S.; Li, C.; Yang, L.; You, H.; Li, L. Essential roles of sphingosine 1-phosphate receptor types 1 and 3 in human hepatic stellate cells motility and activation. J. Cell. Physiol. 2011, 226, 2370–2377. [Google Scholar] [CrossRef]
- Wang, Y.; Aoki, H.; Yang, J.; Peng, K.; Liu, R.; Li, X.; Qiang, X.; Sun, L.; Gurley, E.C.; Lai, G.; et al. The role of sphingosine 1-phosphate receptor 2 in bile-acid-induced cholangiocyte proliferation and cholestasis-induced liver injury in mice. Hepatology 2017, 65, 2005–2018. [Google Scholar] [CrossRef] [Green Version]
- Hou, L.; Yang, L.; Chang, N.; Zhao, X.; Zhou, X.; Dong, C.; Liu, F.; Yang, L.; Li, L. Macrophage sphingosine 1-phosphate receptor 2 blockade attenuates liver inflammation and fibrogenesis triggered by NLRP3 inflammasome. Front. Immunol. 2020, 11, 1149. [Google Scholar] [CrossRef]
- Yang, L.; Han, Z.; Tian, L.; Mai, P.; Zhang, Y.; Wang, L.; Li, L. Sphingosine 1-phosphate receptor 2 and 3 mediate bone marrow-derived monocyte/macrophage motility in cholestatic liver injury in mice. Sci Rep. 2015, 5, 13423. [Google Scholar] [CrossRef]
- Barrasa, J.I.; Olmo, N.; Lizarbe, M.A.; Turnay, J. Bile acids in the colon, from healthy to cytotoxic molecules. Toxicol. Vitr. Int. J. Publ. Assoc. Bibra 2013, 27, 964–977. [Google Scholar] [CrossRef]
- Pols, T.W.; Noriega, L.G.; Nomura, M.; Auwerx, J.; Schoonjans, K. The bile acid membrane receptor TGR5 as an emerging target in metabolism and inflammation. J. Hepatol. 2011, 54, 1263–1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiorucci, S.; Mencarelli, A.; Palladino, G.; Cipriani, S. Bile-acid-activated receptors: Targeting TGR5 and farnesoid-X-receptor in lipid and glucose disorders. Trends Pharmacol. Sci. 2009, 30, 570–580. [Google Scholar] [CrossRef] [PubMed]
- Lieu, T.; Jayaweera, G.; Bunnett, N.W. GPBA: A GPCR for bile acids and an emerging therapeutic target for disorders of digestion and sensation. Br. J. Pharmacol. 2014, 171, 1156–1166. [Google Scholar] [CrossRef] [PubMed]
- de Aguiar Vallim, T.Q.; Tarling, E.J.; Edwards, P.A. Pleiotropic roles of bile acids in metabolism. Cell Metab. 2013, 17, 657–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawamata, Y.; Fujii, R.; Hosoya, M.; Harada, M.; Yoshida, H.; Miwa, M.; Fukusumi, S.; Habata, Y.; Itoh, T.; Shintani, Y.; et al. A G protein-coupled receptor responsive to bile acids. J. Biol. Chem. 2003, 278, 9435–9440. [Google Scholar] [CrossRef] [Green Version]
- Keitel, V.; Donner, M.; Winandy, S.; Kubitz, R.; Haussinger, D. Expression and function of the bile acid receptor TGR5 in Kupffer cells. Biochem. Biophys. Res. Commun. 2008, 372, 78–84. [Google Scholar] [CrossRef]
- Merlen, G.; Kahale, N.; Ursic-Bedoya, J.; Bidault-Jourdainne, V.; Simerabet, H.; Doignon, I.; Tanfin, Z.; Garcin, I.; Pean, N.; Gautherot, J.; et al. TGR5-dependent hepatoprotection through the regulation of biliary epithelium barrier function. Gut 2020, 69, 146–157. [Google Scholar] [CrossRef]
- Maruyama, T.; Tanaka, K.; Suzuki, J.; Miyoshi, H.; Harada, N.; Nakamura, T.; Miyamoto, Y.; Kanatani, A.; Tamai, Y. Targeted disruption of G protein-coupled bile acid receptor 1 (Gpbar1/M-Bar) in mice. J. Endocrinol. 2006, 191, 197–205. [Google Scholar] [CrossRef] [Green Version]
- Thomas, C.; Pellicciari, R.; Pruzanski, M.; Auwerx, J.; Schoonjans, K. Targeting bile-acid signalling for metabolic diseases. Nat. Rev. Drug Discov. 2008, 7, 678–693. [Google Scholar] [CrossRef]
- Sato, H.; Macchiarulo, A.; Thomas, C.; Gioiello, A.; Une, M.; Hofmann, A.F.; Saladin, R.; Schoonjans, K.; Pellicciari, R.; Auwerx, J. Novel potent and selective bile acid derivatives as TGR5 agonists: Biological screening, structure-activity relationships, and molecular modeling studies. J. Med. Chem. 2008, 51, 1831–1841. [Google Scholar] [CrossRef]
- Hong, J.; Behar, J.; Wands, J.; Resnick, M.; Wang, L.J.; DeLellis, R.A.; Lambeth, D.; Souza, R.F.; Spechler, S.J.; Cao, W. Role of a novel bile acid receptor TGR5 in the development of oesophageal adenocarcinoma. Gut 2010, 59, 170–180. [Google Scholar] [CrossRef] [PubMed]
- Keitel, V.; Gorg, B.; Bidmon, H.J.; Zemtsova, I.; Spomer, L.; Zilles, K.; Haussinger, D. The bile acid receptor TGR5 (Gpbar-1) acts as a neurosteroid receptor in brain. Glia 2010, 58, 1794–1805. [Google Scholar] [CrossRef] [PubMed]
- Iracheta-Vellve, A.; Calenda, C.D.; Petrasek, J.; Ambade, A.; Kodys, K.; Adorini, L.; Szabo, G. FXR and TGR5 agonists ameliorate liver injury, steatosis, and inflammation after binge or prolonged alcohol feeding in mice. Hepatol. Commun. 2018, 2, 1379–1391. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.D.; Chen, W.D.; Yu, D.; Forman, B.M.; Huang, W. The G-protein-coupled bile acid receptor, Gpbar1 (TGR5), negatively regulates hepatic inflammatory response through antagonizing nuclear factor kappa light-chain enhancer of activated B cells (NF-kappaB) in mice. Hepatology 2011, 54, 1421–1432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, H.; Sun, X.; Liu, R.; Chen, Z.; Lin, Z.; Yang, Y.; Zhang, M.; Liu, P.; Quan, S.; Huang, H. Gentiopicroside activates the bile acid receptor Gpbar1 (TGR5) to repress NF-kappaB pathway and ameliorate diabetic nephropathy. Pharmacol. Res. Off. J. Ital. Pharmacol. Soc. 2020, 151, 104559. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; Zhang, Q.; Qi, H.; Wu, L.; Li, Y.; Yu, D.; Huang, W.; Chen, W.D.; Wang, Y.D. The G-protein-coupled bile acid receptor Gpbar1 (TGR5) protects against renal inflammation and renal cancer cell proliferation and migration through antagonizing NF-kappaB and STAT3 signaling pathways. Oncotarget 2017, 8, 54378–54387. [Google Scholar] [CrossRef] [Green Version]
