High-Sugar Diet Disrupts Hypothalamic but Not Cerebral Cortex Redox Homeostasis




Abstract
1. Introduction
2. Materials and Methods
2.1. Animal Treatment
2.2. Plasma Insulin and Glucose Determination
2.3. Prooxidant Enzyme Activity Determination
2.4. Enzymatic and Nonenzymatic Antioxidant Determination
2.5. Glutathione Metabolism Determination
2.6. Redox Status Determination
2.7. Oxidative Damage Determination
2.8. Apoptosis Marker Determination
2.9. Statistical Analysis
3. Results
3.1. General Characteristics of the Animals
3.2. Enzymatic and Non-Enzymatic Antioxidants, Glutathione Metabolism, Redox Status, Oxidative Damage and Apoptosis Markers in Plasma/Serum
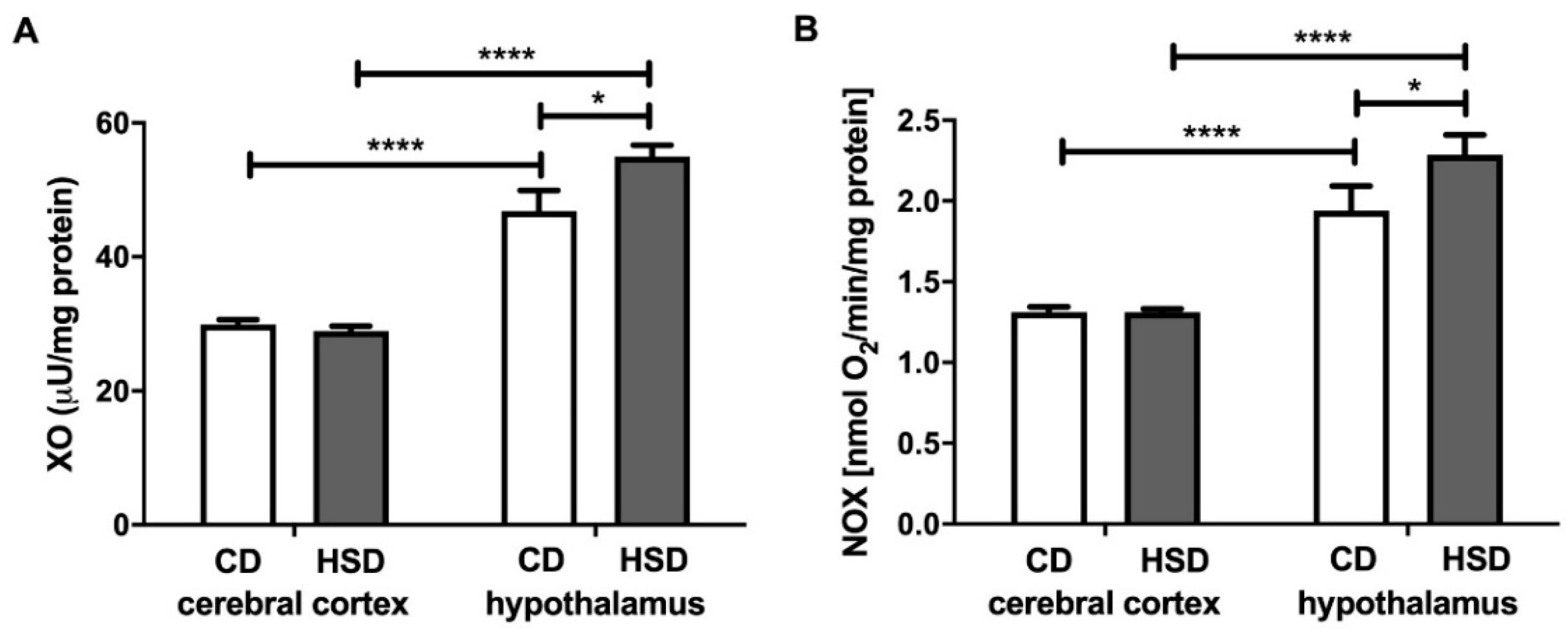
3.3. Prooxidant Brain Enzymes
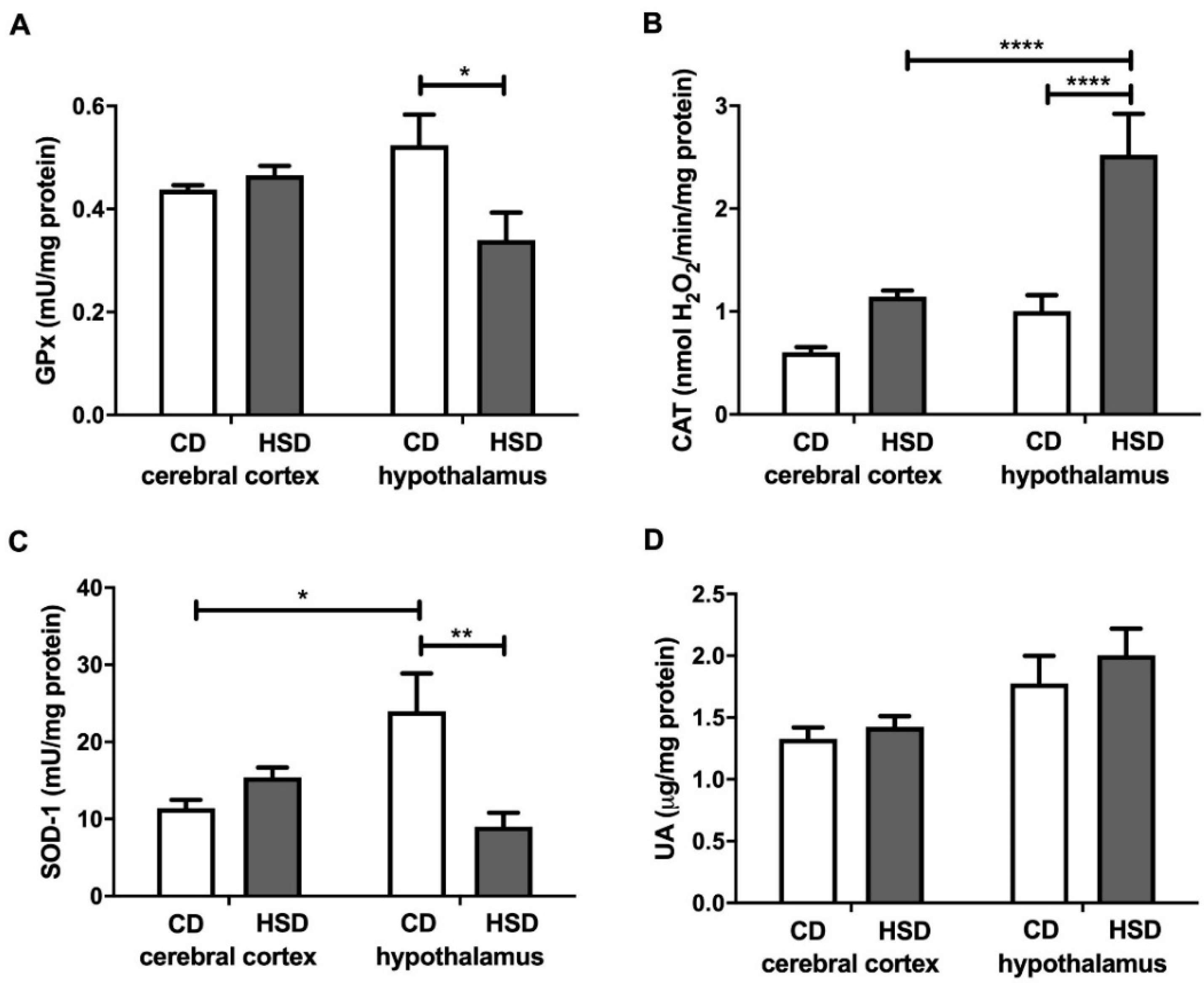
3.4. Enzymatic and Non-Enzymatic Brain Antioxidants
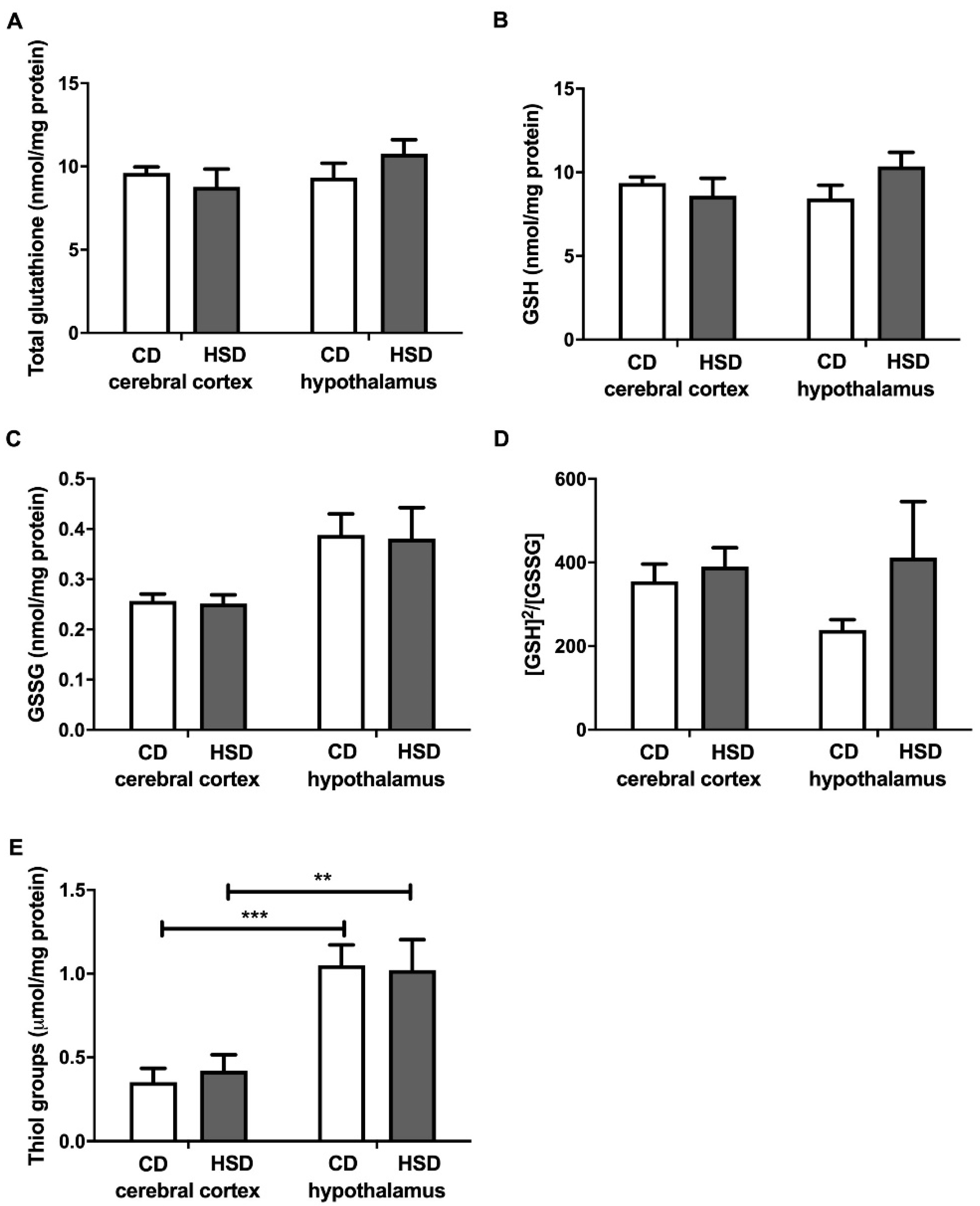
3.5. Brain Glutathione Metabolism
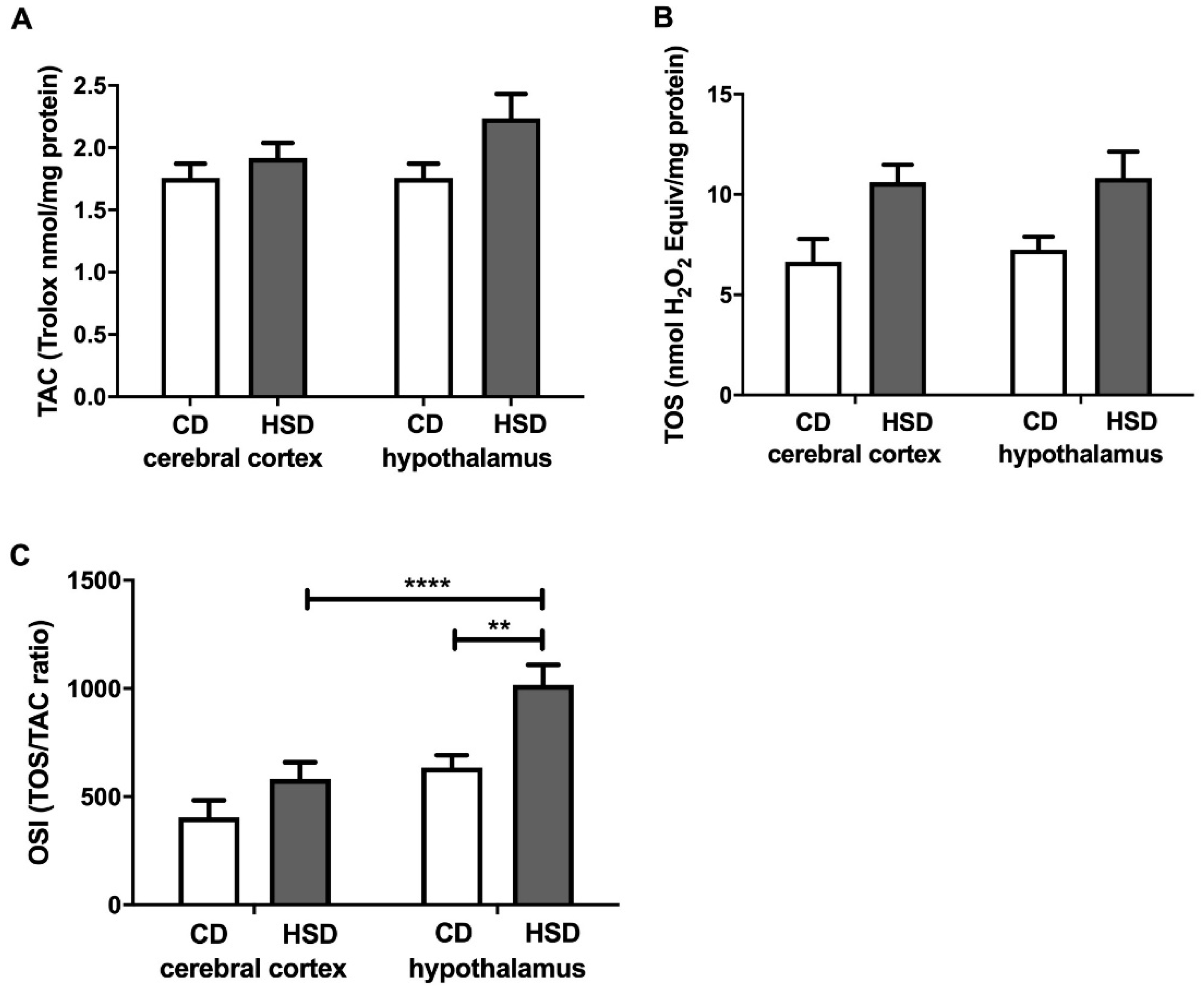
3.6. Brain Redox Status
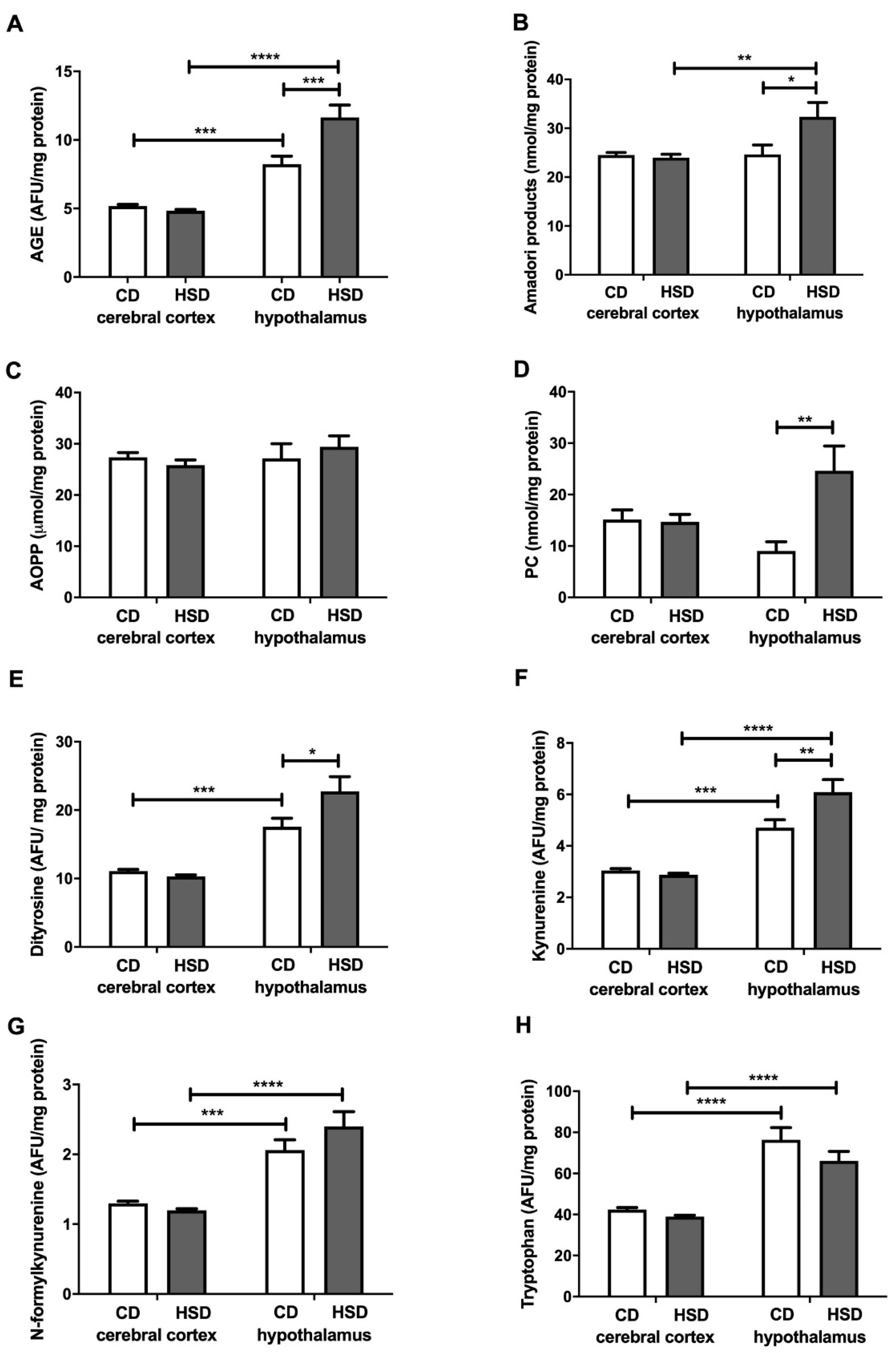
3.7. Brain Oxidative Damage Markers
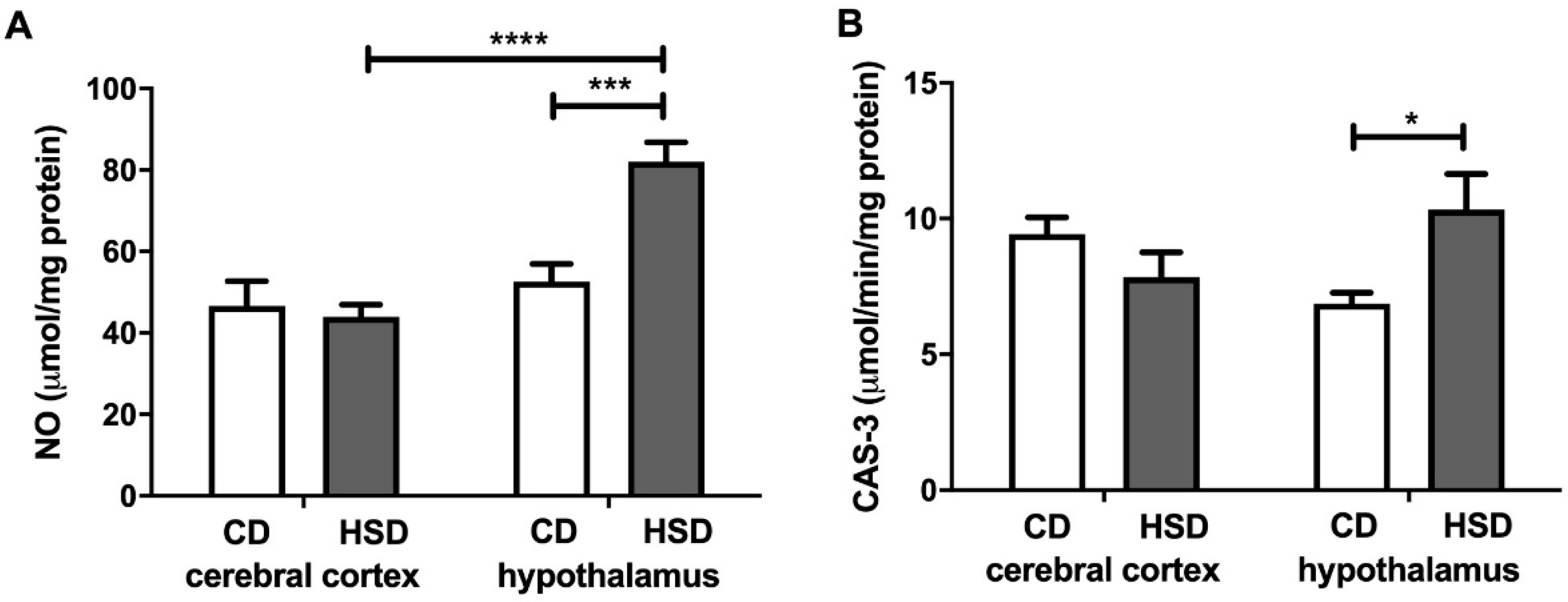
3.8. Brain Apoptosis Markers
3.9. Correlations
