Cytochrome P450 Metabolism of Polyunsaturated Fatty Acids and Neurodegeneration


Abstract
1. Introduction
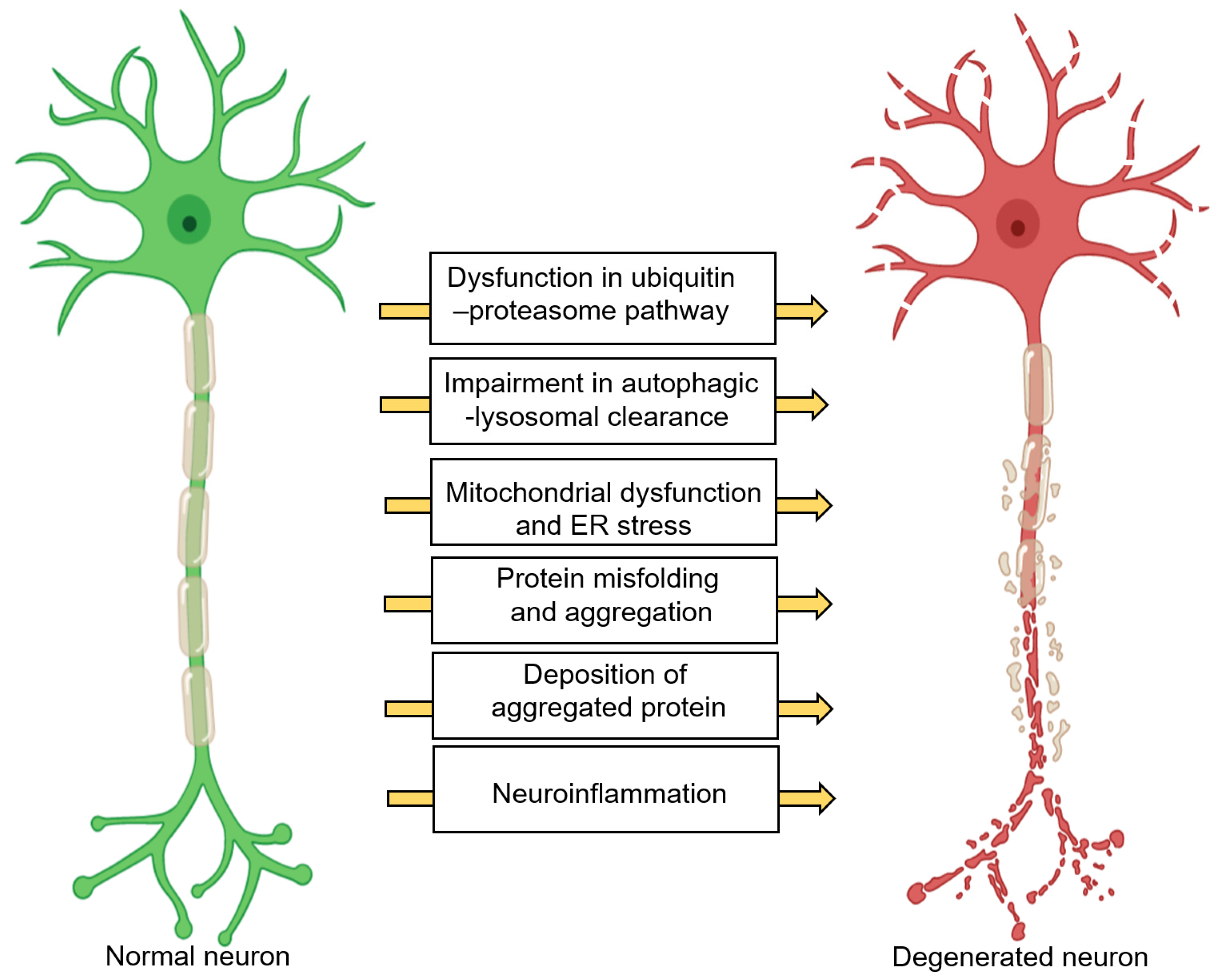
2. Neurodegeneration: A Brief Overview
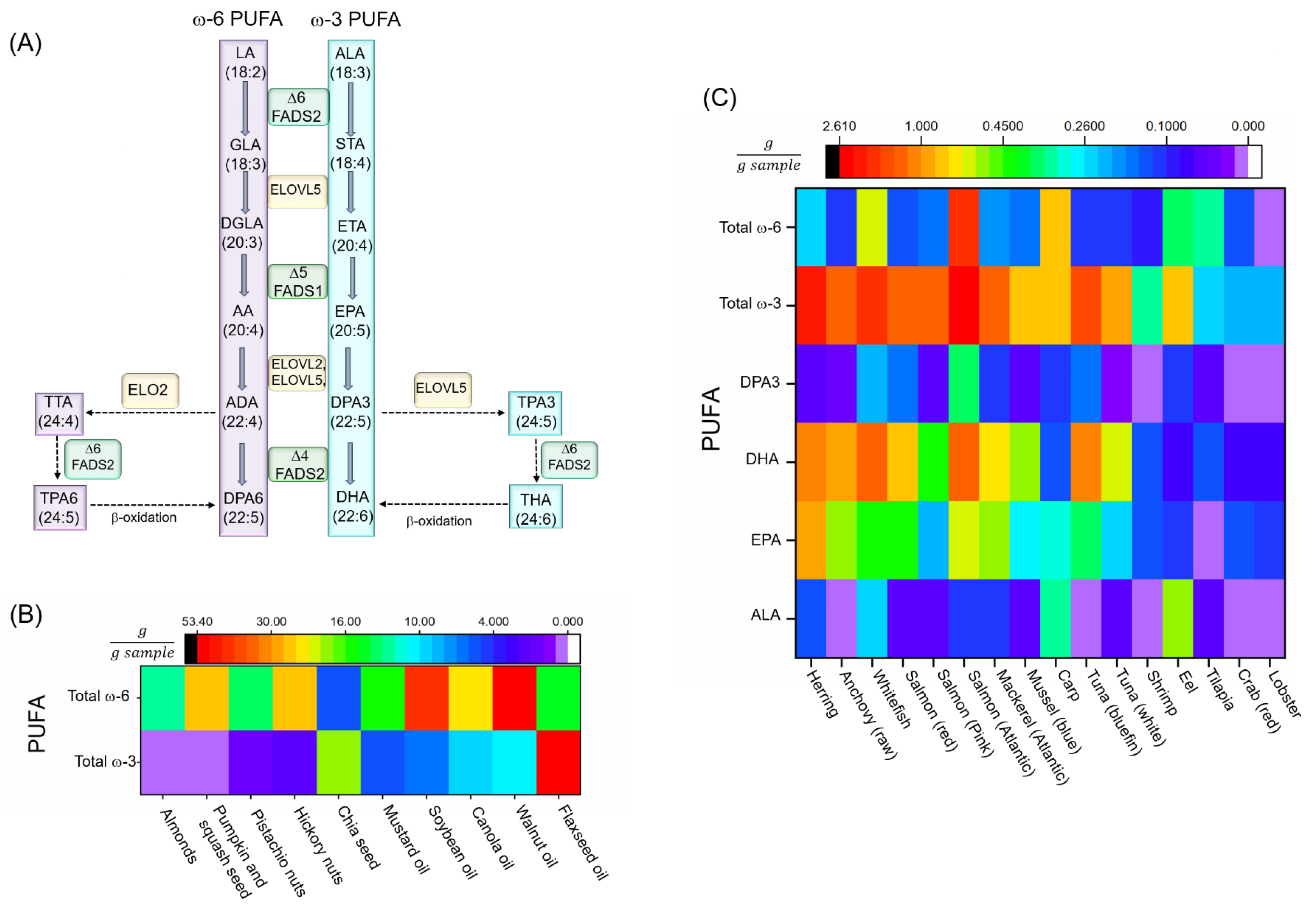
3. Overview of PUFAs
3.1. Biosynthesis of PUFAs and Neurodegenerative Diseases
3.2. Dietary PUFA

4. CYP: A Key Monooxygenase Enzyme in PUFAs Metabolism
4.1. Characteristics of CYP
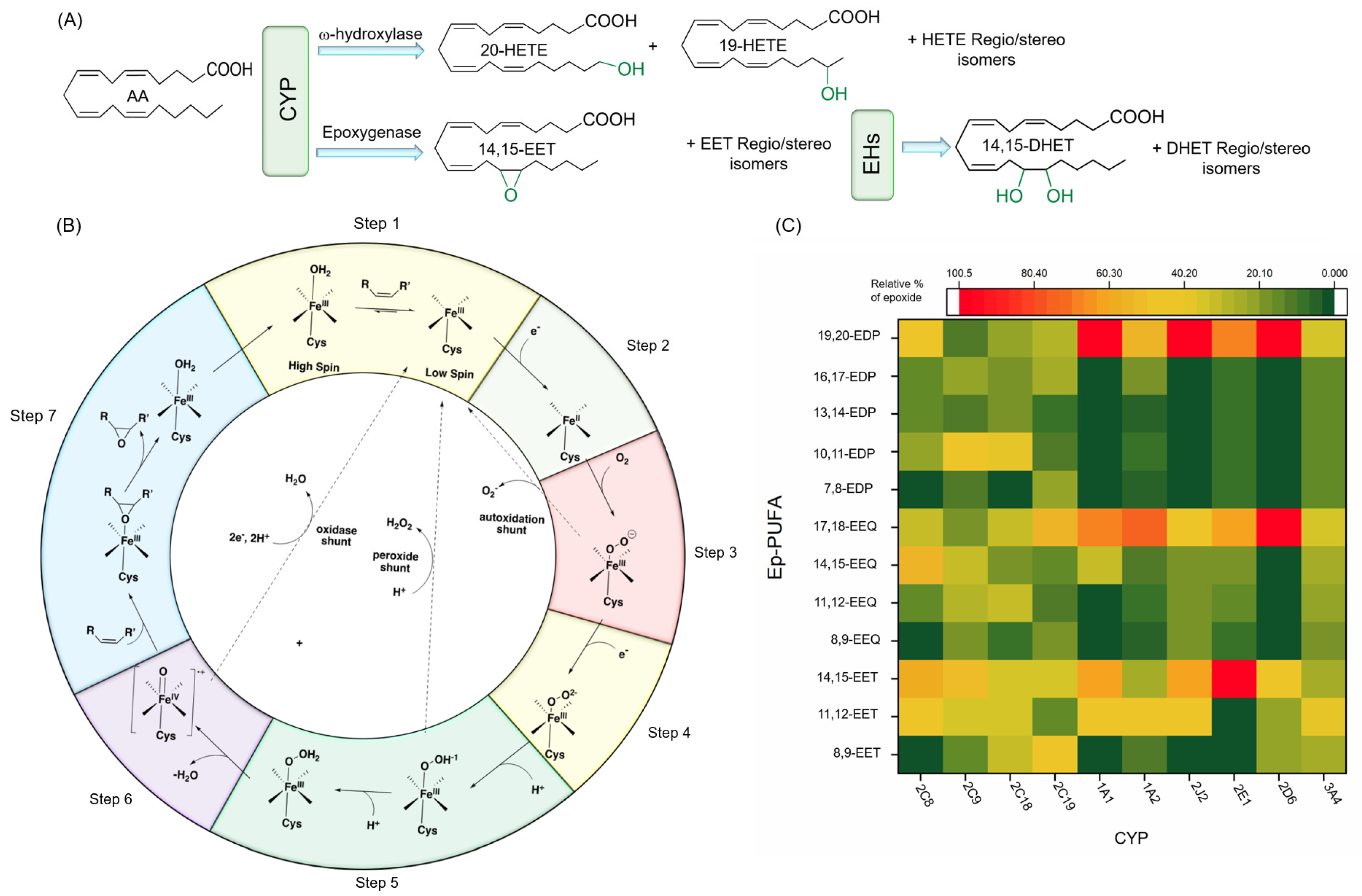
4.2. Catalytic Function and Mechanism of CYP
4.3. Major CYP Responsible for PUFA Metabolism
4.3.1. Regio- and Stereoselective Epoxidation by Major CYPs

4.3.2. Regio- and Stereoselective Hydroxylation of PUFAs by CYPs
4.3.3. Physiological Functions of Regio-/Stereoisomers of CYP Products: Ep-PUFAs and Hydroxy-PUFAs
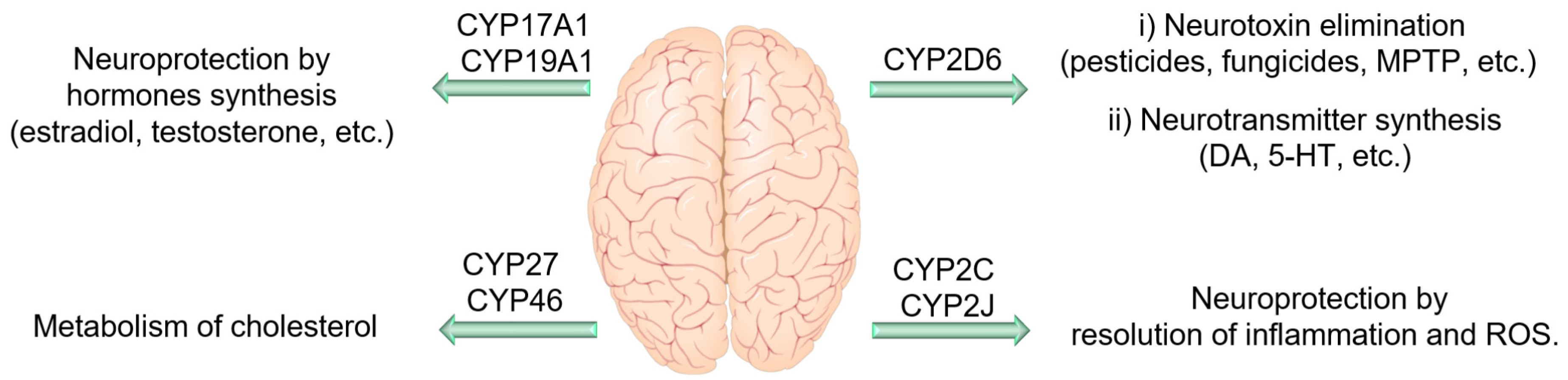
4.4. CYP Enzymes in the Central Nervous System
5. EH: Epoxy Hydrolase, a Critical Member in Ep-PUFA Metabolism
5.1. Characteristics of the Main EH Enzymes
5.2. Catalytic Function and Mechanism of EHs
5.3. Major EHs Responsible for Ep-PUFA Metabolism
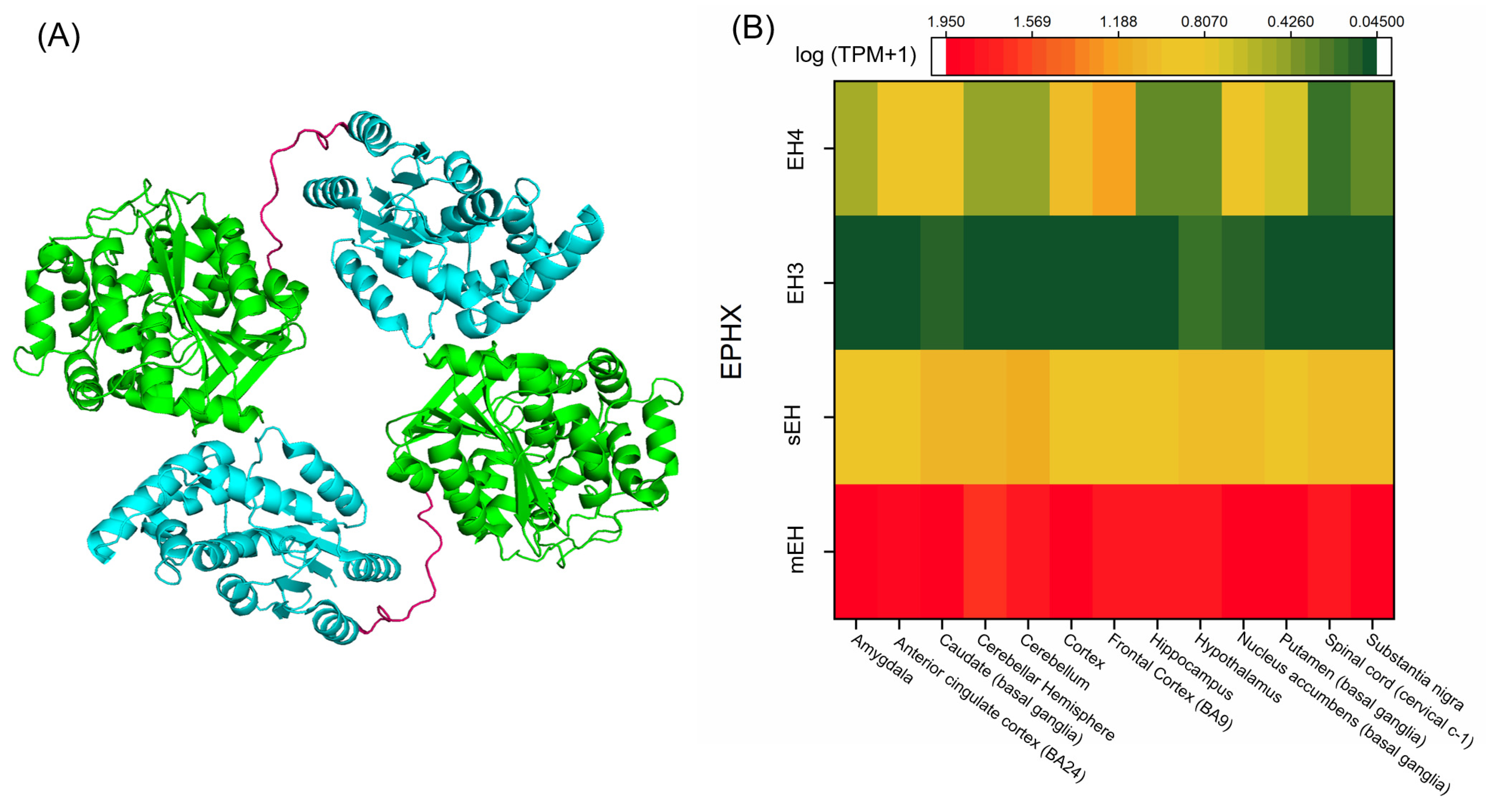
5.4. EHs in the Brain
5.4.1. mEH in the Brain
5.4.2. sEH in the Brain
5.4.3. EH3 and Other EHs in the Brain
5.5. Possible Physiological Roles of Dihydroxy-PUFAs
6. CYP PUFA Metabolism and the Nervous System
7. Ep-PUFAs and Neuroinflammation
7.1. Acute Neuroinflammation
7.2. Chronic Neuroinflammation
7.2.1. Alzheimer’s Disease
7.2.2. Parkinson Disease
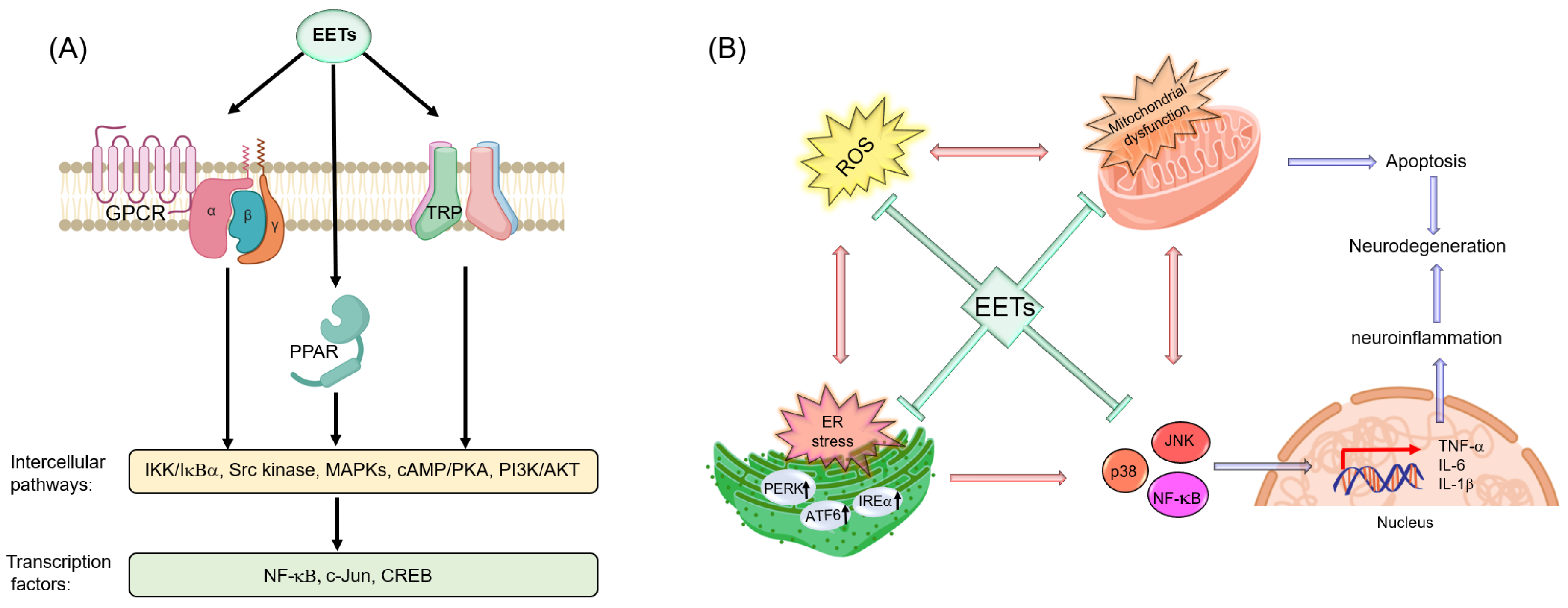
7.3. Potential Molecular Targets of Ep-PUFAs in the Nervous System
7.3.1. PPARs
7.3.2. TRP Channels
7.3.3. GPCRs
7.3.4. ER
7.3.5. Mitochondria
8. Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Gan, L.; Cookson, M.R.; Petrucelli, L.; La Spada, A.R. Converging pathways in neurodegeneration, from genetics to mechanisms. Nat. Neurosci. 2018, 21, 1300–1309. [Google Scholar] [CrossRef] [PubMed]
- Bishop, N.A.; Lu, T.; Yankner, B.A. Neural mechanisms of ageing and cognitive decline. Nature 2010, 464, 529–535. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef] [PubMed]
- Migliore, L.; Coppedè, F. Genetics, environmental factors and the emerging role of epigenetics in neurodegenerative diseases. Mutat. Res. 2009, 667, 82–97. [Google Scholar] [CrossRef]
- Coppedè, F.; Mancuso, M.; Siciliano, G.; Migliore, L.; Murri, L. Genes and the environment in neurodegeneration. Biosci. Rep. 2006, 26, 341–367. [Google Scholar] [CrossRef]
- Moore, K.; Hughes, C.F.; Ward, M.; Hoey, L.; McNulty, H. Diet, nutrition and the ageing brain: Current evidence and new directions. Proc. Nutr. Soc. 2018, 77, 152–163. [Google Scholar] [CrossRef]
- Shahidi, F.; Ambigaipalan, P. Omega-3 polyunsaturated fatty acids and their health benefits. Annu. Rev. Food Sci. Technol. 2018, 9, 345–381. [Google Scholar] [CrossRef]
- Bishop-Bailey, D.; Thomson, S.; Askari, A.; Faulkner, A.; Wheeler-Jones, C. Lipid-metabolizing CYPs in the regulation and dysregulation of metabolism. Annu. Rev. Nutr. 2014, 34, 261–279. [Google Scholar] [CrossRef]
- Wall, R.; Ross, R.P.; Fitzgerald, G.F.; Stanton, C. Fatty acids from fish: The anti-inflammatory potential of long-chain omega-3 fatty acids. Nutr. Rev. 2010, 68, 280–289. [Google Scholar] [CrossRef]
- de Lau, L.M.L.; Bornebroek, M.; Witteman, J.C.M.; Hofman, A.; Koudstaal, P.J.; Breteler, M.M.B. Dietary fatty acids and the risk of Parkinson disease: The Rotterdam study. Neurology 2005, 64, 2040–2045. [Google Scholar] [CrossRef]
- Cardoso, H.D.; dos Santos Junior, E.F.; de Santana, D.F.; Gonçalves-Pimentel, C.; Angelim, M.K.; Isaac, A.R.; Lagranha, C.J.; Guedes, R.C.A.; Beltrão, E.I.; Morya, E.; et al. Omega-3 deficiency and neurodegeneration in the substantia nigra: Involvement of increased nitric oxide production and reduced BDNF expression. Biochim. Biophys. Acta 2014, 1840, 1902–1912. [Google Scholar] [CrossRef] [PubMed]
- Healy-Stoffel, M.; Levant, B. N-3 (omega-3) fatty acids: Effects on brain dopamine systems and potential role in the etiology and treatment of neuropsychiatric disorders. CNS Neurol. Disord. Drug Targets 2018, 17, 216–232. [Google Scholar] [CrossRef] [PubMed]
- Bazinet, R.P.; Layé, S. Polyunsaturated fatty acids and their metabolites in brain function and disease. Nat. Rev. Neurosci. 2014, 15, 771–785. [Google Scholar] [CrossRef] [PubMed]
- Ferdouse, A.; Leng, S.; Winter, T.; Aukema, H.M. The brain oxylipin profile is resistant to modulation by dietary n-6 and n-3 polyunsaturated fatty acids in male and female rats. Lipids 2019, 54, 67–80. [Google Scholar] [CrossRef]
- Freitas, H.R.; Isaac, A.R.; Malcher-Lopes, R.; Diaz, B.L.; Trevenzoli, I.H.; De Melo Reis, R.A. Polyunsaturated fatty acids and endocannabinoids in health and disease. Nutr. Neurosci. 2018, 21, 695–714. [Google Scholar] [CrossRef]
- Inceoglu, B.; Zolkowska, D.; Yoo, H.J.; Wagner, K.M.; Yang, J.; Hackett, E.; Hwang, S.H.; Lee, K.S.S.; Rogawski, M.A.; Morisseau, C.; et al. Epoxy fatty acids and inhibition of the soluble epoxide hydrolase selectively modulate GABA mediated neurotransmission to delay onset of seizures. PLoS ONE 2013, 8, e80922. [Google Scholar] [CrossRef]
- Harris, T.R.; Aronov, P.A.; Jones, P.D.; Tanaka, H.; Arand, M.; Hammock, B.D. Identification of two epoxide hydrolases in Caenorhabditis elegans that metabolize mammalian lipid signaling molecules. Arch. Biochem. Biophys. 2008, 472, 139–149. [Google Scholar] [CrossRef]
- Navarro-Mabarak, C.; Camacho-Carranza, R.; Espinosa-Aguirre, J.J. Cytochrome P450 in the central nervous system as a therapeutic target in neurodegenerative diseases. Drug Metab. Rev. 2018, 50, 95–108. [Google Scholar] [CrossRef]
- Ren, Q.; Ma, M.; Yang, J.; Nonaka, R.; Yamaguchi, A.; Ishikawa, K.-I.; Kobayashi, K.; Murayama, S.; Hwang, S.H.; Saiki, S.; et al. Soluble epoxide hydrolase plays a key role in the pathogenesis of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2018, 115, E5815–E5823. [Google Scholar] [CrossRef]
- Morris, J.K.; Piccolo, B.D.; John, C.S.; Green, Z.D.; Thyfault, J.P.; Adams, S.H. Oxylipin profiling of Alzheimer’s disease in nondiabetic and type 2 diabetic elderly. Metabolites 2019, 9, 177. [Google Scholar] [CrossRef]
- Wagner, K.; Vito, S.; Inceoglu, B.; Hammock, B.D. The role of long chain fatty acids and their epoxide metabolites in nociceptive signaling. Prostaglandins Other Lipid Mediat. 2014, 113–115, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Konkel, A.; Schunck, W.-H. Role of cytochrome P450 enzymes in the bioactivation of polyunsaturated fatty acids. Biochim. Biophys. Acta 2011, 1814, 210–222. [Google Scholar] [CrossRef] [PubMed]
- Spector, A.A.; Kim, H.-Y. Cytochrome P450 epoxygenase pathway of polyunsaturated fatty acid metabolism. Biochim. Biophys. Acta 2015, 1851, 356–365. [Google Scholar] [CrossRef] [PubMed]
- Soto, C.; Pritzkow, S. Protein misfolding, aggregation, and conformational strains in neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1332–1340. [Google Scholar] [CrossRef] [PubMed]
- Dugger, B.N.; Dickson, D.W. Pathology of neurodegenerative diseases. Cold Spring Harb. Perspect. Biol. 2017, 9, a028035. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Campisi, J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69 (Suppl. 1), S4–S9. [Google Scholar] [CrossRef] [PubMed]
- Guzman-Martinez, L.; Maccioni, R.B.; Andrade, V.; Navarrete, L.P.; Pastor, M.G.; Ramos-Escobar, N. Neuroinflammation as a common feature of neurodegenerative disorders. Front. Pharmacol. 2019, 10, 1008. [Google Scholar] [CrossRef] [PubMed]
- Peters, R. Ageing and the brain. Postgrad. Med. J. 2006, 82, 84–88. [Google Scholar] [CrossRef]
- Svennerholm, L.; Boström, K.; Jungbjer, B. Changes in weight and compositions of major membrane components of human brain during the span of adult human life of Swedes. Acta Neuropathol. 1997, 94, 345–352. [Google Scholar] [CrossRef]
- Elobeid, A.; Libard, S.; Leino, M.; Popova, S.N.; Alafuzoff, I. Altered proteins in the aging brain. J. Neuropathol. Exp. Neurol. 2016, 75, 316–325. [Google Scholar] [CrossRef]
- Naoe, S.; Tsugawa, H.; Takahashi, M.; Ikeda, K.; Arita, M. Characterization of lipid profiles after dietary intake of polyunsaturated fatty acids using integrated untargeted and targeted lipidomics. Metabolites 2019, 9, 241. [Google Scholar] [CrossRef] [PubMed]
- Ferdouse, A.; Leng, S.; Winter, T.; Aukema, H.M. Dietary n-6 and n-3 PUFA alter the free oxylipin profile differently in male and female rat hearts. Br. J. Nutr. 2019, 122, 252–261. [Google Scholar] [CrossRef] [PubMed]
- Monroig, Ó.; Tocher, D.R.; Navarro, J.C. Biosynthesis of polyunsaturated fatty acids in marine invertebrates: Recent advances in molecular mechanisms. Mar. Drugs 2013, 11, 3998–4018. [Google Scholar] [CrossRef] [PubMed]
- Mokoena, N.Z.; Sebolai, O.M.; Albertyn, J.; Pohl, C.H. Synthesis and function of fatty acids and oxylipins, with a focus on Caenorhabditis elegans. Prostaglandins Other Lipid Mediat. 2020, 148, 106426. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.T.; Nara, T.Y. Structure, function, and dietary regulation of delta6, delta5, and delta9 desaturases. Annu. Rev. Nutr. 2004, 24, 345–376. [Google Scholar] [CrossRef]
- Ameur, A.; Enroth, S.; Johansson, A.; Zaboli, G.; Igl, W.; Johansson, A.C.V.; Rivas, M.A.; Daly, M.J.; Schmitz, G.; Hicks, A.A.; et al. Genetic adaptation of fatty-acid metabolism: A human-specific haplotype increasing the biosynthesis of long-chain omega-3 and omega-6 fatty acids. Am. J. Hum. Genet. 2012, 90, 809–820. [Google Scholar] [CrossRef]
- Koletzko, B.; Reischl, E.; Tanjung, C.; Gonzalez-Casanova, I.; Ramakrishnan, U.; Meldrum, S.; Simmer, K.; Heinrich, J.; Demmelmair, H. FADS1 and FADS2 polymorphisms modulate fatty acid metabolism and dietary impact on health. Annu. Rev. Nutr. 2019, 39, 21–44. [Google Scholar] [CrossRef]