- Guo, C.; Qi, H.; Yu, Y.; Zhang, Q.; Su, J.; Yu, D.; Huang, W.; Chen, W.D.; Wang, Y.D. The G-protein-coupled bile acid receptor Gpbar1 (TGR5) inhibits gastric inflammation through antagonizing NF-kappaB signaling pathway. Front. Pharm. 2015, 6, 287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, D.P.; Rajagopal, S.; Mahavadi, S.; Mirshahi, F.; Grider, J.R.; Murthy, K.S.; Sanyal, A.J. Activation of transmembrane bile acid receptor TGR5 stimulates insulin secretion in pancreatic β cells. Biochem. Biophys. Res. Commun. 2012, 427, 600–605. [Google Scholar] [CrossRef] [Green Version]
- Kumar, D.P.; Asgharpour, A.; Mirshahi, F.; Park, S.H.; Liu, S.; Imai, Y.; Nadler, J.L.; Grider, J.R.; Murthy, K.S.; Sanyal, A.J. Activation of transmembrane bile acid receptor TGR5 modulates pancreatic islet α cells to promote glucose homeostasis. J. Biol. Chem. 2016, 291, 6626–6640. [Google Scholar] [CrossRef] [Green Version]
- Ding, L.; Sousa, K.M.; Jin, L.; Dong, B.; Kim, B.W.; Ramirez, R.; Xiao, Z.; Gu, Y.; Yang, Q.; Wang, J.; et al. Vertical sleeve gastrectomy activates GPBAR-1/TGR5 to sustain weight loss, improve fatty liver, and remit insulin resistance in mice. Hepatology 2016, 64, 760–773. [Google Scholar] [CrossRef] [Green Version]
- McGavigan, A.K.; Garibay, D.; Henseler, Z.M.; Chen, J.; Bettaieb, A.; Haj, F.G.; Ley, R.E.; Chouinard, M.L.; Cummings, B.P. TGR5 contributes to glucoregulatory improvements after vertical sleeve gastrectomy in mice. Gut 2017, 66, 226–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, M.; Houten, S.M.; Mataki, C.; Christoffolete, M.A.; Kim, B.W.; Sato, H.; Messaddeq, N.; Harney, J.W.; Ezaki, O.; Kodama, T.; et al. Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature 2006, 439, 484–489. [Google Scholar] [CrossRef] [PubMed]
- Velazquez-Villegas, L.A.; Perino, A.; Lemos, V.; Zietak, M.; Nomura, M.; Pols, T.W.H.; Schoonjans, K. TGR5 signalling promotes mitochondrial fission and beige remodelling of white adipose tissue. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, S.Y.; Ouyang, X.; Chen, Y.; Soroka, C.J.; Wang, J.; Mennone, A.; Wang, Y.; Mehal, W.Z.; Jain, D.; Boyer, J.L. Bile acids initiate cholestatic liver injury by triggering a hepatocyte-specific inflammatory response. JCI Insight 2017, 2, e90780. [Google Scholar] [CrossRef] [Green Version]
- Schaap, F.G.; Trauner, M.; Jansen, P.L. Bile acid receptors as targets for drug development. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 55–67. [Google Scholar] [CrossRef]
- Thulesen, J. Glucagon-like peptide 2 (GLP-2), an intestinotrophic mediator. Curr. Protein Pept. Sci. 2004, 5, 51–65. [Google Scholar] [CrossRef]
- Liu, N.; Zhao, J.; Wang, J.; Teng, H.; Fu, Y.; Yuan, H. Farnesoid X receptor ligand CDCA suppresses human prostate cancer cells growth by inhibiting lipid metabolism via targeting sterol response element binding protein 1. Am. J. Transl. Res. 2016, 8, 5118–5124. [Google Scholar]
- Baghdasaryan, A.; Claudel, T.; Gumhold, J.; Silbert, D.; Adorini, L.; Roda, A.; Vecchiotti, S.; Gonzalez, F.J.; Schoonjans, K.; Strazzabosco, M.; et al. Dual farnesoid X receptor/TGR5 agonist INT-767 reduces liver injury in the Mdr2-/- (Abcb4-/-) mouse cholangiopathy model by promoting biliary HCO(-)(3) output. Hepatology 2011, 54, 1303–1312. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Holmstrom, S.R.; Kir, S.; Umetani, M.; Schmidt, D.R.; Kliewer, S.A.; Mangelsdorf, D.J. The G protein-coupled bile acid receptor, TGR5, stimulates gallbladder filling. Mol. Endocrinol 2011, 25, 1066–1071. [Google Scholar] [CrossRef] [Green Version]
- Vassileva, G.; Golovko, A.; Markowitz, L.; Abbondanzo, S.J.; Zeng, M.; Yang, S.; Hoos, L.; Tetzloff, G.; Levitan, D.; Murgolo, N.J.; et al. Targeted deletion of Gpbar1 protects mice from cholesterol gallstone formation. Biochem. J. 2006, 398, 423–430. [Google Scholar] [CrossRef] [Green Version]
- Donepudi, A.C.; Boehme, S.; Li, F.; Chiang, J.Y. G-protein-coupled bile acid receptor plays a key role in bile acid metabolism and fasting-induced hepatic steatosis in mice. Hepatology 2017, 65, 813–827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pathak, P.; Xie, C.; Nichols, R.G.; Ferrell, J.M.; Boehme, S.; Krausz, K.W.; Patterson, A.D.; Gonzalez, F.J.; Chiang, J.Y.L. Intestine farnesoid X receptor agonist and the gut microbiota activate G-protein bile acid receptor-1 signaling to improve metabolism. Hepatology 2018, 68, 1574–1588. [Google Scholar] [CrossRef] [PubMed]
- Keitel, V.; Cupisti, K.; Ullmer, C.; Knoefel, W.T.; Kubitz, R.; Haussinger, D. The membrane-bound bile acid receptor TGR5 is localized in the epithelium of human gallbladders. Hepatology 2009, 50, 861–870. [Google Scholar] [CrossRef] [PubMed]
- Debray, D.; Rainteau, D.; Barbu, V.; Rouahi, M.; El Mourabit, H.; Lerondel, S.; Rey, C.; Humbert, L.; Wendum, D.; Cottart, C.H.; et al. Defects in gallbladder emptying and bile Acid homeostasis in mice with cystic fibrosis transmembrane conductance regulator deficiencies. Gastroenterology 2012, 142, 1581–1591.e6. [Google Scholar] [CrossRef] [Green Version]
- Roda, A.; Pellicciari, R.; Gioiello, A.; Neri, F.; Camborata, C.; Passeri, D.; De Franco, F.; Spinozzi, S.; Colliva, C.; Adorini, L.; et al. Semisynthetic bile acid FXR and TGR5 agonists: Physicochemical properties, pharmacokinetics, and metabolism in the rat. J. Pharmacol. Exp. Ther. 2014, 350, 56–68. [Google Scholar] [CrossRef] [Green Version]
- Hendrick, S.M.; Mroz, M.S.; Greene, C.M.; Keely, S.J.; Harvey, B.J. Bile acids stimulate chloride secretion through CFTR and calcium-activated Cl- channels in Calu-3 airway epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 307, 407–418. [Google Scholar] [CrossRef]