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Barazzoni, R.; Cappellari, G.G.; Ragni, M.; Nisoli, E. Insulin resistance in obesity: An overview of fundamental alterations. Eat. Weight. Disord. Stud. Anorexia, Bulim. Obes. 2018, 23, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Wu, J.; Jing, S.; Yan, L.-J. Hyperglycemic Stress and Carbon Stress in Diabetic Glucotoxicity. Aging Dis. 2016, 7, 90–110. [Google Scholar] [CrossRef] [PubMed]
- Giacco, F.; Brownlee, M. Oxidative stress and diabetic complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef]
- Ott, C.; Jacobs, K.; Haucke, E.; Santos, A.N.; Grune, T.; Simm, A. Role of advanced glycation end products in cellular signaling. Redox Biol. 2014, 2, 411–429. [Google Scholar] [CrossRef]
- Wautier, M.-P.; Chappey, O.; Corda, S.; Stern, D.M.; Schmidt, A.M.; Wautier, J.-L. Activation of NADPH oxidase by AGE links oxidant stress to altered gene expression via RAGE. Am. J. Physiol. Metab. 2001, 280, E685–E694. [Google Scholar] [CrossRef] [PubMed]
- Del Prato, S. Role of glucotoxicity and lipotoxicity in the pathophysiology of Type 2 diabetes mellitus and emerging treatment strategies. Diabet. Med. 2009, 26, 1185–1192. [Google Scholar] [CrossRef] [PubMed]