- Steer, C.D.; Davey Smith, G.; Emmett, P.M.; Hibbeln, J.R.; Golding, J. FADS2 polymorphisms modify the effect of breastfeeding on child IQ. PLoS ONE 2010, 5, e11570. [Google Scholar] [CrossRef]
- Moltó-Puigmartí, C.; Plat, J.; Mensink, R.P.; Müller, A.; Jansen, E.; Zeegers, M.P.; Thijs, C. FADS1 FADS2 gene variants modify the association between fish intake and the docosahexaenoic acid proportions in human milk. Am. J. Clin. Nutr. 2010, 91, 1368–1376. [Google Scholar] [CrossRef]
- Xie, L.; Innis, S.M. Genetic variants of the FADS1 FADS2 gene cluster are associated with altered (n-6) and (n-3) essential fatty acids in plasma and erythrocyte phospholipids in women during pregnancy and in breast milk during lactation. J. Nutr. 2008, 138, 2222–2228. [Google Scholar] [CrossRef]
- Hernell, O.; Holmgren, G.; Jagell, S.F.; Johnson, S.B.; Holman, R.T. Suspected faulty essential fatty acid metabolism in Sjögren-Larsson syndrome. Pediatr. Res. 1982, 16, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Brookes, K.J.; Chen, W.; Xu, X.; Taylor, E.; Asherson, P. Association of fatty acid desaturase genes with attention-deficit/hyperactivity disorder. Biol. Psychiatry 2006, 60, 1053–1061. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.-Y.; Monk, J.M.; Hou, T.Y.; Callway, E.; Vincent, L.; Weeks, B.; Yang, P.; Chapkin, R.S. Characterization of an arachidonic acid-deficient (Fads1 knockout) mouse model. J. Lipid Res. 2012, 53, 1287–1295. [Google Scholar] [CrossRef]
- Stoffel, W.; Holz, B.; Jenke, B.; Binczek, E.; Günter, R.H.; Kiss, C.; Karakesisoglou, I.; Thevis, M.; Weber, A.-A.; Arnhold, S.; et al. Delta6-desaturase (FADS2) deficiency unveils the role of omega3- and omega6-polyunsaturated fatty acids. EMBO J. 2008, 27, 2281–2292. [Google Scholar] [CrossRef]
- Gromovsky, A.D.; Schugar, R.C.; Brown, A.L.; Helsley, R.N.; Burrows, A.C.; Ferguson, D.; Zhang, R.; Sansbury, B.E.; Lee, R.G.; Morton, R.E.; et al. Δ-5 fatty acid desaturase FADS1 impacts metabolic disease by balancing proinflammatory and proresolving lipid mediators. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 218–231. [Google Scholar] [CrossRef]
- Ghasemi Fard, S.; Wang, F.; Sinclair, A.J.; Elliott, G.; Turchini, G.M. How does high DHA fish oil affect health? A systematic review of evidence. Crit. Rev. Food Sci. Nutr. 2019, 59, 1684–1727. [Google Scholar] [CrossRef] [PubMed]
- The Canadian Nutrient File (CNF). 2015. Available online: https://www.canada.ca/en/health-canada/services/food-nutrition/healthy-eating/nutrient-data/canadian-nutrient-file-2015-download-files.html (accessed on 11 July 2020).
- Avallone, R.; Vitale, G.; Bertolotti, M. Omega-3 fatty acids and neurodegenerative diseases: New evidence in clinical trials. Int. J. Mol. Sci. 2019, 20, 4256. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.C.; Evans, D.A.; Bienias, J.L.; Tangney, C.C.; Bennett, D.A.; Wilson, R.S.; Aggarwal, N.; Schneider, J. Consumption of fish and n-3 fatty acids and risk of incident Alzheimer disease. Arch. Neurol. 2003, 60, 940–946. [Google Scholar] [CrossRef] [PubMed]
- Tapiero, H.; Ba, G.N.; Couvreur, P.; Tew, K.D. Polyunsaturated fatty acids (PUFA) and eicosanoids in human health and pathologies. Biomed. Pharmacother. 2002, 56, 215–222. [Google Scholar] [CrossRef]
- Christie, W.W.; Harwood, J.L. Oxidation of polyunsaturated fatty acids to produce lipid mediators. Essays Biochem. 2020. [Google Scholar] [CrossRef]
- de Bus, I.; Witkamp, R.; Zuilhof, H.; Albada, B.; Balvers, M. The role of n-3 PUFA-derived fatty acid derivatives and their oxygenated metabolites in the modulation of inflammation. Prostaglandins Other Lipid Mediat. 2019, 144, 106351. [Google Scholar] [CrossRef] [PubMed]
- Czapski, G.A.; Czubowicz, K.; Strosznajder, J.B.; Strosznajder, R.P. The lipoxygenases: Their regulation and implication in Alzheimer’s disease. Neurochem. Res. 2016, 41, 243–257. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Jiang, Y.; Guengerich, F.P.; Ma, L.; Li, S.; Zhang, W. Engineering cytochrome P450 enzyme systems for biomedical and biotechnological applications. J. Biol. Chem. 2020, 295, 833–849. [Google Scholar] [CrossRef] [PubMed]
- Omura, T.; Sato, R. A new cytochrome in liver microsomes. J. Biol. Chem. 1962, 237, 1375–1376. [Google Scholar] [PubMed]
- Guengerich, F.P.; Waterman, M.R.; Egli, M. Recent structural insights into cytochrome P450 function. Trends Pharmacol. Sci. 2016, 37, 625–640. [Google Scholar] [CrossRef]
- Crane, B.R.; Arvai, A.S.; Ghosh, D.K.; Wu, C.; Getzoff, E.D.; Stuehr, D.J.; Tainer, J.A. Structure of nitric oxide synthase oxygenase dimer with pterin and substrate. Science 1998, 279, 2121–2126. [Google Scholar] [CrossRef]
- Sundaramoorthy, M.; Terner, J.; Poulos, T.L. The crystal structure of chloroperoxidase: A heme peroxidase-cytochrome P450 functional hybrid. J. Inorg. Biochem. 1995, 59, 427. [Google Scholar] [CrossRef]
- Ingelman-Sundberg, M. The human genome project and novel aspects of cytochrome P450 research. Toxicol. Appl. Pharmacol. 2005, 207, 52–56. [Google Scholar] [CrossRef]
- Nelson, D.R. The cytochrome p450 homepage. Hum. Genom. 2009, 4, 59–65. [Google Scholar] [CrossRef]
- Black, S.D. Membrane topology of the mammalian P450 cytochromes. FASEB J. 1992, 6, 680–685. [Google Scholar] [CrossRef]
- Šrejber, M.; Navrátilová, V.; Paloncýová, M.; Bazgier, V.; Berka, K.; Anzenbacher, P.; Otyepka, M. Membrane-attached mammalian cytochromes P450: An overview of the membrane’s effects on structure, drug binding, and interactions with redox partners. J. Inorg. Biochem. 2018, 183, 117–136. [Google Scholar] [CrossRef] [PubMed]
- Guengerich, F.P. Human Cytochrome P450 Enzymes. In Cytochrome P450; Springer International Publishing: Cham, Switzerland, 2015; pp. 523–785. ISBN 9783319121079. [Google Scholar]
- Preissner, S.C.; Hoffmann, M.F.; Preissner, R.; Dunkel, M.; Gewiess, A.; Preissner, S. Polymorphic cytochrome P450 enzymes (CYPs) and their role in personalized therapy. PLoS ONE 2013, 8, e82562. [Google Scholar] [CrossRef]
- Klingenberg, M. Pigments of rat liver microsomes. Arch. Biochem. Biophys. 1958, 75, 376–386. [Google Scholar] [CrossRef]
- Garfinkel, D. Studies on pig liver microsomes. I. Enzymic and pigment composition of different microsomal fractions. Arch. Biochem. Biophys. 1958, 77, 493–509. [Google Scholar] [CrossRef]
- Pessayre, D.; Mazel, P.; Descatoire, V.; Rogier, E.; Feldmann, G.; Benhamou, J.P. Inhibition of hepatic drug-metabolizing enzymes by arachidonic acid. Xenobiotica 1979, 9, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Ortiz de Montellano, P.R. Substrate Oxidation by Cytochrome P450 Enzymes. In Cytochrome P450; Springer International Publishing: Cham, Switzerland, 2015; pp. 111–176. ISBN 9783319121079. [Google Scholar]
- Guengerich, F.P. Common and uncommon cytochrome P450 reactions related to metabolism and chemical toxicity. Chem. Res. Toxicol. 2001, 14, 611–650. [Google Scholar] [CrossRef]
- Isin, E.M.; Guengerich, F.P. Complex reactions catalyzed by cytochrome P450 enzymes. Biochim. Biophys. Acta 2007, 1770, 314–329. [Google Scholar] [CrossRef]
- Guengerich, F.P. Mechanisms of cytochrome P450-catalyzed oxidations. ACS Catal. 2018, 8, 10964–10976. [Google Scholar] [CrossRef]
- Meunier, B.; de Visser, S.P.; Shaik, S. Mechanism of oxidation reactions catalyzed by cytochrome p450 enzymes. Chem. Rev. 2004, 104, 3947–3980. [Google Scholar] [CrossRef]
- Denisov, I.G.; Makris, T.M.; Sligar, S.G.; Schlichting, I. Structure and chemistry of cytochrome P450. Chem. Rev. 2005, 105, 2253–2277. [Google Scholar] [CrossRef]
- Xu, L.-H.; Fushinobu, S.; Ikeda, H.; Wakagi, T.; Shoun, H. Crystal structures of cytochrome P450 105P1 from Streptomyces avermitilis: Conformational flexibility and histidine ligation state. J. Bacteriol. 2009, 191, 1211–1219. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.-H.; Du, Y.-L. Rational and semi-rational engineering of cytochrome P450s for biotechnological applications. Synth. Syst. Biotechnol. 2018, 3, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Shoieb, S.M.; El-Ghiaty, M.A.; Alqahtani, M.A.; El-Kadi, A.O.S. Cytochrome P450-derived eicosanoids and inflammation in liver diseases. Prostaglandins Other Lipid Mediat. 2020, 147, 106400. [Google Scholar] [CrossRef] [PubMed]
- El-Sherbeni, A.A.; Aboutabl, M.E.; Zordoky, B.N.M.; Anwar-Mohamed, A.; El-Kadi, A.O.S. Determination of the dominant arachidonic acid cytochrome p450 monooxygenases in rat heart, lung, kidney, and liver: Protein expression and metabolite kinetics. AAPS J. 2013, 15, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Isobe, Y.; Itagaki, M.; Ito, Y.; Naoe, S.; Kojima, K.; Ikeguchi, M.; Arita, M. Comprehensive analysis of the mouse cytochrome P450 family responsible for omega-3 epoxidation of eicosapentaenoic acid. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Zeldin, D.C.; DuBois, R.N.; Falck, J.R.; Capdevila, J.H. Molecular cloning, expression and characterization of an endogenous human cytochrome P450 arachidonic acid epoxygenase isoform. Arch. Biochem. Biophys. 1995, 322, 76–86. [Google Scholar] [CrossRef]
- Daikh, B.E.; Lasker, J.M.; Raucy, J.L.; Koop, D.R. Regio- and stereoselective epoxidation of arachidonic acid by human cytochromes P450 2C8 and 2C9. J. Pharmacol. Exp. Ther. 1994, 271, 1427–1433. [Google Scholar]
- Wu, S.; Moomaw, C.R.; Tomer, K.B.; Falck, J.R.; Zeldin, D.C. Molecular cloning and expression of CYP2J2, a human cytochrome P450 arachidonic acid epoxygenase highly expressed in heart. J. Biol. Chem. 1996, 271, 3460–3468. [Google Scholar] [CrossRef]
- Karara, A.; Makita, K.; Jacobson, H.R.; Falck, J.R.; Guengerich, F.P.; DuBois, R.N.; Capdevila, J.H. Molecular cloning, expression, and enzymatic characterization of the rat kidney cytochrome P-450 arachidonic acid epoxygenase. J. Biol. Chem. 1993, 268, 13565–13570. [Google Scholar]