- Li, S.; Qiu, M.; Kong, Y.; Zhao, X.; Choi, H.J.; Reich, M.; Bunkelman, B.H.; Liu, Q.; Hu, S.; Han, M.; et al. Bile acid G protein-coupled membrane receptor TGR5 modulates aquaporin 2-mediated water homeostasis. J. Am. Soc. Nephrol. 2018, 29, 2658–2670. [Google Scholar] [CrossRef] [Green Version]
- Masyuk, A.I.; Huang, B.Q.; Radtke, B.N.; Gajdos, G.B.; Splinter, P.L.; Masyuk, T.V.; Gradilone, S.A.; LaRusso, N.F. Ciliary subcellular localization of TGR5 determines the cholangiocyte functional response to bile acid signaling. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 304, 1013–1024. [Google Scholar] [CrossRef]
- Keitel, V.; Ullmer, C.; Haussinger, D. The membrane-bound bile acid receptor TGR5 (Gpbar-1) is localized in the primary cilium of cholangiocytes. Biol. Chem. 2010, 391, 785–789. [Google Scholar] [CrossRef]
- Gradilone, S.A.; Masyuk, A.I.; Splinter, P.L.; Banales, J.M.; Huang, B.Q.; Tietz, P.S.; Masyuk, T.V.; Larusso, N.F. Cholangiocyte cilia express TRPV4 and detect changes in luminal tonicity inducing bicarbonate secretion. Proc. Natl. Acad. Sci. USA 2007, 104, 19138–19143. [Google Scholar] [CrossRef] [Green Version]
- Hohenester, S.; Wenniger, L.M.; Paulusma, C.C.; van Vliet, S.J.; Jefferson, D.M.; Elferink, R.P.; Beuers, U. A biliary HCO3- umbrella constitutes a protective mechanism against bile acid-induced injury in human cholangiocytes. Hepatology 2012, 55, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Morimoto, K.; Houten, S.M.; Kaneko-Iwasaki, N.; Sugizaki, T.; Horai, Y.; Mataki, C.; Sato, H.; Murahashi, K.; Arita, E.; et al. Bile acid binding resin improves metabolic control through the induction of energy expenditure. PLoS ONE 2012, 7, e38286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Ciaula, A.; Wang, D.Q.; Molina-Molina, E.; Lunardi Baccetto, R.; Calamita, G.; Palmieri, V.O.; Portincasa, P. Bile acids and cancer: Direct and environmental-dependent effects. Ann. Hepatol. 2017, 16, s87–s105. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Ma, K.; Zhang, J.; Qatanani, M.; Cuvillier, J.; Liu, J.; Dong, B.; Huang, X.; Moore, D.D. Nuclear receptor-dependent bile acid signaling is required for normal liver regeneration. Science 2006, 312, 233–236. [Google Scholar] [CrossRef]
- Meng, Z.; Wang, Y.; Wang, L.; Jin, W.; Liu, N.; Pan, H.; Liu, L.; Wagman, L.; Forman, B.M.; Huang, W. FXR regulates liver repair after CCl4-induced toxic injury. Mol. Endocrinol. 2010, 24, 886–897. [Google Scholar] [CrossRef] [Green Version]
- Doignon, I.; Julien, B.; Serriere-Lanneau, V.; Garcin, I.; Alonso, G.; Nicou, A.; Monnet, F.; Gigou, M.; Humbert, L.; Rainteau, D.; et al. Immediate neuroendocrine signaling after partial hepatectomy through acute portal hyperpressure and cholestasis. J. Hepatol. 2011, 54, 481–488. [Google Scholar] [CrossRef]
- Naugler, W.E. Bile acid flux is necessary for normal liver regeneration. PLoS ONE 2014, 9, e97426. [Google Scholar] [CrossRef] [Green Version]
- Hoekstra, L.T.; Rietkerk, M.; van Lienden, K.P.; van den Esschert, J.W.; Schaap, F.G.; van Gulik, T.M. Bile salts predict liver regeneration in rabbit model of portal vein embolization. J. Surg. Res. 2012, 178, 773–778. [Google Scholar] [CrossRef] [Green Version]
- Hoekstra, L.T.; van Lienden, K.P.; Schaap, F.G.; Chamuleau, R.A.; Bennink, R.J.; van Gulik, T.M. Can plasma bile salt, triglycerides, and apoA-V levels predict liver regeneration? World J. Surg. 2012, 36, 2901–2908. [Google Scholar] [CrossRef]
- Woolbright, B.L.; Jaeschke, H. Inflammation and cell death during cholestasis: The evolving role of bile acids. Gene Expr. 2019, 19, 215–228. [Google Scholar] [CrossRef]
- Maillette de Buy Wenniger, L.; Beuers, U. Bile salts and cholestasis. Dig. Liver Dis. 2010, 42, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Yerushalmi, B.; Dahl, R.; Devereaux, M.W.; Gumpricht, E.; Sokol, R.J. Bile acid-induced rat hepatocyte apoptosis is inhibited by antioxidants and blockers of the mitochondrial permeability transition. Hepatology 2001, 33, 616–626. [Google Scholar] [CrossRef] [PubMed]
- Grattagliano, I.; Bonfrate, L.; Diogo, C.V.; Wang, H.H.; Wang, D.Q.; Portincasa, P. Biochemical mechanisms in drug-induced liver injury: Certainties and doubts. World J. Gastroenterol. 2009, 15, 4865–4876. [Google Scholar] [CrossRef] [PubMed]
- Grattagliano, I.; De Bari, O.; Di Palo, D.; Montecucco, F.; Carbone, F.; Oliveira, P.; Wang, D.Q.H.; Portincasa, P. Mitochondria in liver diseases. In Mitochondrial Biology and Experimental Therapeutics; Oliveira, P., Ed.; Springer Nature: Cham, Switzerland, 2018; pp. 91–126. [Google Scholar] [CrossRef]
- Grattagliano, I.; Lauterburg, B.H.; Portincasa, P.; Caruso, M.L.; Vendemiale, G.; Valentini, A.M.; Palmieri, V.O.; Palasciano, G. Mitochondrial glutathione content determines the rate of liver regeneration after partial hepatectomy in eu- and hypothyroid rats. J. Hepatol. 2003, 39, 571–579. [Google Scholar] [CrossRef]
- Grattagliano, I.; Montezinho, L.P.; Oliveira, P.J.; Fruhbeck, G.; Gomez-Ambrosi, J.; Montecucco, F.; Carbone, F.; Wieckowski, M.R.; Wang, D.Q.; Portincasa, P. Targeting mitochondria to oppose the progression of nonalcoholic fatty liver disease. Biochem. Pharmacol. 2019, 160, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Reinehr, R.; Graf, D.; Haussinger, D. Bile salt-induced hepatocyte apoptosis involves epidermal growth factor receptor-dependent CD95 tyrosine phosphorylation. Gastroenterology 2003, 125, 839–853. [Google Scholar] [CrossRef]
- Miyoshi, H.; Rust, C.; Roberts, P.J.; Burgart, L.J.; Gores, G.J. Hepatocyte apoptosis after bile duct ligation in the mouse involves Fas. Gastroenterology 1999, 117, 669–677. [Google Scholar] [CrossRef]
- Allen, K.; Jaeschke, H.; Copple, B.L. Bile acids induce inflammatory genes in hepatocytes: A novel mechanism of inflammation during obstructive cholestasis. Am. J. Pathol. 2011, 178, 175–186. [Google Scholar] [CrossRef]