- De La Monte, S.M. Type 3 diabetes is sporadic Alzheimer’s disease: Mini-review. Eur. Neuropsychopharmacol. 2014, 24, 1954–1960. [Google Scholar] [CrossRef] [PubMed]
- Biessels, G.J.; Reagan, L.P. Hippocampal insulin resistance and cognitive dysfunction. Nat. Rev. Neurosci. 2015, 16, 660–671. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Begley, P.; Church, S.J.; Patassini, S.; McHarg, S.; Kureishy, N.; Hollywood, K.A.; Waldvogel, H.J.; Liu, H.; Zhang, S.; et al. Elevation of brain glucose and polyol-pathway intermediates with accompanying brain-copper deficiency in patients with Alzheimer’s disease: Metabolic basis for dementia. Sci. Rep. 2016, 6, 27524. [Google Scholar] [CrossRef] [PubMed]
- Maciejczyk, M.; Żebrowska, E.; Chabowski, A. Insulin Resistance and Oxidative Stress in the Brain: What’s New? Int. J. Mol. Sci. 2019, 20, 874. [Google Scholar] [CrossRef]
- Sripetchwandee, J.; Chattipakorn, N.; Chattipakorn, S.C. Links Between Obesity-Induced Brain Insulin Resistance, Brain Mitochondrial Dysfunction, and Dementia. Front. Endocrinol. 2018, 9, 496. [Google Scholar] [CrossRef] [PubMed]