- DeLozier, T.C.; Tsao, C.-C.; Coulter, S.J.; Foley, J.; Bradbury, J.A.; Zeldin, D.C.; Goldstein, J.A. CYP2C44, a new murine CYP2C that metabolizes arachidonic acid to unique stereospecific products. J. Pharmacol. Exp. Ther. 2004, 310, 845–854. [Google Scholar] [CrossRef]
- Draper, A.J.; Hammock, B.D. Identification of CYP2C9 as a human liver microsomal linoleic acid epoxygenase. Arch. Biochem. Biophys. 2000, 376, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Bylund, J.; Kunz, T.; Valmsen, K.; Oliw, E.H. Cytochromes P450 with bisallylic hydroxylation activity on arachidonic and linoleic acids studied with human recombinant enzymes and with human and rat liver microsomes. J. Pharmacol. Exp. Ther. 1998, 284, 51–60. [Google Scholar] [PubMed]
- Daikh, B.E.; Laethem, R.M.; Koop, D.R. Stereoselective epoxidation of arachidonic acid by cytochrome P-450s 2CAA and 2C2. J. Pharmacol. Exp. Ther. 1994, 269, 1130–1135. [Google Scholar] [PubMed]
- Moran, J.H.; Mitchell, L.A.; Bradbury, J.A.; Qu, W.; Zeldin, D.C.; Schnellmann, R.G.; Grant, D.F. Analysis of the cytotoxic properties of linoleic acid metabolites produced by renal and hepatic P450s. Toxicol. Appl. Pharmacol. 2000, 168, 268–279. [Google Scholar] [CrossRef] [PubMed]
- Fer, M.; Dréano, Y.; Lucas, D.; Corcos, L.; Salaün, J.-P.; Berthou, F.; Amet, Y. Metabolism of eicosapentaenoic and docosahexaenoic acids by recombinant human cytochromes P450. Arch. Biochem. Biophys. 2008, 471, 116–125. [Google Scholar] [CrossRef]
- Westphal, C.; Konkel, A.; Schunck, W.-H. Cytochrome p450 enzymes in the bioactivation of polyunsaturated Fatty acids and their role in cardiovascular disease. Adv. Exp. Med. Biol. 2015, 851, 151–187. [Google Scholar] [CrossRef]
- Arnold, C.; Markovic, M.; Blossey, K.; Wallukat, G.; Fischer, R.; Dechend, R.; Konkel, A.; von Schacky, C.; Luft, F.C.; Muller, D.N.; et al. Arachidonic acid-metabolizing cytochrome P450 enzymes are targets of ω-3 fatty acids. J. Biol. Chem. 2010, 285, 32720–32733. [Google Scholar] [CrossRef]
- Barbosa-Sicard, E.; Markovic, M.; Honeck, H.; Christ, B.; Muller, D.N.; Schunck, W.-H. Eicosapentaenoic acid metabolism by cytochrome P450 enzymes of the CYP2C subfamily. Biochem. Biophys. Res. Commun. 2005, 329, 1275–1281. [Google Scholar] [CrossRef]
- Lucas, D.; Goulitquer, S.; Marienhagen, J.; Fer, M.; Dreano, Y.; Schwaneberg, U.; Amet, Y.; Corcos, L. Stereoselective epoxidation of the last double bond of polyunsaturated fatty acids by human cytochromes P450. J. Lipid Res. 2010, 51, 1125–1133. [Google Scholar] [CrossRef]
- Gainer, J.V.; Bellamine, A.; Dawson, E.P.; Womble, K.E.; Grant, S.W.; Wang, Y.; Cupples, L.A.; Guo, C.-Y.; Demissie, S.; O’Donnell, C.J.; et al. Functional variant of CYP4A11 20-hydroxyeicosatetraenoic acid synthase is associated with essential hypertension. Circulation 2005, 111, 63–69. [Google Scholar] [CrossRef]
- Nguyen, X.; Wang, M.-H.; Reddy, K.M.; Falck, J.R.; Schwartzman, M.L. Kinetic profile of the rat CYP4A isoforms: Arachidonic acid metabolism and isoform-specific inhibitors. Am. J. Physiol. Regul. Integr. Comp. Physiol. 1999, 276, R1691–R1700. [Google Scholar] [CrossRef] [PubMed]
- Muller, D.N.; Schmidt, C.; Barbosa-Sicard, E.; Wellner, M.; Gross, V.; Hercule, H.; Markovic, M.; Honeck, H.; Luft, F.C.; Schunck, W.-H. Mouse Cyp4a isoforms: Enzymatic properties, gender- and strain-specific expression, and role in renal 20-hydroxyeicosatetraenoic acid formation. Biochem. J. 2007, 403, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Fer, M.; Corcos, L.; Dréano, Y.; Plée-Gautier, E.; Salaün, J.-P.; Berthou, F.; Amet, Y. Cytochromes P450 from family 4 are the main omega hydroxylating enzymes in humans: CYP4F3B is the prominent player in PUFA metabolism. J. Lipid Res. 2008, 49, 2379–2389. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Chen, W.; Murphy, E.; Gabel, S.; Tomer, K.B.; Foley, J.; Steenbergen, C.; Falck, J.R.; Moomaw, C.R.; Zeldin, D.C. Molecular cloning, expression, and functional significance of a cytochrome P450 highly expressed in rat heart myocytes. J. Biol. Chem. 1997, 272, 12551–12559. [Google Scholar] [CrossRef] [PubMed]
- Adas, F.; Salaün, J.P.; Berthou, F.; Picart, D.; Simon, B.; Amet, Y. Requirement for omega and (omega;-1)-hydroxylations of fatty acids by human cytochromes P450 2E1 and 4A11. J. Lipid Res. 1999, 40, 1990–1997. [Google Scholar] [PubMed]
- Harmon, S.D.; Fang, X.; Kaduce, T.L.; Hu, S.; Raj Gopal, V.; Falck, J.R.; Spector, A.A. Oxygenation of omega-3 fatty acids by human cytochrome P450 4F3B: Effect on 20-hydroxyeicosatetraenoic acid production. Prostaglandins Leukot. Essent. Fat. Acids 2006, 75, 169–177. [Google Scholar] [CrossRef]
- Lauterbach, B.; Barbosa-Sicard, E.; Wang, M.-H.; Honeck, H.; Kärgel, E.; Theuer, J.; Schwartzman, M.L.; Haller, H.; Luft, F.C.; Gollasch, M.; et al. Cytochrome P450-dependent eicosapentaenoic acid metabolites are novel BK channel activators. Hypertension 2002, 39, 609–613. [Google Scholar] [CrossRef]
- Schwarz, D.; Kisselev, P.; Ericksen, S.S.; Szklarz, G.D.; Chernogolov, A.; Honeck, H.; Schunck, W.-H.; Roots, I. Arachidonic and eicosapentaenoic acid metabolism by human CYP1A1: Highly stereoselective formation of 17(R),18(S)-epoxyeicosatetraenoic acid. Biochem. Pharmacol. 2004, 67, 1445–1457. [Google Scholar] [CrossRef]
- Stark, K.; Wongsud, B.; Burman, R.; Oliw, E.H. Oxygenation of polyunsaturated long chain fatty acids by recombinant CYP4F8 and CYP4F12 and catalytic importance of Tyr-125 and Gly-328 of CYP4F8. Arch. Biochem. Biophys. 2005, 441, 174–181. [Google Scholar] [CrossRef]
- Chuang, S.S.; Helvig, C.; Taimi, M.; Ramshaw, H.A.; Collop, A.H.; Amad, M.; White, J.A.; Petkovich, M.; Jones, G.; Korczak, B. CYP2U1, a novel human thymus- and brain-specific cytochrome P450, catalyzes omega- and (omega-1)-hydroxylation of fatty acids. J. Biol. Chem. 2004, 279, 6305–6314. [Google Scholar] [CrossRef]
- Karlgren, M.; Backlund, M.; Johansson, I.; Oscarson, M.; Ingelman-Sundberg, M. Characterization and tissue distribution of a novel human cytochrome P450-CYP2U1. Biochem. Biophys. Res. Commun. 2004, 315, 679–685. [Google Scholar] [CrossRef] [PubMed]
- Node, K.; Huo, Y.; Ruan, X.; Yang, B.; Spiecker, M.; Ley, K.; Zeldin, D.C.; Liao, J.K. Anti-inflammatory properties of cytochrome P450 epoxygenase-derived eicosanoids. Science 1999, 285, 1276–1279. [Google Scholar] [CrossRef] [PubMed]
- Wray, J.A.; Sugden, M.C.; Zeldin, D.C.; Greenwood, G.K.; Samsuddin, S.; Miller-Degraff, L.; Bradbury, J.A.; Holness, M.J.; Warner, T.D.; Bishop-Bailey, D. The epoxygenases CYP2J2 activates the nuclear receptor PPARalpha in vitro and in vivo. PLoS ONE 2009, 4, e7421. [Google Scholar] [CrossRef] [PubMed]
- Morisseau, C.; Inceoglu, B.; Schmelzer, K.; Tsai, H.-J.; Jinks, S.L.; Hegedus, C.M.; Hammock, B.D. Naturally occurring monoepoxides of eicosapentaenoic acid and docosahexaenoic acid are bioactive antihyperalgesic lipids. J. Lipid Res. 2010, 51, 3481–3490. [Google Scholar] [CrossRef]
- Ding, Y.; Frömel, T.; Popp, R.; Falck, J.R.; Schunck, W.-H.; Fleming, I. The biological actions of 11,12-epoxyeicosatrienoic acid in endothelial cells are specific to the R/S-enantiomer and require the G(s) protein. J. Pharmacol. Exp. Ther. 2014, 350, 14–21. [Google Scholar] [CrossRef]
- Zou, A.P.; Fleming, J.T.; Falck, J.R.; Jacobs, E.R.; Gebremedhin, D.; Harder, D.R.; Roman, R.J. Stereospecific effects of epoxyeicosatrienoic acids on renal vascular tone and K(+)-channel activity. Am. J. Physiol. 1996, 270, F822-32. [Google Scholar] [CrossRef]
- Falck, J.R.; Krishna, U.M.; Reddy, Y.K.; Kumar, P.S.; Reddy, K.M.; Hittner, S.B.; Deeter, C.; Sharma, K.K.; Gauthier, K.M.; Campbell, W.B. Comparison of vasodilatory properties of 14,15-EET analogs: Structural requirements for dilation. Am. J. Physiol. Heart Circ. Physiol. 2003, 284, H337–H349. [Google Scholar] [CrossRef]
- Fitzpatrick, F.A.; Ennis, M.D.; Baze, M.E.; Wynalda, M.A.; McGee, J.E.; Liggett, W.F. Inhibition of cyclooxygenase activity and platelet aggregation by epoxyeicosatrienoic acids. Influence of stereochemistry. J. Biol. Chem. 1986, 261, 15334–15338. [Google Scholar]
- Carroll, M.A.; Balazy, M.; Margiotta, P.; Huang, D.D.; Falck, J.R.; McGiff, J.C. Cytochrome P-450-dependent HETEs: Profile of biological activity and stimulation by vasoactive peptides. Am. J. Physiol. 1996, 271, R863-9. [Google Scholar] [CrossRef]
- Cheng, J.; Ou, J.-S.; Singh, H.; Falck, J.R.; Narsimhaswamy, D.; Pritchard, K.A., Jr.; Schwartzman, M.L. 20-hydroxyeicosatetraenoic acid causes endothelial dysfunction via eNOS uncoupling. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H1018–H1026. [Google Scholar] [CrossRef]
- Alonso-Galicia, M.; Falck, J.R.; Reddy, K.M.; Roman, R.J. 20-HETE agonists and antagonists in the renal circulation. Am. J. Physiol. 1999, 277, F790–F796. [Google Scholar] [CrossRef] [PubMed]
- Shoieb, S.M.; El-Sherbeni, A.A.; El-Kadi, A.O.S. Identification of 19-(S/R)hydroxyeicosatetraenoic acid as the first endogenous noncompetitive inhibitor of cytochrome P450 1B1 with enantioselective activity. Drug Metab. Dispos. 2019, 47, 67–70. [Google Scholar] [CrossRef] [PubMed]
- Cinelli, M.A.; Lee, K.S.S. Asymmetric total synthesis of 19,20-epoxydocosapentaenoic acid, a bioactive metabolite of docosahexaenoic acid. J. Org. Chem. 2019, 84, 15362–15372. [Google Scholar] [CrossRef] [PubMed]
- Cinelli, M.A.; Yang, J.; Scharmen, A.; Woodman, J.; Karchalla, L.M.; Lee, K.S.S. Enzymatic synthesis and chemical inversion provide both enantiomers of bioactive epoxydocosapentaenoic acids. J. Lipid Res. 2018, 59, 2237–2252. [Google Scholar] [CrossRef] [PubMed]