- Fahrner, R.; Patsenker, E.; de Gottardi, A.; Stickel, F.; Montani, M.; Stroka, D.; Candinas, D.; Beldi, G. Elevated liver regeneration in response to pharmacological reduction of elevated portal venous pressure by terlipressin after partial hepatectomy. Transplantation 2014, 97, 892–900. [Google Scholar] [CrossRef] [Green Version]
- Miura, T.; Kimura, N.; Yamada, T.; Shimizu, T.; Nanashima, N.; Yamana, D.; Hakamada, K.; Tsuchida, S. Sustained repression and translocation of Ntcp and expression of Mrp4 for cholestasis after rat 90% partial hepatectomy. J. Hepatol. 2011, 55, 407–414. [Google Scholar] [CrossRef] [Green Version]
- Starkel, P.; Shindano, T.; Horsmans, Y.; Gigot, J.F.; Fernandez-Tagarro, M.; Marin, J.J.; Monte, M.J. Foetal ‘flat’ bile acids reappear during human liver regeneration after surgery. Eur. J. Clin. Investig. 2009, 39, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Bhushan, B.; Borude, P.; Edwards, G.; Walesky, C.; Cleveland, J.; Li, F.; Ma, X.; Apte, U. Role of bile acids in liver injury and regeneration following acetaminophen overdose. Am. J. Pathol. 2013, 183, 1518–1526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naugler, W.E.; Tarlow, B.D.; Fedorov, L.M.; Taylor, M.; Pelz, C.; Li, B.; Darnell, J.; Grompe, M. Fibroblast growth factor signaling controls liver size in mice with humanized livers. Gastroenterology 2015, 149, 728–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, B.; Zhang, M.; Huang, M.; Rizzolo, D.; Armstrong, L.E.; Schumacher, J.D.; Chow, M.D.; Lee, Y.H.; Guo, G.L. FXR deficiency alters bile acid pool composition and exacerbates chronic alcohol induced liver injury. Dig. Liver Dis 2019, 51, 570–576. [Google Scholar] [CrossRef] [PubMed]
- Kong, B.; Huang, J.; Zhu, Y.; Li, G.; Williams, J.; Shen, S.; Aleksunes, L.M.; Richardson, J.R.; Apte, U.; Rudnick, D.A.; et al. Fibroblast growth factor 15 deficiency impairs liver regeneration in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 306, 893–902. [Google Scholar] [CrossRef] [Green Version]
- Uriarte, I.; Fernandez-Barrena, M.G.; Monte, M.J.; Latasa, M.U.; Chang, H.C.; Carotti, S.; Vespasiani-Gentilucci, U.; Morini, S.; Vicente, E.; Concepcion, A.R.; et al. Identification of fibroblast growth factor 15 as a novel mediator of liver regeneration and its application in the prevention of post-resection liver failure in mice. Gut 2013, 62, 899–910. [Google Scholar] [CrossRef] [PubMed]
- Kong, B.; Sun, R.; Huang, M.; Chow, M.D.; Zhong, X.B.; Xie, W.; Lee, Y.H.; Guo, G.L. Fibroblast growth factor 15-dependent and bile acid-independent promotion of liver regeneration in mice. Hepatology 2018, 68, 1961–1976. [Google Scholar] [CrossRef] [Green Version]
- Schumacher, J.D.; Kong, B.; Pan, Y.; Zhan, L.; Sun, R.; Aa, J.; Rizzolo, D.; Richardson, J.R.; Chen, A.; Goedken, M.; et al. The effect of fibroblast growth factor 15 deficiency on the development of high fat diet induced non-alcoholic steatohepatitis. Toxicol. Appl. Pharmacol. 2017, 330, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Cai, S.Y.; Boyer, J.L. Mechanisms of bile acid mediated inflammation in the liver. Mol. Asp. Med. 2017, 56, 45–53. [Google Scholar] [CrossRef]
- Carulli, N.; Bertolotti, M.; Carubbi, F.; Concari, M.; Martella, P.; Carulli, L.; Loria, P. Review article: Effect of bile salt pool composition on hepatic and biliary functions. Aliment. Pharmacol. Ther. 2000, 14, 14–18. [Google Scholar] [CrossRef]
- Ma, C.; Han, M.; Heinrich, B.; Fu, Q.; Zhang, Q.; Sandhu, M.; Agdashian, D.; Terabe, M.; Berzofsky, J.A.; Fako, V.; et al. Gut microbiome-mediated bile acid metabolism regulates liver cancer via NKT cells. Science 2018, 360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Rodriguez, J.L.; Barbier-Torres, L.; Fernandez-Alvarez, S.; Gutierrez-de Juan, V.; Monte, M.J.; Halilbasic, E.; Herranz, D.; Alvarez, L.; Aspichueta, P.; Marin, J.J.; et al. SIRT1 controls liver regeneration by regulating bile acid metabolism through farnesoid X receptor and mammalian target of rapamycin signaling. Hepatology 2014, 59, 1972–1983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilgenkrantz, H.; Tordjmann, T. Bile acids and FGF receptors: Orchestrators of optimal liver regeneration. Gut 2015, 64, 1351–1352. [Google Scholar] [CrossRef] [PubMed]
- Padrissa-Altes, S.; Bachofner, M.; Bogorad, R.L.; Pohlmeier, L.; Rossolini, T.; Bohm, F.; Liebisch, G.; Hellerbrand, C.; Koteliansky, V.; Speicher, T.; et al. Control of hepatocyte proliferation and survival by Fgf receptors is essential for liver regeneration in mice. Gut 2015, 64, 1444–1453. [Google Scholar] [CrossRef]
- Park, Y.J.; Qatanani, M.; Chua, S.S.; LaRey, J.L.; Johnson, S.A.; Watanabe, M.; Moore, D.D.; Lee, Y.K. Loss of orphan receptor small heterodimer partner sensitizes mice to liver injury from obstructive cholestasis. Hepatology 2008, 47, 1578–1586. [Google Scholar] [CrossRef]
- Modica, S.; Petruzzelli, M.; Bellafante, E.; Murzilli, S.; Salvatore, L.; Celli, N.; Di Tullio, G.; Palasciano, G.; Moustafa, T.; Halilbasic, E.; et al. Selective activation of nuclear bile acid receptor FXR in the intestine protects mice against cholestasis. Gastroenterology 2012, 142, 355–365. [Google Scholar] [CrossRef]
- Fickert, P.; Fuchsbichler, A.; Marschall, H.U.; Wagner, M.; Zollner, G.; Krause, R.; Zatloukal, K.; Jaeschke, H.; Denk, H.; Trauner, M. Lithocholic acid feeding induces segmental bile duct obstruction and destructive cholangitis in mice. Am. J. Pathol. 2006, 168, 410–422. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, S.; Fukami, T.; Masuo, Y.; Brocker, C.N.; Xie, C.; Krausz, K.W.; Wolf, C.R.; Henderson, C.J.; Gonzalez, F.J. Cyp2c70 is responsible for the species difference in bile acid metabolism between mice and humans. J. Lipid Res. 2016, 57, 2130–2137. [Google Scholar] [CrossRef] [Green Version]