- De La Monte, S.M.; Longato, L.; Tong, M.; Wands, J.R. Insulin resistance and neurodegeneration: Roles of obesity, type 2 diabetes mellitus and non-alcoholic steatohepatitis. Curr. Opin. Investig. Drugs (Lond. Engl. 2000) 2009, 10, 1049–1060. [Google Scholar]
- Maciejczyk, M.; Zalewska, A.; Gerreth, K. Salivary Redox Biomarkers in Selected Neurodegenerative Diseases. J. Clin. Med. 2020, 9, 497. [Google Scholar] [CrossRef]
- Zalewska, A.; Ziembicka, D.; Żendzian-Piotrowska, M.; Maciejczyk, M. The Impact of High-Fat Diet on Mitochondrial Function, Free Radical Production, and Nitrosative Stress in the Salivary Glands of Wistar Rats. Oxidative Med. Cell. Longev. 2019, 2019, 1–15. [Google Scholar] [CrossRef]
- Cobley, J.N.; Fiorello, M.L.; Bailey, D.M. 13 reasons why the brain is susceptible to oxidative stress. Redox Biol. 2018, 15, 490–503. [Google Scholar] [CrossRef] [PubMed]
- Żebrowska, E.; Maciejczyk, M.; Żendzian-Piotrowska, M.; Zalewska, A.; Chabowski, A. High Protein Diet Induces Oxidative Stress in Rat Cerebral Cortex and Hypothalamus. Int. J. Mol. Sci. 2019, 20, 1547. [Google Scholar] [CrossRef] [PubMed]
- Maciejczyk, M.; Matczuk, J.; Żendzian-Piotrowska, M.; Niklińska, W.; Fejfer, K.; Szarmach, I.; Ładny, J.R.; Zieniewska, I.; Zalewska, A. Eight-Week Consumption of High-Sucrose Diet Has a Pro-Oxidant Effect and Alters the Function of the Salivary Glands of Rats. Nutrients 2018, 10, 1530. [Google Scholar] [CrossRef]