- Sasame, H.A.; Ames, M.M.; Nelson, S.D. Cytochrome P-450 and NADPH cytochrome c reductase in rat brain: Formation of catechols and reactive catechol metabolites. Biochem. Biophys. Res. Commun. 1977, 78, 919–926. [Google Scholar] [CrossRef]
- Dutheil, F.; Beaune, P.; Loriot, M.-A. Xenobiotic metabolizing enzymes in the central nervous system: Contribution of cytochrome P450 enzymes in normal and pathological human brain. Biochimie 2008, 90, 426–436. [Google Scholar] [CrossRef]
- Dutheil, F.; Dauchy, S.; Diry, M.; Sazdovitch, V.; Cloarec, O.; Mellottée, L.; Bièche, I.; Ingelman-Sundberg, M.; Flinois, J.-P.; de Waziers, I.; et al. Xenobiotic-metabolizing enzymes and transporters in the normal human brain: Regional and cellular mapping as a basis for putative roles in cerebral function. Drug Metab. Dispos. 2009, 37, 1528–1538. [Google Scholar] [CrossRef]
- Miksys, S.; Tyndale, R.F. Cytochrome P450-mediated drug metabolism in the brain. J. Psychiatry Neurosci. 2013, 38, 152–163. [Google Scholar] [CrossRef]
- Petrov, A.M.; Pikuleva, I.A. Cholesterol 24-hydroxylation by CYP46A1: Benefits of modulation for brain diseases. Neurotherapeutics 2019, 16, 635–648. [Google Scholar] [CrossRef]
- Cheng, J.; Zhen, Y.; Miksys, S.; Beyoğlu, D.; Krausz, K.W.; Tyndale, R.F.; Yu, A.; Idle, J.R.; Gonzalez, F.J. Potential role of CYP2D6 in the central nervous system. Xenobiotica 2013, 43, 973–984. [Google Scholar] [CrossRef]
- Guengerich, F.P. Cytochrome p450 and chemical toxicology. Chem. Res. Toxicol. 2008, 21, 70–83. [Google Scholar] [CrossRef] [PubMed]
- Mann, A.; Miksys, S.L.; Gaedigk, A.; Kish, S.J.; Mash, D.C.; Tyndale, R.F. The neuroprotective enzyme CYP2D6 increases in the brain with age and is lower in Parkinson’s disease patients. Neurobiol. Aging 2012, 33, 2160–2171. [Google Scholar] [CrossRef] [PubMed]
- Garcia, A.N.M.; Muniz, M.T.C.; Souza e Silva, H.R.; da Silva, H.A.; Athayde-Junior, L. Cyp46 polymorphisms in Alzheimer’s disease: A review. J. Mol. Neurosci. 2009, 39, 342–345. [Google Scholar] [CrossRef] [PubMed]
- Boussicault, L.; Alves, S.; Lamazière, A.; Planques, A.; Heck, N.; Moumné, L.; Despres, G.; Bolte, S.; Hu, A.; Pagès, C.; et al. CYP46A1, the rate-limiting enzyme for cholesterol degradation, is neuroprotective in Huntington’s disease. Brain 2016, 139, 953–970. [Google Scholar] [CrossRef] [PubMed]
- Brown, J., 3rd; Theisler, C.; Silberman, S.; Magnuson, D.; Gottardi-Littell, N.; Lee, J.M.; Yager, D.; Crowley, J.; Sambamurti, K.; Rahman, M.M.; et al. Differential expression of cholesterol hydroxylases in Alzheimer’s disease. J. Biol. Chem. 2004, 279, 34674–34681. [Google Scholar] [CrossRef]
- Huang, R.; Poduslo, S.E. CYP19 haplotypes increase risk for Alzheimer’s disease. J. Med. Genet. 2006, 43, e42. [Google Scholar] [CrossRef]
- Solanki, M.; Pointon, A.; Jones, B.; Herbert, K. Cytochrome P450 2J2: Potential role in drug metabolism and cardiotoxicity. Drug Metab. Dispos. 2018, 46, 1053–1065. [Google Scholar] [CrossRef]
- Yan, H.; Kong, Y.; He, B.; Huang, M.; Li, J.; Zheng, J.; Liang, L.; Bi, J.; Zhao, S.; Shi, L. CYP2J2 rs890293 polymorphism is associated with susceptibility to Alzheimer’s disease in the Chinese Han population. Neurosci. Lett. 2015, 593, 56–60. [Google Scholar] [CrossRef]
- Parker, R.E.; Isaacs, N.S. Mechanisms of epoxide reactions. Chem. Rev. 1959, 59, 737–799. [Google Scholar] [CrossRef]
- Arand, M.; Cronin, A.; Oesch, F.; Mowbray, S.L.; Jones, T.A. The telltale structures of epoxide hydrolases. Drug Metab. Rev. 2003, 35, 365–383. [Google Scholar] [CrossRef]
- Moore, M.M.; Pottenger, L.H.; House-Knight, T. Critical review of styrene genotoxicity focused on the mutagenicity/clastogenicity literature and using current organization of economic cooperation and development guidance. Environ. Mol. Mutagen. 2019, 60, 624–663. [Google Scholar] [CrossRef] [PubMed]
- Morisseau, C.; Hammock, B.D. Epoxide hydrolases: Mechanisms, inhibitor designs, and biological roles. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 311–333. [Google Scholar] [CrossRef] [PubMed]
- Sevanian, A.; McLeod, L.L. Catalytic properties and inhibition of hepatic cholesterol-epoxide hydrolase. J. Biol. Chem. 1986, 261, 54–59. [Google Scholar]
- Haeggström, J.Z.; Tholander, F.; Wetterholm, A. Structure and catalytic mechanisms of leukotriene A4 hydrolase. Prostaglandins Other Lipid Mediat. 2007, 83, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Pace-Asciak, C.R.; Lee, W.S. Purification of hepoxilin epoxide hydrolase from rat liver. J. Biol. Chem. 1989, 264, 9310–9313. [Google Scholar] [PubMed]
- Nigam, S.; Zafiriou, M.-P.; Deva, R.; Ciccoli, R.; Roux-Van der Merwe, R. Structure, biochemistry and biology of hepoxilins: An update. FEBS J. 2007, 274, 3503–3512. [Google Scholar] [CrossRef] [PubMed]
- Harris, T.R.; Hammock, B.D. Soluble epoxide hydrolase: Gene structure, expression and deletion. Gene 2013, 526, 61–74. [Google Scholar] [CrossRef]
- Decker, M.; Arand, M.; Cronin, A. Mammalian epoxide hydrolases in xenobiotic metabolism and signalling. Arch. Toxicol. 2009, 83, 297–318. [Google Scholar] [CrossRef]
- Oesch, F. Purification and specificity of a human microsomal epoxide hydratase. Biochem. J. 1974, 139, 77–88. [Google Scholar] [CrossRef]
- Porter, T.D.; Beck, T.W.; Kasper, C.B. Complementary DNA and amino acid sequence of rat liver microsomal, xenobiotic epoxide hydrolase. Arch. Biochem. Biophys. 1986, 248, 121–129. [Google Scholar] [CrossRef]
- Holler, R.; Arand, M.; Mecky, A.; Oesch, F.; Friedberg, T. Erratum to “the membrane anchor of microsomal epoxide hydrolase from human, rat, and rabbit displays an unexpected membrane topology”. Biochem. Biophys. Res. Commun. 1997, 236, 754–759, reprinted in Biochem. Biophys. Res. Commun. 2003, 306, 797. [Google Scholar] [CrossRef] [PubMed]
- Friedberg, T.; Löllmann, B.; Becker, R.; Holler, R.; Oesch, F. The microsomal epoxide hydrolase has a single membrane signal anchor sequence which is dispensable for the catalytic activity of this protein. Biochem. J. 1994, 303 Pt 3, 967–972. [Google Scholar] [CrossRef]
- Zou, J.; Hallberg, B.M.; Bergfors, T.; Oesch, F.; Arand, M.; Mowbray, S.L.; Jones, T.A. Structure of Aspergillus niger epoxide hydrolase at 1.8 Å resolution: Implications for the structure and function of the mammalian microsomal class of epoxide hydrolases. Structure 2000, 8, 111–122. [Google Scholar] [CrossRef]
- Arand, M.; Hemmer, H.; Dürk, H.; Baratti, J.; Archelas, A.; Furstoss, R.; Oesch, F. Cloning and molecular characterization of a soluble epoxide hydrolase from Aspergillus niger that is related to mammalian microsomal epoxide hydrolase. Biochem. J. 1999, 344 Pt 1, 273–280. [Google Scholar] [CrossRef]
- Orjuela Leon, A.C.; Marwosky, A.; Arand, M. Evidence for a complex formation between CYP2J5 and mEH in living cells by FRET analysis of membrane protein interaction in the endoplasmic reticulum (FAMPIR). Arch. Toxicol. 2017, 91, 3561–3570. [Google Scholar] [CrossRef] [PubMed]
- Hammock, B.D.; Loury, D.N.; Moody, D.E.; Ruebner, B.; Baselt, R.; Milam, K.M.; Volberding, P.; Ketterman, A.; Talcott, R. A methodology for the analysis of the preneoplastic antigen. Carcinogenesis 1984, 5, 1467–1473. [Google Scholar] [CrossRef] [PubMed]
- Gomez, G.A.; Morisseau, C.; Hammock, B.D.; Christianson, D.W. Structure of human epoxide hydrolase reveals mechanistic inferences on bifunctional catalysis in epoxide and phosphate ester hydrolysis. Biochemistry 2004, 43, 4716–4723. [Google Scholar] [CrossRef]
- Václavíková, R.; Hughes, D.J.; Souček, P. Microsomal epoxide hydrolase 1 (EPHX1): Gene, structure, function, and role in human disease. Gene 2015, 571, 1–8. [Google Scholar] [CrossRef]
- The Genotype-Tissue Expression (GTEx) Project Protal. Available online: https://www.gtexportal.org/home/gene/EPHX (accessed on 11 November 2020).
- Lord, C.C.; Thomas, G.; Brown, J.M. Mammalian alpha beta hydrolase domain (ABHD) proteins: Lipid metabolizing enzymes at the interface of cell signaling and energy metabolism. Biochim. Biophys. Acta 2013, 1831, 792–802. [Google Scholar] [CrossRef]
- Decker, M.; Adamska, M.; Cronin, A.; Di Giallonardo, F.; Burgener, J.; Marowsky, A.; Falck, J.R.; Morisseau, C.; Hammock, B.D.; Gruzdev, A.; et al. EH3 (ABHD9): The first member of a new epoxide hydrolase family with high activity for fatty acid epoxides. J. Lipid Res. 2012, 53, 2038–2045. [Google Scholar] [CrossRef]
- Yamanashi, H.; Boeglin, W.E.; Morisseau, C.; Davis, R.W.; Sulikowski, G.A.; Hammock, B.D.; Brash, A.R. Catalytic activities of mammalian epoxide hydrolases with cis and trans fatty acid epoxides relevant to skin barrier function. J. Lipid Res. 2018, 59, 684–695. [Google Scholar] [CrossRef] [PubMed]
- Moghaddam, M.F.; Grant, D.F.; Cheek, J.M.; Greene, J.F.; Williamson, K.C.; Hammock, B.D. Bioactivation of leukotoxins to their toxic diols by epoxide hydrolase. Nat. Med. 1997, 3, 562–566. [Google Scholar] [CrossRef] [PubMed]
- Cottrell, S.; Jung, K.; Kristiansen, G.; Eltze, E.; Semjonow, A.; Ittmann, M.; Hartmann, A.; Stamey, T.; Haefliger, C.; Weiss, G. Discovery and validation of 3 novel DNA methylation markers of prostate cancer prognosis. J. Urol. 2007, 177, 1753–1758. [Google Scholar] [CrossRef] [PubMed]
- Oster, B.; Thorsen, K.; Lamy, P.; Wojdacz, T.K.; Hansen, L.L.; Birkenkamp-Demtröder, K.; Sørensen, K.D.; Laurberg, S.; Orntoft, T.F.; Andersen, C.L. Identification and validation of highly frequent CpG island hypermethylation in colorectal adenomas and carcinomas. Int. J. Cancer 2011, 129, 2855–2866. [Google Scholar] [CrossRef]
- Yamashita, S.; Tsujino, Y.; Moriguchi, K.; Tatematsu, M.; Ushijima, T. Chemical genomic screening for methylation-silenced genes in gastric cancer cell lines using 5-aza-2′-deoxycytidine treatment and oligonucleotide microarray. Cancer Sci. 2006, 97, 64–71. [Google Scholar] [CrossRef]