- Honda, A.; Miyazaki, T.; Iwamoto, J.; Hirayama, T.; Morishita, Y.; Monma, T.; Ueda, H.; Mizuno, S.; Sugiyama, F.; Takahashi, S.; et al. Regulation of bile acid metabolism in mouse models with hydrophobic bile acid composition. J. Lipid Res. 2020, 61, 54–69. [Google Scholar] [CrossRef]
- de Boer, J.F.; Verkade, E.; Mulder, N.L.; de Vries, H.D.; Huijkman, N.; Koehorst, M.; Boer, T.; Wolters, J.C.; Bloks, V.W.; van de Sluis, B.; et al. A human-like bile acid pool induced by deletion of hepatic Cyp2c70 modulates effects of FXR activation in mice. J. Lipid Res. 2020, 61, 291–305. [Google Scholar] [CrossRef] [Green Version]
- Hrycay, E.; Forrest, D.; Liu, L.; Wang, R.; Tai, J.; Deo, A.; Ling, V.; Bandiera, S. Hepatic bile acid metabolism and expression of cytochrome P450 and related enzymes are altered in Bsep (-/-) mice. Mol. Cell. Biochem. 2014, 389, 119–132. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A. Progressive familial intrahepatic cholestasis. J. Clin. Exp. Hepatol. 2014, 4, 25–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuchs, C.D.; Paumgartner, G.; Wahlstrom, A.; Schwabl, P.; Reiberger, T.; Leditznig, N.; Stojakovic, T.; Rohr-Udilova, N.; Chiba, P.; Marschall, H.U.; et al. Metabolic preconditioning protects BSEP/ABCB11(-/-) mice against cholestatic liver injury. J. Hepatol. 2017, 66, 95–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.S.; Kimura, A.; Wu, J.F.; Ni, Y.H.; Hsu, H.Y.; Chang, M.H.; Nittono, H.; Chen, H.L. Prognostic roles of tetrahydroxy bile acids in infantile intrahepatic cholestasis. J. Lipid Res. 2017, 58, 607–614. [Google Scholar] [CrossRef] [Green Version]
- Lee, N.P. The blood-biliary barrier, tight junctions and human liver diseases. Adv. Exp. Med. Biol. 2012, 763, 171–185. [Google Scholar] [CrossRef]
- Sato, K.; Meng, F.; Giang, T.; Glaser, S.; Alpini, G. Mechanisms of cholangiocyte responses to injury. Biochim. Biophys Acta Mol. Basis Dis. 2018, 1864, 1262–1269. [Google Scholar] [CrossRef]
- Anderson, J.M.; Van Itallie, C.M. Physiology and function of the tight junction. Cold Spring Harb. Perspect. Biol. 2009, 1, a002584. [Google Scholar] [CrossRef]
- Van Itallie, C.M.; Anderson, J.M. Architecture of tight junctions and principles of molecular composition. Semin. Cell Dev. Biol. 2014, 36, 157–165. [Google Scholar] [CrossRef] [Green Version]
- Iden, S.; Misselwitz, S.; Peddibhotla, S.S.; Tuncay, H.; Rehder, D.; Gerke, V.; Robenek, H.; Suzuki, A.; Ebnet, K. aPKC phosphorylates JAM-A at Ser285 to promote cell contact maturation and tight junction formation. J. Cell Biol. 2012, 196, 623–639. [Google Scholar] [CrossRef] [Green Version]
- Shigetomi, K.; Ikenouchi, J. Regulation of the epithelial barrier by post-translational modifications of tight junction membrane proteins. J. Biochem. 2018, 163, 265–272. [Google Scholar] [CrossRef] [Green Version]
- Rahner, C.; Stieger, B.; Landmann, L. Structure-function correlation of tight junctional impairment after intrahepatic and extrahepatic cholestasis in rat liver. Gastroenterology 1996, 110, 1564–1578. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Imasato, M.; Yamazaki, Y.; Matsumoto, K.; Kunimoto, K.; Delpierre, J.; Meyer, K.; Zerial, M.; Kitamura, N.; Watanabe, M.; et al. Claudin-3 regulates bile canalicular paracellular barrier and cholesterol gallstone core formation in mice. J. Hepatol. 2018, 69, 1308–1316. [Google Scholar] [CrossRef] [PubMed]
- Boyer, J.L. The hepatobiliary paracellular pathway: A paradigm revisited. Gastroenterology 2014, 147, 965–968. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Imasato, M.; Yamazaki, Y.; Tanaka, H.; Watanabe, M.; Eguchi, H.; Nagano, H.; Hikita, H.; Tatsumi, T.; Takehara, T.; et al. Claudin 2 deficiency reduces bile flow and increases susceptibility to cholesterol gallstone disease in mice. Gastroenterology 2014, 147, 1134–1145. [Google Scholar] [CrossRef] [Green Version]
- Fickert, P.; Fuchsbichler, A.; Wagner, M.; Zollner, G.; Kaser, A.; Tilg, H.; Krause, R.; Lammert, F.; Langner, C.; Zatloukal, K.; et al. Regurgitation of bile acids from leaky bile ducts causes sclerosing cholangitis in Mdr2 (Abcb4) knockout mice. Gastroenterology 2004, 127, 261–274. [Google Scholar] [CrossRef]
- Sakisaka, S.; Kawaguchi, T.; Taniguchi, E.; Hanada, S.; Sasatomi, K.; Koga, H.; Harada, M.; Kimura, R.; Sata, M.; Sawada, N.; et al. Alterations in tight junctions differ between primary biliary cirrhosis and primary sclerosing cholangitis. Hepatology 2001, 33, 1460–1468. [Google Scholar] [CrossRef]
- Sambrotta, M.; Thompson, R.J. Mutations in TJP2, encoding zona occludens 2, and liver disease. Tissue Barriers 2015, 3, e1026537. [Google Scholar] [CrossRef] [Green Version]
- Grosse, B.; Cassio, D.; Yousef, N.; Bernardo, C.; Jacquemin, E.; Gonzales, E. Claudin-1 involved in neonatal ichthyosis sclerosing cholangitis syndrome regulates hepatic paracellular permeability. Hepatology 2012, 55, 1249–1259. [Google Scholar] [CrossRef]
- Hadj-Rabia, S.; Baala, L.; Vabres, P.; Hamel-Teillac, D.; Jacquemin, E.; Fabre, M.; Lyonnet, S.; De Prost, Y.; Munnich, A.; Hadchouel, M.; et al. Claudin-1 gene mutations in neonatal sclerosing cholangitis associated with ichthyosis: A tight junction disease. Gastroenterology 2004, 127, 1386–1390. [Google Scholar] [CrossRef]
- Muller, T.; Beutler, C.; Pico, A.H.; Otten, M.; Durr, A.; Al-Abadi, H.; Guckelberger, O.; Meyer Zum Buschenfelde, D.; Johrens, K.; Volkmann, M.; et al. Increased T-helper 2 cytokines in bile from patients with IgG4-related cholangitis disrupt the tight junction-associated biliary epithelial cell barrier. Gastroenterology 2013, 144, 1116–1128. [Google Scholar] [CrossRef]