- Novelli, E.L.; Diniz, Y.S.; Galhardi, C.M.; Ebaid, G.M.X.; Rodrigues, H.G.; Mani, F.; Fernandes, A.A.H.; Cicogna, A.C.; Filho, J.L.V.B.N. Anthropometrical parameters and markers of obesity in rats. Lab. Anim. 2007, 41, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Kołodziej, U.; Maciejczyk, M.; Miąsko, A.; Matczuk, J.; Knaś, M.; Żukowski, P.; Żendzian-Piotrowska, M.; Borys, J.; Zalewska, A. Oxidative Modification in the Salivary Glands of High Fat-Diet Induced Insulin Resistant Rats. Front. Physiol. 2017, 8, 20. [Google Scholar] [CrossRef] [PubMed]
- Żukowski, P.; Maciejczyk, M.; Matczuk, J.; Kurek, K.; Waszkiel, D.; Żendzian-Piotrowska, M.; Zalewska, A. Effect of N-Acetylcysteine on Antioxidant Defense, Oxidative Modification, and Salivary Gland Function in a Rat Model of Insulin Resistance. Oxidative Med. Cell. Longev. 2018, 2018, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Griendling, K.K.; Minieri, C.A.; Ollerenshaw, J.D.; Alexander, R.W. Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ. Res. 1994, 74, 1141–1148. [Google Scholar] [CrossRef]