- Furuta, J.; Nobeyama, Y.; Umebayashi, Y.; Otsuka, F.; Kikuchi, K.; Ushijima, T. Silencing of Peroxiredoxin 2 and aberrant methylation of 33 CpG islands in putative promoter regions in human malignant melanomas. Cancer Res. 2006, 66, 6080–6086. [Google Scholar] [CrossRef]
- Krig, S.R.; Jin, V.X.; Bieda, M.C.; O’Geen, H.; Yaswen, P.; Green, R.; Farnham, P.J. Identification of genes directly regulated by the oncogene ZNF217 using chromatin immunoprecipitation (ChIP)-chip assays. J. Biol. Chem. 2007, 282, 9703–9712. [Google Scholar] [CrossRef]
- Schnekenburger, M.; Diederich, M. Epigenetics offer New Horizons for colorectal cancer prevention. Curr. Colorectal Cancer Rep. 2012, 8, 66–81. [Google Scholar] [CrossRef]
- Oliw, E.H.; Guengerich, F.P.; Oates, J.A. Oxygenation of arachidonic acid by hepatic monooxygenases. Isolation and metabolism of four epoxide intermediates. J. Biol. Chem. 1982, 257, 3771–3781. [Google Scholar]
- Zeldin, D.C.; Wei, S.; Falck, J.R.; Hammock, B.D.; Snapper, J.R.; Capdevila, J.H. Metabolism of epoxyeicosatrienoic acids by cytosolic epoxide hydrolase: Substrate structural determinants of asymmetric catalysis. Arch. Biochem. Biophys. 1995, 316, 443–451. [Google Scholar] [CrossRef]
- Marowsky, A.; Burgener, J.; Falck, J.R.; Fritschy, J.-M.; Arand, M. Distribution of soluble and microsomal epoxide hydrolase in the mouse brain and its contribution to cerebral epoxyeicosatrienoic acid metabolism. Neuroscience 2009, 163, 646–661. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, R.N.; Cassidy, C.S. New structural and chemical insight into the catalytic mechanism of epoxide hydrolases. Drug Metab. Rev. 2000, 32, 327–338. [Google Scholar] [CrossRef] [PubMed]
- Farin, F.M.; Omiecinski, C.J. Regiospecific expression of cytochrome P-450s and microsomal epoxide hydrolase in human brain tissue. J. Toxicol. Environ. Health 1993, 40, 317–335. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Sun, A.; Shin, E.-J.; Liu, X.; Kim, S.-G.; Runyons, C.R.; Markesbery, W.; Kim, H.-C.; Bing, G. Expression of microsomal epoxide hydrolase is elevated in Alzheimer’s hippocampus and induced by exogenous beta-amyloid and trimethyl-tin. Eur. J. Neurosci. 2006, 23, 2027–2034. [Google Scholar] [CrossRef] [PubMed]
- Shin, E.-J.; Bing, G.; Chae, J.S.; Kim, T.W.; Bach, J.-H.; Park, D.H.; Yamada, K.; Nabeshima, T.; Kim, H.-C. Role of microsomal epoxide hydrolase in methamphetamine-induced drug dependence in mice. J. Neurosci. Res. 2009, 87, 3679–3686. [Google Scholar] [CrossRef] [PubMed]
- Kessler, R.; Hamou, M.F.; Albertoni, M.; de Tribolet, N.; Arand, M.; Van Meir, E.G. Identification of the putative brain tumor antigen BF7/GE2 as the (de)toxifying enzyme microsomal epoxide hydrolase. Cancer Res. 2000, 60, 1403–1409. [Google Scholar]
- Rawal, S.; Morisseau, C.; Hammock, B.D.; Shivachar, A.C. Differential subcellular distribution and colocalization of the microsomal and soluble epoxide hydrolases in cultured neonatal rat brain cortical astrocytes. J. Neurosci. Res. 2009, 87, 218–227. [Google Scholar] [CrossRef]
- Kohn, M.C.; Melnick, R.L. Physiological modeling of butadiene disposition in mice and rats. Chem. Biol. Interact. 2001, 135–136, 285–301. [Google Scholar] [CrossRef]
- Sura, P.; Sura, R.; Enayetallah, A.E.; Grant, D.F. Distribution and expression of soluble epoxide hydrolase in human brain. J. Histochem. Cytochem. 2008, 56, 551–559. [Google Scholar] [CrossRef]
- Atone, J.; Wagner, K.; Hashimoto, K.; Hammock, B.D. Cytochrome P450 derived epoxidized fatty acids as a therapeutic tool against neuroinflammatory diseases. Prostaglandins Other Lipid Mediat. 2020, 147, 106385. [Google Scholar] [CrossRef]
- Hung, T.-H.; Shyue, S.-K.; Wu, C.-H.; Chen, C.-C.; Lin, C.-C.; Chang, C.-F.; Chen, S.-F. Deletion or inhibition of soluble epoxide hydrolase protects against brain damage and reduces microglia-mediated neuroinflammation in traumatic brain injury. Oncotarget 2017, 8, 103236–103260. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Hong, G.; Lee, K.S.S.; Hammock, B.D.; Gebremedhin, D.; Harder, D.R.; Koehler, R.C.; Sapirstein, A. Inhibition of soluble epoxide hydrolase augments astrocyte release of vascular endothelial growth factor and neuronal recovery after oxygen-glucose deprivation. J. Neurochem. 2017, 140, 814–825. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.M.; McReynolds, C.B.; Schmidt, W.K.; Hammock, B.D. Soluble epoxide hydrolase as a therapeutic target for pain, inflammatory and neurodegenerative diseases. Pharmacol. Ther. 2017, 180, 62–76. [Google Scholar] [CrossRef] [PubMed]
- Abdu, E.; Bruun, D.A.; Yang, D.; Yang, J.; Inceoglu, B.; Hammock, B.D.; Alkayed, N.J.; Lein, P.J. Epoxyeicosatrienoic acids enhance axonal growth in primary sensory and cortical neuronal cell cultures. J. Neurochem. 2011, 117, 632–642. [Google Scholar] [CrossRef][Green Version]
- Hoopes, S.L.; Gruzdev, A.; Edin, M.L.; Graves, J.P.; Bradbury, J.A.; Flake, G.P.; Lih, F.B.; DeGraff, L.M.; Zeldin, D.C. Generation and characterization of epoxide hydrolase 3 (EPHX3)-deficient mice. PLoS ONE 2017, 12, e0175348. [Google Scholar] [CrossRef]
- Marowsky, A.; Arand, M. Mammalian Epoxide Hydrolases. In Comprehensive Toxicology; Elsevier: Amsterdam, The Netherlands, 2018; pp. 308–325. ISBN 9780081006016. [Google Scholar]
- Weintraub, N.L.; Fang, X.; Kaduce, T.L.; VanRollins, M.; Chatterjee, P.; Spector, A.A. Potentiation of endothelium-dependent relaxation by epoxyeicosatrienoic acids. Circ. Res. 1997, 81, 258–267. [Google Scholar] [CrossRef]
- Oltman, C.L.; Weintraub, N.L.; VanRollins, M.; Dellsperger, K.C. Epoxyeicosatrienoic acids and dihydroxyeicosatrienoic acids are potent vasodilators in the canine coronary microcirculation. Circ. Res. 1998, 83, 932–939. [Google Scholar] [CrossRef]
- Larsen, B.T.; Miura, H.; Hatoum, O.A.; Campbell, W.B.; Hammock, B.D.; Zeldin, D.C.; Falck, J.R.; Gutterman, D.D. Epoxyeicosatrienoic and dihydroxyeicosatrienoic acids dilate human coronary arterioles via BK(Ca) channels: Implications for soluble epoxide hydrolase inhibition. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H491–H499. [Google Scholar] [CrossRef]
- Abukhashim, M.; Wiebe, G.J.; Seubert, J.M. Regulation of forskolin-induced cAMP production by cytochrome P450 epoxygenase metabolites of arachidonic acid in HEK293 cells. Cell Biol. Toxicol. 2011, 27, 321–332. [Google Scholar] [CrossRef]
- Fang, X.; Hu, S.; Xu, B.; Snyder, G.D.; Harmon, S.; Yao, J.; Liu, Y.; Sangras, B.; Falck, J.R.; Weintraub, N.L.; et al. 14,15-Dihydroxyeicosatrienoic acid activates peroxisome proliferator-activated receptor-alpha. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H55–H63. [Google Scholar] [CrossRef]
- Ng, V.Y.; Huang, Y.; Reddy, L.M.; Falck, J.R.; Lin, E.T.; Kroetz, D.L. Cytochrome P450 eicosanoids are activators of peroxisome proliferator-activated receptor alpha. Drug Metab. Dispos. 2007, 35, 1126–1134. [Google Scholar] [CrossRef] [PubMed]
- Schepmann, H.G.; Pang, J.; Matsuda, S.P. Cloning and characterization of Ginkgo biloba levopimaradiene synthase which catalyzes the first committed step in ginkgolide biosynthesis. Arch. Biochem. Biophys. 2001, 392, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Moran, J.H.; Mon, T.; Hendrickson, T.L.; Mitchell, L.A.; Grant, D.F. Defining mechanisms of toxicity for linoleic acid monoepoxides and diols in Sf-21 cells. Chem. Res. Toxicol. 2001, 14, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Stanford, K.I.; Lynes, M.D.; Takahashi, H.; Baer, L.A.; Arts, P.J.; May, F.J.; Lehnig, A.C.; Middelbeek, R.J.W.; Richard, J.J.; So, K.; et al. 12,13-diHOME: An Exercise-Induced Lipokine that Increases Skeletal Muscle Fatty Acid Uptake. Cell Metab. 2018, 27, 1111–1120.e3. [Google Scholar] [CrossRef] [PubMed]
- Kundu, S.; Roome, T.; Bhattacharjee, A.; Carnevale, K.A.; Yakubenko, V.P.; Zhang, R.; Hwang, S.H.; Hammock, B.D.; Cathcart, M.K. Metabolic products of soluble epoxide hydrolase are essential for monocyte chemotaxis to MCP-1 in vitro and in vivo. J. Lipid Res. 2013, 54, 436–447. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.; Inceoglu, B.; Hammock, B.D. Soluble epoxide hydrolase inhibition, epoxygenated fatty acids and nociception. Prostaglandins Other Lipid Mediat. 2011, 96, 76–83. [Google Scholar] [CrossRef]
- Spector, A.A. Arachidonic acid cytochrome P450 epoxygenase pathway. J. Lipid Res. 2009, 50, S52–S56. [Google Scholar] [CrossRef]
- Iliff, J.J.; Jia, J.; Nelson, J.; Goyagi, T.; Klaus, J.; Alkayed, N.J. Epoxyeicosanoid signaling in CNS function and disease. Prostaglandins Other Lipid Mediat. 2010, 91, 68–84. [Google Scholar] [CrossRef]
- Spector, A.A.; Norris, A.W. Action of epoxyeicosatrienoic acids on cellular function. Am. J. Physiol. Cell Physiol. 2007, 292, C996–C1012. [Google Scholar] [CrossRef]
- Hashimoto, M.; Katakura, M.; Tanabe, Y.; Al Mamun, A.; Inoue, T.; Hossain, S.; Arita, M.; Shido, O. N-3 fatty acids effectively improve the reference memory-related learning ability associated with increased brain docosahexaenoic acid-derived docosanoids in aged rats. Biochim. Biophys. Acta 2015, 1851, 203–209. [Google Scholar] [CrossRef]
- Ostermann, A.I.; Waindok, P.; Schmidt, M.J.; Chiu, C.-Y.; Smyl, C.; Rohwer, N.; Weylandt, K.-H.; Schebb, N.H. Modulation of the endogenous omega-3 fatty acid and oxylipin profile in vivo-A comparison of the fat-1 transgenic mouse with C57BL/6 wildtype mice on an omega-3 fatty acid enriched diet. PLoS ONE 2017, 12, e0184470. [Google Scholar] [CrossRef] [PubMed]