- Pradhan-Sundd, T.; Zhou, L.; Vats, R.; Jiang, A.; Molina, L.; Singh, S.; Poddar, M.; Russell, J.; Stolz, D.B.; Oertel, M.; et al. Dual catenin loss in murine liver causes tight junctional deregulation and progressive intrahepatic cholestasis. Hepatology 2018, 67, 2320–2337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raimondi, F.; Santoro, P.; Barone, M.V.; Pappacoda, S.; Barretta, M.L.; Nanayakkara, M.; Apicella, C.; Capasso, L.; Paludetto, R. Bile acids modulate tight junction structure and barrier function of Caco-2 monolayers via EGFR activation. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, 906–913. [Google Scholar] [CrossRef] [PubMed]
- Su, K.C.; Wu, Y.C.; Chen, C.S.; Hung, M.H.; Hsiao, Y.H.; Tseng, C.M.; Chang, S.C.; Lee, Y.C.; Perng, D.W. Bile acids increase alveolar epithelial permeability via mitogen-activated protein kinase, cytosolic phospholipase A2, cyclooxygenase-2, prostaglandin E2 and junctional proteins. Respirology 2013, 18, 848–856. [Google Scholar] [CrossRef] [PubMed]
- Cipriani, S.; Mencarelli, A.; Chini, M.G.; Distrutti, E.; Renga, B.; Bifulco, G.; Baldelli, F.; Donini, A.; Fiorucci, S. The bile acid receptor GPBAR-1 (TGR5) modulates integrity of intestinal barrier and immune response to experimental colitis. PLoS ONE 2011, 6, e25637. [Google Scholar] [CrossRef] [PubMed]
- Hao, H.; Cao, L.; Jiang, C.; Che, Y.; Zhang, S.; Takahashi, S.; Wang, G.; Gonzalez, F.J. Farnesoid X receptor regulation of the NLRP3 inflammasome underlies cholestasis-associated sepsis. Cell Metab. 2017, 25, 856–867.e5. [Google Scholar] [CrossRef] [Green Version]
- Gong, Z.; Zhou, J.; Zhao, S.; Tian, C.; Wang, P.; Xu, C.; Chen, Y.; Cai, W.; Wu, J. Chenodeoxycholic acid activates NLRP3 inflammasome and contributes to cholestatic liver fibrosis. Oncotarget 2016, 7, 83951–83963. [Google Scholar] [CrossRef] [Green Version]
- Misawa, T.; Takahama, M.; Kozaki, T.; Lee, H.; Zou, J.; Saitoh, T.; Akira, S. Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nature Immunology 2013, 14, 454–460. [Google Scholar] [CrossRef]
- Spirli, C.; Nathanson, M.H.; Fiorotto, R.; Duner, E.; Denson, L.A.; Sanz, J.M.; Di Virgilio, F.; Okolicsanyi, L.; Casagrande, F.; Strazzabosco, M. Proinflammatory cytokines inhibit secretion in rat bile duct epithelium. Gastroenterology 2001, 121, 156–169. [Google Scholar] [CrossRef]
- Mazidi, M.; de Caravatto, P.P.; Speakman, J.R.; Cohen, R.V. Mechanisms of action of surgical interventions on weight-related diseases: The potential role of bile acids. Obes. Surg. 2017, 27, 826–836. [Google Scholar] [CrossRef]
- Wahlström, A.; Sayin, S.I.; Marschall, H.-U.; Bäckhed, F. Intestinal crosstalk between bile acids and microbiota and its impact on host metabolism. Cell Metab. 2016, 24, 41–50. [Google Scholar] [CrossRef] [Green Version]
- Merlen, G.; Bidault-Jourdainne, V.; Kahale, N.; Glenisson, M.; Ursic-Bedoya, J.; Doignon, I.; Garcin, I.; Humbert, L.; Rainteau, D.; Tordjmann, T. Hepatoprotective impact of the bile acid receptor TGR5. Liver Int. 2020, 40, 1005–1015. [Google Scholar] [CrossRef] [PubMed]
- Perino, A.; Pols, T.W.; Nomura, M.; Stein, S.; Pellicciari, R.; Schoonjans, K. TGR5 reduces macrophage migration through mTOR-induced C/EBPbeta differential translation. J. Clin. Investig. 2014, 124, 5424–5436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagathihalli, N.S.; Beesetty, Y.; Lee, W.; Washington, M.K.; Chen, X.; Lockhart, A.C.; Merchant, N.B. Novel mechanistic insights into ectodomain shedding of EGFR Ligands Amphiregulin and TGF-alpha: Impact on gastrointestinal cancers driven by secondary bile acids. Cancer Res. 2014, 74, 2062–2072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, W.; Tian, W.; Hong, J.; Li, D.; Tavares, R.; Noble, L.; Moss, S.F.; Resnick, M.B. Expression of bile acid receptor TGR5 in gastric adenocarcinoma. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 304, 322–327. [Google Scholar] [CrossRef] [PubMed]
- Casaburi, I.; Avena, P.; Lanzino, M.; Sisci, D.; Giordano, F.; Maris, P.; Catalano, S.; Morelli, C.; Ando, S. Chenodeoxycholic acid through a TGR5-dependent CREB signaling activation enhances cyclin D1 expression and promotes human endometrial cancer cell proliferation. Cell Cycle 2012, 11, 2699–2710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasuda, H.; Hirata, S.; Inoue, K.; Mashima, H.; Ohnishi, H.; Yoshiba, M. Involvement of membrane-type bile acid receptor M-BAR/TGR5 in bile acid-induced activation of epidermal growth factor receptor and mitogen-activated protein kinases in gastric carcinoma cells. Biochem. Biophys. Res. Commun. 2007, 354, 154–159. [Google Scholar] [CrossRef]
- Jensen, D.D.; Godfrey, C.B.; Niklas, C.; Canals, M.; Kocan, M.; Poole, D.P.; Murphy, J.E.; Alemi, F.; Cottrell, G.S.; Korbmacher, C.; et al. The bile acid receptor TGR5 does not interact with beta-arrestins or traffic to endosomes but transmits sustained signals from plasma membrane rafts. J. Biol. Chem. 2013, 288, 22942–22960. [Google Scholar] [CrossRef] [Green Version]
- Deutschmann, K.; Reich, M.; Krieg, A.; Knoefel, W.; Häussinger, D.; Keitel, V. Role of the bile acid receptor TGR5 (Gpbar-1) in gastrointestinal tumors. Zeitschrift für Gastroenterologie 2015, 53, A4_29. [Google Scholar] [CrossRef]
- Di Ciaula, A.; Portincasa, P. Fat, epigenome and pancreatic diseases. Interplay and common pathways from a toxic and obesogenic environment. Eur. J. Intern. Med. 2014, 25, 865–873. [Google Scholar] [CrossRef]
- Di Ciaula, A.; Portincasa, P. The role of environmental pollution in endocrine diseases. Endocrinology. In Endocrinology and Systemic Diseases; Portincasa, P., Fruhbeck, G., Nathoe, H.N., Eds.; Springer Nature: Basel, Switzerland, 2020; pp. 1–31. [Google Scholar] [CrossRef]