- Prajda, N.; Weber, G. Malignant transformation-linked imbalance: Decreased xanthine oxidase activity in hepatomas. FEBS Lett. 1975, 59, 245–249. [Google Scholar] [CrossRef]
- Walker, J.M. The Bicinchoninic Acid (BCA) Assay for Protein Quantitation. In The Protein Protocols Handbook; Humana Press: Totowa, NJ, USA, 1994. [Google Scholar]
- Paglia, D.E.; Valentine, W.N. Studies on the quantitative and qualitative characterization of erythrocyte glutathione peroxidase. J. Lab. Clin. Med. 1967, 70, 158–169. [Google Scholar] [PubMed]
- Misra, H.P.; Fridovich, I. The role of superoxide anion in the autoxidation of epinephrine and a simple assay for superoxide dismutase. J. Biol. Chem. 1972, 247, 3170–3175. [Google Scholar] [PubMed]
- Aebi, H. Catalase in vitro. In Methods in Enzymology; Elsevier BV: Amsterdam, The Netherlands, 1984; Volume 105, pp. 121–126. [Google Scholar]
- Moron, M.S.; DePIERRE, J.W.; Mannervik, B. Levels of glutathione, glutathione reductase and glutathione S-transferase activities in rat lung and liver. Biochim. Biophys. Acta (BBA) Gen. Subj. 1979, 582, 67–78. [Google Scholar] [CrossRef]