- Ostermann, A.I.; Reutzel, M.; Hartung, N.; Franke, N.; Kutzner, L.; Schoenfeld, K.; Weylandt, K.-H.; Eckert, G.P.; Schebb, N.H. A diet rich in omega-3 fatty acids enhances expression of soluble epoxide hydrolase in murine brain. Prostaglandins Other Lipid Mediat. 2017, 133, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Rey, C.; Delpech, J.C.; Madore, C.; Nadjar, A.; Greenhalgh, A.D.; Amadieu, C.; Aubert, A.; Pallet, V.; Vaysse, C.; Layé, S.; et al. Dietary n-3 long chain PUFA supplementation promotes a pro-resolving oxylipin profile in the brain. Brain Behav. Immun. 2019, 76, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Taha, A.Y.; Hennebelle, M.; Yang, J.; Zamora, D.; Rapoport, S.I.; Hammock, B.D.; Ramsden, C.E. Regulation of rat plasma and cerebral cortex oxylipin concentrations with increasing levels of dietary linoleic acid. Prostaglandins Leukot. Essent. Fat. Acids 2018, 138, 71–80. [Google Scholar] [CrossRef]
- Taha, A.Y.; Blanchard, H.C.; Cheon, Y.; Ramadan, E.; Chen, M.; Chang, L.; Rapoport, S.I. Dietary linoleic acid lowering reduces lipopolysaccharide-induced increase in brain arachidonic acid metabolism. Mol. Neurobiol. 2017, 54, 4303–4315. [Google Scholar] [CrossRef]
- Leng, S.; Winter, T.; Aukema, H.M. Dietary LA and sex effects on oxylipin profiles in rat kidney, liver, and serum differ from their effects on PUFAs. J. Lipid Res. 2017, 58, 1702–1712. [Google Scholar] [CrossRef]
- Leng, S.; Winter, T.; Aukema, H.M. Dietary ALA, EPA and DHA have distinct effects on oxylipin profiles in female and male rat kidney, liver and serum. J. Nutr. Biochem. 2018, 57, 228–237. [Google Scholar] [CrossRef]
- Mendonça, A.M.; Cayer, L.G.J.; Pauls, S.D.; Winter, T.; Leng, S.; Taylor, C.G.; Zahradka, P.; Aukema, H.M. Distinct effects of dietary ALA, EPA and DHA on rat adipose oxylipins vary by depot location and sex. Prostaglandins Leukot. Essent. Fat. Acids 2018, 129, 13–24. [Google Scholar] [CrossRef]
- Sochocka, M.; Diniz, B.S.; Leszek, J. Inflammatory response in the CNS: Friend or foe? Mol. Neurobiol. 2017, 54, 8071–8089. [Google Scholar] [CrossRef]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef]
- Simon, D.W.; McGeachy, M.J.; Bayır, H.; Clark, R.S.B.; Loane, D.J.; Kochanek, P.M. The far-reaching scope of neuroinflammation after traumatic brain injury. Nat. Rev. Neurol. 2017, 13, 171–191. [Google Scholar] [CrossRef]
- Zhang, J.-M.; An, J. Cytokines, inflammation, and pain. Int. Anesthesiol. Clin. 2007, 45, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Liu, Y.; Zhou, J. Neuroinflammation in Parkinson’s disease and its potential as therapeutic target. Transl. Neurodegener. 2015, 4, 19. [Google Scholar] [CrossRef]
- Kodani, S.D.; Morisseau, C. Role of epoxy-fatty acids and epoxide hydrolases in the pathology of neuro-inflammation. Biochimie 2019, 159, 59–65. [Google Scholar] [CrossRef]
- Vezzani, A.; Balosso, S.; Ravizza, T. Neuroinflammatory pathways as treatment targets and biomarkers in epilepsy. Nat. Rev. Neurol. 2019, 15, 459–472. [Google Scholar] [CrossRef]
- Taguchi, N.; Nakayama, S.; Tanaka, M. Single administration of soluble epoxide hydrolase inhibitor suppresses neuroinflammation and improves neuronal damage after cardiac arrest in mice. Neurosci. Res. 2016, 111, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Pallàs, M.; Vázquez, S.; Sanfeliu, C.; Galdeano, C.; Griñán-Ferré, C. Soluble epoxide hydrolase inhibition to face neuroinflammation in Parkinson’s disease: A new therapeutic strategy. Biomolecules 2020, 10, 703. [Google Scholar] [CrossRef] [PubMed]
- Vito, S.T.; Austin, A.T.; Banks, C.N.; Inceoglu, B.; Bruun, D.A.; Zolkowska, D.; Tancredi, D.J.; Rogawski, M.A.; Hammock, B.D.; Lein, P.J. Post-exposure administration of diazepam combined with soluble epoxide hydrolase inhibition stops seizures and modulates neuroinflammation in a murine model of acute TETS intoxication. Toxicol. Appl. Pharmacol. 2014, 281, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Hung, Y.-W.; Hung, S.-W.; Wu, Y.-C.; Wong, L.-K.; Lai, M.-T.; Shih, Y.-H.; Lee, T.-S.; Lin, Y.-Y. Soluble epoxide hydrolase activity regulates inflammatory responses and seizure generation in two mouse models of temporal lobe epilepsy. Brain Behav. Immun. 2015, 43, 118–129. [Google Scholar] [CrossRef] [PubMed]
- Birnie, M.; Morrison, R.; Camara, R.; Strauss, K.I. Temporal changes of cytochrome P450 (Cyp) and eicosanoid-related gene expression in the rat brain after traumatic brain injury. BMC Genom. 2013, 14, 303. [Google Scholar] [CrossRef]
- Yuan, L.; Liu, J.; Dong, R.; Zhu, J.; Tao, C.; Zheng, R.; Zhu, S. 14,15-epoxyeicosatrienoic acid promotes production of brain derived neurotrophic factor from astrocytes and exerts neuroprotective effects during ischaemic injury. Neuropathol. Appl. Neurobiol. 2016, 42, 607–620. [Google Scholar] [CrossRef] [PubMed]
- Oguro, A.; Inoue, T.; Kudoh, S.N.; Imaoka, S. 14,15-epoxyeicosatrienoic acid produced by cytochrome P450s enhances neurite outgrowth of PC12 and rat hippocampal neuronal cells. Pharmacol. Res. Perspect. 2018, 6, e00428. [Google Scholar] [CrossRef] [PubMed]
- Simpkins, A.N.; Rudic, R.D.; Schreihofer, D.A.; Roy, S.; Manhiani, M.; Tsai, H.-J.; Hammock, B.D.; Imig, J.D. Soluble epoxide inhibition is protective against cerebral ischemia via vascular and neural protection. Am. J. Pathol. 2009, 174, 2086–2095. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Otsuka, T.; Sugo, N.; Ardeshiri, A.; Alhadid, Y.K.; Iliff, J.J.; DeBarber, A.E.; Koop, D.R.; Alkayed, N.J. Soluble epoxide hydrolase gene deletion is protective against experimental cerebral ischemia. Stroke 2008, 39, 2073–2078. [Google Scholar] [CrossRef]
- Reitz, C.; Mayeux, R. Alzheimer disease: Epidemiology, diagnostic criteria, risk factors and biomarkers. Biochem. Pharmacol. 2014, 88, 640–651. [Google Scholar] [CrossRef]
- Tönnies, E.; Trushina, E. Oxidative stress, synaptic dysfunction, and Alzheimer’s disease. J. Alzheimers. Dis. 2017, 57, 1105–1121. [Google Scholar] [CrossRef]
- Tarasoff-Conway, J.M.; Carare, R.O.; Osorio, R.S.; Glodzik, L.; Butler, T.; Fieremans, E.; Axel, L.; Rusinek, H.; Nicholson, C.; Zlokovic, B.V.; et al. Clearance systems in the brain-implications for Alzheimer disease. Nat. Rev. Neurol. 2015, 11, 457–470. [Google Scholar] [CrossRef]
- Casley, C.S.; Land, J.M.; Sharpe, M.A.; Clark, J.B.; Duchen, M.R.; Canevari, L. Beta-amyloid fragment 25-35 causes mitochondrial dysfunction in primary cortical neurons. Neurobiol. Dis. 2002, 10, 258–267. [Google Scholar] [CrossRef]
- Van Giau, V.; An, S.S.A.; Hulme, J.P. Mitochondrial therapeutic interventions in Alzheimer’s disease. J. Neurol. Sci. 2018, 395, 62–70. [Google Scholar] [CrossRef]
- Sarkar, P.; Zaja, I.; Bienengraeber, M.; Rarick, K.R.; Terashvili, M.; Canfield, S.; Falck, J.R.; Harder, D.R. Epoxyeicosatrienoic acids pretreatment improves amyloid β-induced mitochondrial dysfunction in cultured rat hippocampal astrocytes. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H475–H484. [Google Scholar] [CrossRef]
- Sarkar, P.; Narayanan, J.; Harder, D.R. Differential effect of amyloid β on the cytochrome P450 epoxygenase activity in rat brain. Neuroscience 2011, 194, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Mejia, R.O.; Newman, J.W.; Toh, S.; Yu, G.-Q.; Zhou, Y.; Halabisky, B.; Cissé, M.; Scearce-Levie, K.; Cheng, I.H.; Gan, L.; et al. Phospholipase A2 reduction ameliorates cognitive deficits in a mouse model of Alzheimer’s disease. Nat. Neurosci. 2008, 11, 1311–1318. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Comerota, M.E.; Wan, D.; Chen, F.; Propson, N.E.; Hwang, S.H.; Hammock, B.D.; Zheng, H. Epoxy fatty acid dysregulation and neuroinflammation in Alzheimer’s disease is resolved by a soluble epoxide hydrolase inhibitor. bioRxiv 2020. [Google Scholar] [CrossRef]
- Lee, H.-T.; Lee, K.-I.; Chen, C.-H.; Lee, T.-S. Genetic deletion of soluble epoxide hydrolase delays the progression of Alzheimer’s disease. J. Neuroinflamm. 2019, 16, 267. [Google Scholar] [CrossRef] [PubMed]
- Griñán-Ferré, C.; Codony, S.; Pujol, E.; Yang, J.; Leiva, R.; Escolano, C.; Puigoriol-Illamola, D.; Companys-Alemany, J.; Corpas, R.; Sanfeliu, C.; et al. Pharmacological inhibition of soluble epoxide hydrolase as a new therapy for Alzheimer’s disease. Neurotherapeutics 2020. [Google Scholar] [CrossRef] [PubMed]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.-E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Primers 2017, 3, 17013. [Google Scholar] [CrossRef] [PubMed]
- Balestrino, R.; Schapira, A.H.V. Parkinson disease. Eur. J. Neurol. 2020, 27, 27–42. [Google Scholar] [CrossRef]
- Qin, X.; Wu, Q.; Lin, L.; Sun, A.; Liu, S.; Li, X.; Cao, X.; Gao, T.; Luo, P.; Zhu, X.; et al. Soluble epoxide hydrolase deficiency or inhibition attenuates MPTP-induced parkinsonism. Mol. Neurobiol. 2015, 52, 187–195. [Google Scholar] [CrossRef]
- Huang, H.-J.; Wang, Y.-T.; Lin, H.-C.; Lee, Y.-H.; Lin, A.M.-Y. Soluble epoxide hydrolase inhibition attenuates MPTP-induced neurotoxicity in the nigrostriatal dopaminergic system: Involvement of α-synuclein aggregation and ER stress. Mol. Neurobiol. 2018, 55, 138–144. [Google Scholar] [CrossRef]
- Borlongan, C.V. Fatty acid chemical mediator provides insights into the pathology and treatment of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2018, 115, 6322–6324. [Google Scholar] [CrossRef]
- Bishop-Bailey, D.; Bystrom, J. Emerging roles of peroxisome proliferator-activated receptor-beta/delta in inflammation. Pharmacol. Ther. 2009, 124, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Moraes, L.A.; Piqueras, L.; Bishop-Bailey, D. Peroxisome proliferator-activated receptors and inflammation. Pharmacol. Ther. 2006, 110, 371–385. [Google Scholar] [CrossRef]
- Bishop-Bailey, D.; Wray, J. Peroxisome proliferator-activated receptors: A critical review on endogenous pathways for ligand generation. Prostaglandins Other Lipid Mediat. 2003, 71, 1–22. [Google Scholar] [CrossRef]