- Di Ciaula, A.; Portincasa, P. Diet and Contaminants: Driving the Rise to Obesity Epidemics? Curr. Med. Chem. 2019, 26, 3471–3482. [Google Scholar] [CrossRef]
- Allais, A.; Albert, O.; Lefevre, P.L.C.; Wade, M.G.; Hales, B.F.; Robaire, B. In utero and lactational exposure to flame retardants disrupts rat ovarian follicular development and advances puberty. Toxicol. Sci. Off. J. Soc. Toxicol. 2020, 175, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Jadhav, R.R.; Santucci-Pereira, J.; Wang, Y.V.; Liu, J.; Nguyen, T.D.; Wang, J.; Jenkins, S.; Russo, J.; Huang, T.H.; Jin, V.X.; et al. DNA methylation targets influenced by bisphenol a and/or genistein are associated with survival outcomes in breast cancer patients. Genes 2017, 8, 144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, S.; Steinmetz, G.; Montillet, G.; Perrard, M.H.; Loundou, A.; Durand, P.; Guichaoua, M.R.; Prat, O. Exposure to low-dose bisphenol A impairs meiosis in the rat seminiferous tubule culture model: A physiotoxicogenomic approach. PLoS ONE 2014, 9, e106245. [Google Scholar] [CrossRef] [PubMed]
- Verbanck, M.; Canouil, M.; Leloire, A.; Dhennin, V.; Coumoul, X.; Yengo, L.; Froguel, P.; Poulain-Godefroy, O. Low-dose exposure to bisphenols A, F and S of human primary adipocyte impacts coding and non-coding RNA profiles. PLoS ONE 2017, 12, e0179583. [Google Scholar] [CrossRef] [Green Version]
- Chu, J.H.; Hart, J.E.; Chhabra, D.; Garshick, E.; Raby, B.A.; Laden, F. Gene expression network analyses in response to air pollution exposures in the trucking industry. Environ. Health A Glob. Access Sci. Source 2016, 15, 101. [Google Scholar] [CrossRef] [Green Version]
- Faienza, M.F.; Wang, D.Q.H.; Frühbeck, G.; Garruti, G.; Portincasa, P. The dangerous link between childhood and adulthood predictors of obesity and metabolic syndrome. Internal and emergency medicine 2016, 11, 175–182. [Google Scholar] [CrossRef]
- Zhuang, L.; Ding, W.; Zhang, Q.; Ding, W.; Xu, X.; Yu, X.; Xi, D. TGR5 attenuated liver ischemia-reperfusion injury by activating the Keap1-Nrf2 signaling pathway in mice. Inflammation 2020, 1–14. [Google Scholar] [CrossRef]
- Susiarjo, M.; Xin, F.; Stefaniak, M.; Mesaros, C.; Simmons, R.A.; Bartolomei, M.S. Bile acids and tryptophan metabolism are novel pathways involved in metabolic abnormalities in bpa-exposed pregnant mice and male offspring. Endocrinology 2017, 158, 2533–2542. [Google Scholar] [CrossRef]
- Gomez, M.V.; Dutta, M.; Suvorov, A.; Shi, X.; Gu, H.; Mani, S.; Cui, J.Y. Early life exposure to environmental contaminants (BDE-47, TBBPA, and BPS) produced persistent alterations in fecal microbiome in adult male mice. Toxicol. Sci. Off. J. Soc. Toxicol. 2020, 1–40. [Google Scholar] [CrossRef]
- Li, C.Y.; Dempsey, J.L.; Wang, D.; Lee, S.; Weigel, K.M.; Fei, Q.; Bhatt, D.K.; Prasad, B.; Raftery, D.; Gu, H.; et al. PBDEs altered gut microbiome and bile acid homeostasis in male C57BL/6 mice. Drug Metab. Dispos. Biol. Fate Chem. 2018, 46, 1226–1240. [Google Scholar] [CrossRef]
- Cheng, S.L.; Li, X.; Lehmler, H.J.; Phillips, B.; Shen, D.; Cui, J.Y. Gut microbiota modulates interactions between polychlorinated biphenyls and bile acid homeostasis. Toxicol. Sci. Off. J. Soc. Toxicol. 2018, 166, 269–287. [Google Scholar] [CrossRef] [PubMed]
- Gioiello, A.; Rosatelli, E.; Nuti, R.; Macchiarulo, A.; Pellicciari, R. Patented TGR5 modulators: A review (2006–present). Expert Opin. Pat. 2012, 22, 1399–1414. [Google Scholar] [CrossRef] [PubMed]
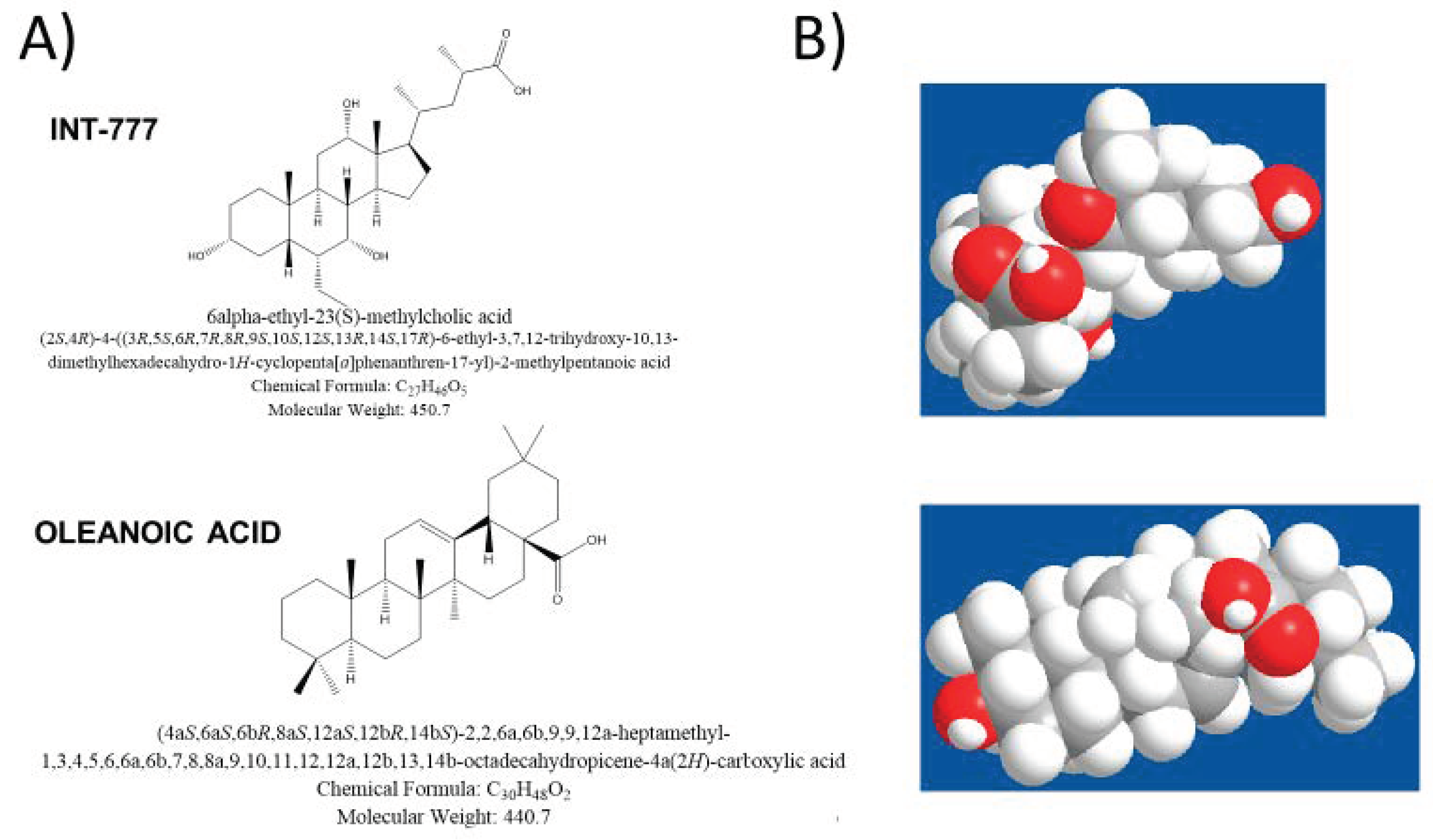
- Pellicciari, R.; Gioiello, A.; Macchiarulo, A.; Thomas, C.; Rosatelli, E.; Natalini, B.; Sardella, R.; Pruzanski, M.; Roda, A.; Pastorini, E.; et al. Discovery of 6alpha-ethyl-23(S)-methylcholic acid (S-EMCA, INT-777) as a potent and selective agonist for the TGR5 receptor, a novel target for diabesity. J. Med. Chem. 2009, 52, 7958–7961. [Google Scholar] [CrossRef] [PubMed]
- Hodge, R.J.; Lin, J.; Vasist Johnson, L.S.; Gould, E.P.; Bowers, G.D.; Nunez, D.J.; Team, S.B.P. Safety, pharmacokinetics, and pharmacodynamic effects of a selective TGR5 agonist, SB-756050, in type 2 diabetes. Clin. Pharm. Drug Dev. 2013, 2, 213–222. [Google Scholar] [CrossRef]
- Genet, C.; Strehle, A.; Schmidt, C.; Boudjelal, G.; Lobstein, A.; Schoonjans, K.; Souchet, M.; Auwerx, J.; Saladin, R.; Wagner, A. Structure-activity relationship study of betulinic acid, a novel and selective TGR5 agonist, and its synthetic derivatives: Potential impact in diabetes. J. Med. Chem. 2010, 53, 178–190. [Google Scholar] [CrossRef]
- Luo, H.; Liu, J.; Ouyang, Q.; Xuan, C.; Wang, L.; Li, T.; Liu, J. The effects of oleanolic acid on atherosclerosis in different animal models. Acta Biochim. Biophys. Sin. 2017, 49, 349–354. [Google Scholar] [CrossRef] [Green Version]
- Shima, K.R.; Ota, T.; Kato, K.I.; Takeshita, Y.; Misu, H.; Kaneko, S.; Takamura, T. Ursodeoxycholic acid potentiates dipeptidyl peptidase-4 inhibitor sitagliptin by enhancing glucagon-like peptide-1 secretion in patients with type 2 diabetes and chronic liver disease: A pilot randomized controlled and add-on study. BMJ Open Diabetes Res. Care 2018, 6, e000469. [Google Scholar] [CrossRef]
- Wu, T.; Bound, M.; Standfield, S.; Gedulin, B.; Jones, K.; Horowitz, M.; Rayner, C. Effects of rectal administration of taurocholic acid on glucagon-like peptide-1 and peptide YY secretion in healthy humans. Diabetes Obes. Metab. 2013, 15, 474–477. [Google Scholar] [CrossRef]
- Hansen, M.; Scheltema, M.J.; Sonne, D.P.; Hansen, J.S.; Sperling, M.; Rehfeld, J.F.; Holst, J.J.; Vilsbøll, T.; Knop, F.K. Effect of chenodeoxycholic acid and the bile acid sequestrant colesevelam on glucagon-like peptide-1 secretion. Diabetes Obes. Metab. 2016, 18, 571–580. [Google Scholar] [CrossRef]