- Zalewska, A.; Szarmach, I.; Żendzian-Piotrowska, M.; Maciejczyk, M. The Effect of N-Acetylcysteine on Respiratory Enzymes, ADP/ATP Ratio, Glutathione Metabolism, and Nitrosative Stress in the Salivary Gland Mitochondria of Insulin Resistant Rats. Nutrients 2020, 12, 458. [Google Scholar] [CrossRef]
- Sedlak, J.; Lindsay, R.H. Estimation of total, protein-bound, and nonprotein sulfhydryl groups in tissue with Ellman’s reagent. Anal. Biochem. 1968, 25, 192–205. [Google Scholar] [CrossRef]
- Erel, O. A novel automated direct measurement method for total antioxidant capacity using a new generation, more stable ABTS radical cation. Clin. Biochem. 2004, 37, 277–285. [Google Scholar] [CrossRef]
- Erel, O. A new automated colorimetric method for measuring total oxidant status. Clin. Biochem. 2005, 38, 1103–1111. [Google Scholar] [CrossRef]
- Heropolitanska-Pliszka, E.; Berk, K.; Maciejczyk, M.; Sawicka-Powierza, J.; Bernatowska, E.; Wolska-Kusnierz, B.; Pac, M.; Dabrowska-Leonik, N.; Piatosa, B.; Lewandowicz-Uszynska, A.; et al. Systemic Redox Imbalance in Patients with Chronic Granulomatous Disease. J. Clin. Med. 2020, 9, 1397. [Google Scholar] [CrossRef]