- Cowart, L.A.; Wei, S.; Hsu, M.-H.; Johnson, E.F.; Krishna, M.U.; Falck, J.R.; Capdevila, J.H. The CYP4A isoforms hydroxylate epoxyeicosatrienoic acids to form high affinity peroxisome proliferator-activated receptor ligands. J. Biol. Chem. 2002, 277, 35105–35112. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.A.; Vanden Heuvel, J.P. Reviews: Current topicsrole of nuclear receptors in the regulation of gene expression by dietary fatty acids (review). J. Nutr. Biochem. 2003, 14, 554–567. [Google Scholar] [CrossRef]
- Navarro-Mabarak, C.; Mitre-Aguilar, I.B.; Camacho-Carranza, R.; Arias, C.; Zentella-Dehesa, A.; Espinosa-Aguirre, J.J. Role of NF-κB in cytochrome P450 epoxygenases down-regulation during an inflammatory process in astrocytes. Neurochem. Int. 2019, 129, 104499. [Google Scholar] [CrossRef]
- Berger, J.; Moller, D.E. The mechanisms of action of PPARs. Annu. Rev. Med. 2002, 53, 409–435. [Google Scholar] [CrossRef]
- Večeřa, R.; Zachařová, A.; Orolin, J.; Strojil, J.; Skottová, N.; Anzenbacher, P. Fenofibrate-induced decrease of expression of CYP2C11 and CYP2C6 in rat. Biopharm. Drug Dispos. 2011, 32, 482–487. [Google Scholar] [CrossRef]
- Shaban, Z.; Soliman, M.; El-Shazly, S.; El-Bohi, K.; Abdelazeez, A.; Kehelo, K.; Kim, H.-S.; Muzandu, K.; Ishizuka, M.; Kazusaka, A.; et al. AhR and PPARalpha: Antagonistic effects on CYP2B and CYP3A, and additive inhibitory effects on CYP2C11. Xenobiotica 2005, 35, 51–68. [Google Scholar] [CrossRef]
- Corton, J.C.; Fan, L.Q.; Brown, S.; Anderson, S.P.; Bocos, C.; Cattley, R.C.; Mode, A.; Gustafsson, J.A. Down-regulation of cytochrome P450 2C family members and positive acute-phase response gene expression by peroxisome proliferator chemicals. Mol. Pharmacol. 1998, 54, 463–473. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, Y.; Schmelzer, K.; Lee, T.-S.; Fang, X.; Zhu, Y.; Spector, A.A.; Gill, S.; Morisseau, C.; Hammock, B.D.; et al. The antiinflammatory effect of laminar flow: The role of PPARgamma, epoxyeicosatrienoic acids, and soluble epoxide hydrolase. Proc. Natl. Acad. Sci. USA 2005, 102, 16747–16752. [Google Scholar] [CrossRef] [PubMed]
- Morin, C.; Sirois, M.; Echavé, V.; Albadine, R.; Rousseau, E. 17,18-epoxyeicosatetraenoic acid targets PPARγ and p38 mitogen-activated protein kinase to mediate its anti-inflammatory effects in the lung: Role of soluble epoxide hydrolase. Am. J. Respir. Cell Mol. Biol. 2010, 43, 564–575. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Hu, S.; Watanabe, T.; Weintraub, N.L.; Snyder, G.D.; Yao, J.; Liu, Y.; Shyy, J.Y.-J.; Hammock, B.D.; Spector, A.A. Activation of peroxisome proliferator-activated receptor alpha by substituted urea-derived soluble epoxide hydrolase inhibitors. J. Pharmacol. Exp. Ther. 2005, 314, 260–270. [Google Scholar] [CrossRef]
- Riehle, M.; Tsvetkov, D.; Gohlke, B.-O.; Preissner, R.; Harteneck, C.; Gollasch, M.; Nürnberg, B. Molecular basis for the sensitivity of TRP channels to polyunsaturated fatty acids. Naunyn Schmiedeberg’s. Arch. Pharmacol. 2018, 391, 833–846. [Google Scholar] [CrossRef]
- Parenti, A.; De Logu, F.; Geppetti, P.; Benemei, S. What is the evidence for the role of TRP channels in inflammatory and immune cells? Br. J. Pharmacol. 2016, 173, 953–969. [Google Scholar] [CrossRef]
- Nilius, B.; Szallasi, A. Transient receptor potential channels as drug targets: From the science of basic research to the art of medicine. Pharmacol. Rev. 2014, 66, 676–814. [Google Scholar] [CrossRef]
- Vennekens, R.; Menigoz, A.; Nilius, B. TRPs in the brain. Rev. Physiol. Biochem. Pharmacol. 2012, 163, 27–64. [Google Scholar] [CrossRef]
- Hardie, R.C. A brief history of trp: Commentary and personal perspective. Pflugers Arch. 2011, 461, 493–498. [Google Scholar] [CrossRef]
- Taberner, F.J.; Fernández-Ballester, G.; Fernández-Carvajal, A.; Ferrer-Montiel, A. TRP channels interaction with lipids and its implications in disease. Biochim. Biophys. Acta 2015, 1848, 1818–1827. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Vriens, J.; Prenen, J.; Droogmans, G.; Voets, T.; Nilius, B. Anandamide and arachidonic acid use epoxyeicosatrienoic acids to activate TRPV4 channels. Nature 2003, 424, 434–438. [Google Scholar] [CrossRef]
- Vriens, J.; Owsianik, G.; Fisslthaler, B.; Suzuki, M.; Janssens, A.; Voets, T.; Morisseau, C.; Hammock, B.D.; Fleming, I.; Busse, R.; et al. Modulation of the Ca2 permeable cation channel TRPV4 by cytochrome P450 epoxygenases in vascular endothelium. Circ. Res. 2005, 97, 908–915. [Google Scholar] [CrossRef] [PubMed]
- Earley, S. Endothelium-dependent cerebral artery dilation mediated by transient receptor potential and Ca2+-activated K+ channels. J. Cardiovasc. Pharmacol. 2011, 57, 148–153. [Google Scholar] [CrossRef] [PubMed]
- Higashimori, H.; Blanco, V.M.; Tuniki, V.R.; Falck, J.R.; Filosa, J.A. Role of epoxyeicosatrienoic acids as autocrine metabolites in glutamate-mediated K+ signaling in perivascular astrocytes. Am. J. Physiol. Cell Physiol. 2010, 299, C1068–C1078. [Google Scholar] [CrossRef] [PubMed]
- Caires, R.; Sierra-Valdez, F.J.; Millet, J.R.M.; Herwig, J.D.; Roan, E.; Vásquez, V.; Cordero-Morales, J.F. Omega-3 fatty acids modulate TRPV4 function through plasma membrane remodeling. Cell Rep. 2017, 21, 246–258. [Google Scholar] [CrossRef]
- Loot, A.E.; Fleming, I. Cytochrome P450-derived epoxyeicosatrienoic acids and pulmonary hypertension: Central role of transient receptor potential C6 channels. J. Cardiovasc. Pharmacol. 2011, 57, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Sudhahar, V.; Shaw, S.; Imig, J. Epoxyeicosatrienoic acid analogs and vascular function. Curr. Med. Chem. 2010, 17, 1181–1190. [Google Scholar] [CrossRef]
- Brierley, S.M.; Page, A.J.; Hughes, P.A.; Adam, B.; Liebregts, T.; Cooper, N.J.; Holtmann, G.; Liedtke, W.; Blackshaw, L.A. Selective role for TRPV4 ion channels in visceral sensory pathways. Gastroenterology 2008, 134, 2059–2069. [Google Scholar] [CrossRef]
- Sipe, W.E.B.; Brierley, S.M.; Martin, C.M.; Phillis, B.D.; Cruz, F.B.; Grady, E.F.; Liedtke, W.; Cohen, D.M.; Vanner, S.; Blackshaw, L.A.; et al. Transient receptor potential vanilloid 4 mediates protease activated receptor 2-induced sensitization of colonic afferent nerves and visceral hyperalgesia. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G1288–G1298. [Google Scholar] [CrossRef]
- Sisignano, M.; Park, C.-K.; Angioni, C.; Zhang, D.D.; von Hehn, C.; Cobos, E.J.; Ghasemlou, N.; Xu, Z.-Z.; Kumaran, V.; Lu, R.; et al. 5,6-EET is released upon neuronal activity and induces mechanical pain hypersensitivity via TRPA1 on central afferent terminals. J. Neurosci. 2012, 32, 6364–6372. [Google Scholar] [CrossRef]
- Brenneis, C.; Sisignano, M.; Coste, O.; Altenrath, K.; Fischer, M.J.; Angioni, C.; Fleming, I.; Brandes, R.P.; Reeh, P.W.; Woolf, C.J.; et al. Soluble epoxide hydrolase limits mechanical hyperalgesia during inflammation. Mol. Pain 2011, 7, 78. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, R.; Li, J.; Rao, J.; Li, W.; Falck, J.R.; Manthati, V.L.; Medhora, M.; Jacobs, E.R.; Zhu, D. Stable EET urea agonist and soluble epoxide hydrolase inhibitor regulate rat pulmonary arteries through TRPCs. Hypertens. Res. 2011, 34, 630–639. [Google Scholar] [CrossRef] [PubMed]
- Fleming, I.; Rueben, A.; Popp, R.; Fisslthaler, B.; Schrodt, S.; Sander, A.; Haendeler, J.; Falck, J.R.; Morisseau, C.; Hammock, B.D.; et al. Epoxyeicosatrienoic acids regulate Trp channel dependent Ca2+ signaling and hyperpolarization in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2612–2618. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Zhang, P.; Li, J.; Lu, J.; Ge, J.; Zhao, Z.; Ma, X.; Wan, S.; Yao, X.; Shen, B. Epoxyeicosatrienoic acids act through TRPV4-TRPC1-KCa1.1 complex to induce smooth muscle membrane hyperpolarization and relaxation in human internal mammary arteries. Biochim. Biophys. Acta 2015, 1852, 552–559. [Google Scholar] [CrossRef] [PubMed]
- Pierce, K.L.; Premont, R.T.; Lefkowitz, R.J. Seven-transmembrane receptors. Nat. Rev. Mol. Cell Biol. 2002, 3, 639–650. [Google Scholar] [CrossRef]
- Heng, B.C.; Aubel, D.; Fussenegger, M. An overview of the diverse roles of G-protein coupled receptors (GPCRs) in the pathophysiology of various human diseases. Biotechnol. Adv. 2013, 31, 1676–1694. [Google Scholar] [CrossRef]
- Vassilatis, D.K.; Hohmann, J.G.; Zeng, H.; Li, F.; Ranchalis, J.E.; Mortrud, M.T.; Brown, A.; Rodriguez, S.S.; Weller, J.R.; Wright, A.C.; et al. The G protein-coupled receptor repertoires of human and mouse. Proc. Natl. Acad. Sci. USA 2003, 100, 4903–4908. [Google Scholar] [CrossRef]
- Guerram, M.; Zhang, L.-Y.; Jiang, Z.-Z. G-protein coupled receptors as therapeutic targets for neurodegenerative and cerebrovascular diseases. Neurochem. Int. 2016, 101, 1–14. [Google Scholar] [CrossRef]
- Huang, Y.; Todd, N.; Thathiah, A. The role of GPCRs in neurodegenerative diseases: Avenues for therapeutic intervention. Curr. Opin. Pharmacol. 2017, 32, 96–110. [Google Scholar] [CrossRef]
- Zhao, J.; Deng, Y.; Jiang, Z.; Qing, H. G protein-coupled receptors (GPCRs) in Alzheimer’s disease: A focus on BACE1 related GPCRs. Front. Aging Neurosci. 2016, 8, 58. [Google Scholar] [CrossRef]
- Wong, P.Y.; Lin, K.T.; Yan, Y.T.; Ahern, D.; Iles, J.; Shen, S.Y.; Bhatt, R.K.; Falck, J.R. 14(R),15(S)-epoxyeicosatrienoic acid (14(R),15(S)-EET) receptor in guinea pig mononuclear cell membranes. J. Lipid Mediat. 1993, 6, 199–208. [Google Scholar]
- Wong, P.Y.; Lai, P.S.; Shen, S.Y.; Belosludtsev, Y.Y.; Falck, J.R. Post-receptor signal transduction and regulation of 14(R),15(S)-epoxyeicosatrienoic acid (14,15-EET) binding in U-937 cells. J. Lipid Mediat. Cell Signal. 1997, 16, 155–169. [Google Scholar] [CrossRef]
- Wong, P.Y.; Lai, P.S.; Falck, J.R. Mechanism and signal transduction of 14 (R), 15 (S)-epoxyeicosatrienoic acid (14,15-EET) binding in guinea pig monocytes. Prostaglandins Other Lipid Mediat. 2000, 62, 321–333. [Google Scholar] [CrossRef]