- Nielsen, S.; Svane, M.S.; Kuhre, R.E.; Clausen, T.R.; Kristiansen, V.B.; Rehfeld, J.F.; Holst, J.J.; Madsbad, S.; Bojsen-Moller, K.N. Chenodeoxycholic acid stimulates glucagon-like peptide-1 secretion in patients after Roux-en-Y gastric bypass. Physiol. Rep. 2017, 5, e13140. [Google Scholar] [CrossRef]
- Teodoro, J.; Zouhar, P.; Flachs, P.; Bardova, K.; Janovska, P.; Gomes, A.; Duarte, F.; Varela, A.; Rolo, A.; Palmeira, C. Enhancement of brown fat thermogenesis using chenodeoxycholic acid in mice. Int. J. Obes. 2014, 38, 1027–1034. [Google Scholar] [CrossRef] [PubMed]
- Broeders, E.P.; Nascimento, E.B.; Havekes, B.; Brans, B.; Roumans, K.H.; Tailleux, A.; Schaart, G.; Kouach, M.; Charton, J.; Deprez, B. The bile acid chenodeoxycholic acid increases human brown adipose tissue activity. Cell Metab. 2015, 22, 418–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Wen, X.; Zhang, W.; Wang, C.; Liu, J.; Liu, C. Hypolipidemic effect of oleanolic acid is mediated by the miR-98-5p/PGC-1β axis in high-fat diet-induced hyperlipidemic mice. FASEB J. 2017, 31, 1085–1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han-Qiong, L.; Jie, S.; Cai-Ping, C.; Xiao, M.; Chao, L.; Ouyang, Q.; Chun-Xiao, X.; Jine, L.; Hong-Bin, S.; Jun, L. Lipid-lowering effects of oleanolic acid in hyperlipidemic patients. Chin. J. Nat. Med. 2018, 16, 339–346. [Google Scholar]
- Biagioli, M.; Carino, A.; Cipriani, S.; Francisci, D.; Marchianò, S.; Scarpelli, P.; Sorcini, D.; Zampella, A.; Fiorucci, S. The bile acid receptor GPBAR1 regulates the M1/M2 phenotype of intestinal macrophages and activation of GPBAR1 rescues mice from murine colitis. J. Immunol. 2017, 199, 718–733. [Google Scholar] [CrossRef] [Green Version]
- Sakanaka, T.; Inoue, T.; Yorifuji, N.; Iguchi, M.; Fujiwara, K.; Narabayashi, K.; Kakimoto, K.; Nouda, S.; Okada, T.; Kuramoto, T.; et al. The effects of a TGR5 agonist and a dipeptidyl peptidase IV inhibitor on dextran sulfate sodium-induced colitis in mice. J. Gastroenterol. Hepatol. 2015, 30, 60–65. [Google Scholar] [CrossRef]
- Fryer, R.M.; Ng, K.J.; Mazurek, S.G.N.; Patnaude, L.; Skow, D.J.; Muthukumarana, A.; Gilpin, K.E.; Dinallo, R.M.; Kuzmich, D.; Lord, J. G Protein–Coupled Bile Acid Receptor 1 Stimulation Mediates Arterial Vasodilation through a KCa1. 1 (BKCa)–Dependent Mechanism. J. Pharmacol. Exp. Ther. 2014, 348, 421–431. [Google Scholar] [CrossRef]
- Perides, G.; Laukkarinen, J.M.; Vassileva, G.; Steer, M.L. Biliary acute pancreatitis in mice is mediated by the G-protein—Coupled cell surface bile acid receptor Gpbar1. Gastroenterology 2010, 138, 715–725. [Google Scholar] [CrossRef] [Green Version]
- Alemi, F.; Kwon, E.; Poole, D.P.; Lieu, T.; Lyo, V.; Cattaruzza, F.; Cevikbas, F.; Steinhoff, M.; Nassini, R.; Materazzi, S.; et al. The TGR5 receptor mediates bile acid-induced itch and analgesia. J. Clin. Investig. 2013, 123, 1513–1530. [Google Scholar] [CrossRef] [Green Version]
- Pang, C.; LaLonde, A.; Godfrey, T.E.; Que, J.; Sun, J.; Wu, T.T.; Zhou, Z. Bile salt receptor TGR5 is highly expressed in esophageal adenocarcinoma and precancerous lesions with significantly worse overall survival and gender differences. Clin. Exp. Gastroenterol. 2017, 10, 29. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Receptor | Mechanism(s) |
| FXR |
|
| GPBAR-1 | |
| S1PR2 |
| Site/Pathway | Mechanism(s) |
|---|---|
| Liver (BA synthesis) |
|
| Intestine (BA biotransformation) |
|
| Enterohepatic cycle, ileum, biliary tract, kidney/transepithelial flux of BA |
|
| Site | Mechanism(s) |
|---|---|
| Endothelium | |
| Portal tract (liver sinusoidal endothelial cells) |
|
| Site | Mechanism |
|---|---|
| |
|
|
| |
|
| Toxic of Environmental Origin | Effect | Model | References |
|---|---|---|---|
| Brominated flame retardants (polybrominated diphenyl ethers, PBDEs) Hexabromocyclododecane (HBCDD) | Increased expression of GPBAR-1 mRNA | Sprague-Dawley rats, ovary | [194] |
| Bisphenol A (BPA) | Decreased methylation of GPBAR-1 gene | Sprague-Dawley rats, mammary gland | [195] |
| Bisphenol A (BPA) | Upregulation of GPBAR-1 | Sprague-Dawley rats, seminiferous tubule | [196] |
| Bisphenol A (BPA), F (BPF), S (BPS) | Altered coding and noncoding RNA profiles; increased expression of GPBAR-1 mRNA | Humans, human primary adipocyte | [197] |
| Elemental carbon (EC) Organic carbon (OC) | Increased expression of GPBAR-1 mRNA | Humans, whole blood | [198] |
| Site | Mechanism(s)/Evidence | Model(s) |
|---|---|---|
| Animal | |
|
| Animal |
| Animal | |
| Animal | |
| Animal | |
|
| Human (in vivo, in vitro) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Portincasa, P.; Di Ciaula, A.; Garruti, G.; Vacca, M.; De Angelis, M.; Wang, D.Q.-H. Bile Acids and GPBAR-1: Dynamic Interaction Involving Genes, Environment and Gut Microbiome. Nutrients 2020, 12, 3709. https://doi.org/10.3390/nu12123709
Portincasa P, Di Ciaula A, Garruti G, Vacca M, De Angelis M, Wang DQ-H. Bile Acids and GPBAR-1: Dynamic Interaction Involving Genes, Environment and Gut Microbiome. Nutrients. 2020; 12(12):3709. https://doi.org/10.3390/nu12123709
Chicago/Turabian StylePortincasa, Piero, Agostino Di Ciaula, Gabriella Garruti, Mirco Vacca, Maria De Angelis, and David Q.-H. Wang. 2020. "Bile Acids and GPBAR-1: Dynamic Interaction Involving Genes, Environment and Gut Microbiome" Nutrients 12, no. 12: 3709. https://doi.org/10.3390/nu12123709
APA StylePortincasa, P., Di Ciaula, A., Garruti, G., Vacca, M., De Angelis, M., & Wang, D. Q.-H. (2020). Bile Acids and GPBAR-1: Dynamic Interaction Involving Genes, Environment and Gut Microbiome. Nutrients, 12(12), 3709. https://doi.org/10.3390/nu12123709