- Campo, G.M.; Avenoso, A.; D’Ascola, A.; Prestipino, V.; Scuruchi, M.; Nastasi, G.; Calatroni, A.; Campo, S. Protein kinase a mediated anti-inflammatory effects exerted by adenosine treatment in mouse chondrocytes stimulated with IL-1β. Biofactors 2012, 38, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.K.; Falck, J.R.; Reddy, K.M.; Capdevila, J.; Harris, R.C. Epoxyeicosatrienoic acids and their sulfonimide derivatives stimulate tyrosine phosphorylation and induce mitogenesis in renal epithelial cells. J. Biol. Chem. 1998, 273, 29254–29261. [Google Scholar] [CrossRef] [PubMed]
- Snyder, G.D.; Krishna, U.M.; Falck, J.R.; Spector, A.A. Evidence for a membrane site of action for 14,15-EET on expression of aromatase in vascular smooth muscle. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H1936–H1942. [Google Scholar] [CrossRef][Green Version]
- Behm, D.J.; Ogbonna, A.; Wu, C.; Burns-Kurtis, C.L.; Douglas, S.A. Epoxyeicosatrienoic acids function as selective, endogenous antagonists of native thromboxane receptors: Identification of a novel mechanism of vasodilation. J. Pharmacol. Exp. Ther. 2009, 328, 231–239. [Google Scholar] [CrossRef]
- Liu, X.; Qian, Z.-Y.; Xie, F.; Fan, W.; Nelson, J.W.; Xiao, X.; Kaul, S.; Barnes, A.P.; Alkayed, N.J. Functional screening for G protein-coupled receptor targets of 14,15-epoxyeicosatrienoic acid. Prostaglandins Other Lipid Mediat. 2017, 132, 31–40. [Google Scholar] [CrossRef]
- Li, P.L.; Chen, C.L.; Bortell, R.; Campbell, W.B. 11,12-Epoxyeicosatrienoic acid stimulates endogenous mono-ADP-ribosylation in bovine coronary arterial smooth muscle. Circ. Res. 1999, 85, 349–356. [Google Scholar] [CrossRef]
- Fukao, M.; Mason, H.S.; Kenyon, J.L.; Horowitz, B.; Keef, K.D. Regulation of BK(Ca) channels expressed in human embryonic kidney 293 cells by epoxyeicosatrienoic acid. Mol. Pharmacol. 2001, 59, 16–23. [Google Scholar] [CrossRef]
- Carroll, M.A.; Doumad, A.B.; Li, J.; Cheng, M.K.; Falck, J.R.; McGiff, J.C. Adenosine2A receptor vasodilation of rat preglomerular microvessels is mediated by EETs that activate the cAMP/PKA pathway. Am. J. Physiol. Renal Physiol. 2006, 291, F155–F161. [Google Scholar] [CrossRef]
- Mule, N.K.; Orjuela Leon, A.C.; Falck, J.R.; Arand, M.; Marowsky, A. 11,12 -Epoxyeicosatrienoic acid (11,12 EET) reduces excitability and excitatory transmission in the hippocampus. Neuropharmacology 2017, 123, 310–321. [Google Scholar] [CrossRef] [PubMed]
- Conroy, J.L.; Fang, C.; Gu, J.; Zeitlin, S.O.; Yang, W.; Yang, J.; VanAlstine, M.A.; Nalwalk, J.W.; Albrecht, P.J.; Mazurkiewicz, J.E.; et al. Opioids activate brain analgesic circuits through cytochrome P450/epoxygenase signaling. Nat. Neurosci. 2010, 13, 284–286. [Google Scholar] [CrossRef] [PubMed]
- Terashvili, M.; Tseng, L.F.; Wu, H.-E.; Narayanan, J.; Hart, L.M.; Falck, J.R.; Pratt, P.F.; Harder, D.R. Antinociception produced by 14,15-epoxyeicosatrienoic acid is mediated by the activation of beta-endorphin and met-enkephalin in the rat ventrolateral periaqueductal gray. J. Pharmacol. Exp. Ther. 2008, 326, 614–622. [Google Scholar] [CrossRef] [PubMed]
- Mercado, G.; Castillo, V.; Soto, P.; Sidhu, A. ER stress and Parkinson’s disease: Pathological inputs that converge into the secretory pathway. Brain Res. 2016, 1648, 626–632. [Google Scholar] [CrossRef]
- Gerakis, Y.; Hetz, C. Emerging roles of ER stress in the etiology and pathogenesis of Alzheimer’s disease. FEBS J. 2018, 285, 995–1011. [Google Scholar] [CrossRef]
- Santos, L.E.; Ferreira, S.T. Crosstalk between endoplasmic reticulum stress and brain inflammation in Alzheimer’s disease. Neuropharmacology 2018, 136, 350–360. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 2010, 140, 900–917. [Google Scholar] [CrossRef]
- Bettaieb, A.; Nagata, N.; AbouBechara, D.; Chahed, S.; Morisseau, C.; Hammock, B.D.; Haj, F.G. Soluble epoxide hydrolase deficiency or inhibition attenuates diet-induced endoplasmic reticulum stress in liver and adipose tissue. J. Biol. Chem. 2013, 288, 14189–14199. [Google Scholar] [CrossRef]
- Dhanasekaran, A.; Gruenloh, S.K.; Buonaccorsi, J.N.; Zhang, R.; Gross, G.J.; Falck, J.R.; Patel, P.K.; Jacobs, E.R.; Medhora, M. Multiple antiapoptotic targets of the PI3K/Akt survival pathway are activated by epoxyeicosatrienoic acids to protect cardiomyocytes from hypoxia/anoxia. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H724–H735. [Google Scholar] [CrossRef]
- Hu, P.; Han, Z.; Couvillon, A.D.; Exton, J.H. Critical role of endogenous Akt/IAPs and MEK1/ERK pathways in counteracting endoplasmic reticulum stress-induced cell death. J. Biol. Chem. 2004, 279, 49420–49429. [Google Scholar] [CrossRef]
- Geng, H.-X.; Li, R.-P.; Li, Y.-G.; Wang, X.-Q.; Zhang, L.; Deng, J.-B.; Wang, L.; Deng, J.-X. 14,15-EET suppresses neuronal apoptosis in ischemia-reperfusion through the mitochondrial pathway. Neurochem. Res. 2017, 42, 2841–2849. [Google Scholar] [CrossRef]
- Rodriguez, D.; Rojas-Rivera, D.; Hetz, C. Integrating stress signals at the endoplasmic reticulum: The BCL-2 protein family rheostat. Biochim. Biophys. Acta 2011, 1813, 564–574. [Google Scholar] [CrossRef] [PubMed]
- Inceoglu, B.; Bettaieb, A.; Trindade da Silva, C.A.; Lee, K.S.S.; Haj, F.G.; Hammock, B.D. Endoplasmic reticulum stress in the peripheral nervous system is a significant driver of neuropathic pain. Proc. Natl. Acad. Sci. USA 2015, 112, 9082–9087. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H.; Burns, J.M.; Khan, S.M. The Alzheimer’s disease mitochondrial cascade hypothesis: Progress and perspectives. Biochim. Biophys. Acta 2014, 1842, 1219–1231. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Zhao, X.; Li, Y.; Li, G.; Liu, X. Damage to dopaminergic neurons by oxidative stress in Parkinson’s disease (Review). Int. J. Mol. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Eckert, G.P.; Lipka, U.; Muller, W.E. Omega-3 fatty acids in neurodegenerative diseases: Focus on mitochondria. Prostaglandins Leukot. Essent. Fat. Acids 2013, 88, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Inceoglu, B.; Bettaieb, A.; Haj, F.G.; Gomes, A.V.; Hammock, B.D. Modulation of mitochondrial dysfunction and endoplasmic reticulum stress are key mechanisms for the wide-ranging actions of epoxy fatty acids and soluble epoxide hydrolase inhibitors. Prostaglandins Other Lipid Mediat. 2017, 133, 68–78. [Google Scholar] [CrossRef]
- Wang, L.; Chen, M.; Yuan, L.; Xiang, Y.; Zheng, R.; Zhu, S. 14,15-EET promotes mitochondrial biogenesis and protects cortical neurons against oxygen/glucose deprivation-induced apoptosis. Biochem. Biophys. Res. Commun. 2014, 450, 604–609. [Google Scholar] [CrossRef]
- Samokhvalov, V.; Alsaleh, N.; El-Sikhry, H.E.; Jamieson, K.L.; Chen, C.B.; Lopaschuk, D.G.; Carter, C.; Light, P.E.; Manne, R.; Falck, J.R.; et al. Epoxyeicosatrienoic acids protect cardiac cells during starvation by modulating an autophagic response. Cell Death Dis. 2013, 4, e885. [Google Scholar] [CrossRef]
- Katragadda, D.; Batchu, S.N.; Cho, W.J.; Chaudhary, K.R.; Falck, J.R.; Seubert, J.M. Epoxyeicosatrienoic acids limit damage to mitochondrial function following stress in cardiac cells. J. Mol. Cell. Cardiol. 2009, 46, 867–875. [Google Scholar] [CrossRef]
- Akao, M.; Ohler, A.; O’Rourke, B.; Marbán, E. Mitochondrial ATP-sensitive potassium channels inhibit apoptosis induced by oxidative stress in cardiac cells. Circ. Res. 2001, 88, 1267–1275. [Google Scholar] [CrossRef] [PubMed]
- Witte, M.E.; Geurts, J.J.G.; de Vries, H.E.; van der Valk, P.; van Horssen, J. Mitochondrial dysfunction: A potential link between neuroinflammation and neurodegeneration? Mitochondrion 2010, 10, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Kodani, S.; Hammock, B.D. Stabilized epoxygenated fatty acids regulate inflammation, pain, angiogenesis and cancer. Prog. Lipid Res. 2014, 53, 108–123. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Chen, C.; Gong, W.; Li, Y.; Edin, M.L.; Zeldin, D.C.; Wang, D.W. Epoxyeicosatrienoic acids attenuate reactive oxygen species level, mitochondrial dysfunction, caspase activation, and apoptosis in carcinoma cells treated with arsenic trioxide. J. Pharmacol. Exp. Ther. 2011, 339, 451–463. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.; Liu, Y.; Zhu, Y.; Chen, L.; Sun, W.; Zhu, Y. Epoxyeicosatrienoic acid inhibits the apoptosis of cerebral microvascular smooth muscle cells by oxygen glucose deprivation via targeting the JNK/c-Jun and mTOR signaling pathways. Mol. Cells 2017, 40, 837–846. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| EH Type | Polarizing Residues | Nucleophilic Residue | Involved in Orientation and Activation of Nucleophilic Residues |
|---|---|---|---|
| mEH | Tyr299 Tyr374 | Asp226 | His431 Glu404 |
| sEH | Tyr383 Tyr466 | Asp335 | His524 Asp496 |
| EH3 | Tyr220 Tyr281 | Asp173 | His337 Asp307 |
| Eh4 | Tyr216 Tyr281 | Asp169 | His336 Asp307 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sarparast, M.; Dattmore, D.; Alan, J.; Lee, K.S.S. Cytochrome P450 Metabolism of Polyunsaturated Fatty Acids and Neurodegeneration. Nutrients 2020, 12, 3523. https://doi.org/10.3390/nu12113523
Sarparast M, Dattmore D, Alan J, Lee KSS. Cytochrome P450 Metabolism of Polyunsaturated Fatty Acids and Neurodegeneration. Nutrients. 2020; 12(11):3523. https://doi.org/10.3390/nu12113523
Chicago/Turabian StyleSarparast, Morteza, Devon Dattmore, Jamie Alan, and Kin Sing Stephen Lee. 2020. "Cytochrome P450 Metabolism of Polyunsaturated Fatty Acids and Neurodegeneration" Nutrients 12, no. 11: 3523. https://doi.org/10.3390/nu12113523
APA StyleSarparast, M., Dattmore, D., Alan, J., & Lee, K. S. S. (2020). Cytochrome P450 Metabolism of Polyunsaturated Fatty Acids and Neurodegeneration. Nutrients, 12(11), 3523. https://doi.org/10.3390/nu12113523