- Borys, J.; Maciejczyk, M.; Antonowicz, B.; Kretowski, A.; Waszkiel, D.; Bortnik, P.; Czarniecka-Bargłowska, K.; Kocisz, M.; Szulimowska, J.; Czajkowski, M.; et al. Exposure to Ti4Al4V Titanium Alloy Leads to Redox Abnormalities, Oxidative Stress, and Oxidative Damage in Patients Treated for Mandible Fractures. Oxidative Med. Cell. Longev. 2018, 2018, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.; Baker, J. Assay of serum fructosamine: Internal vs external standardization. Clin. Chem. 1987, 33, 1955–1956. [Google Scholar] [CrossRef] [PubMed]
- Kalousová, M.; Skrha, J.; Zima, T. Advanced glycation end-products and advanced oxidation protein products in patients with diabetes mellitus. Physiol. Res. 2002, 51, 597–604. [Google Scholar]
- Zalewska, A.; Maciejczyk, M.; Szulimowska, J.; Imierska, M.; Błachnio-Zabielska, A. High-Fat Diet Affects Ceramide Content, Disturbs Mitochondrial Redox Balance, and Induces Apoptosis in the Submandibular Glands of Mice. Biomology 2019, 9, 877. [Google Scholar] [CrossRef] [PubMed]
- Reznick, A.Z.; Packer, L. [38] Oxidative damage to proteins: Spectrophotometric method for carbonyl assay. Methods in Enzymology 1994, 233, 357–363. [Google Scholar] [CrossRef]
- Borys, J.; Maciejczyk, M.; Krȩtowski, A.J.; Antonowicz, B.; Ratajczak-Wrona, W.; Jabłońska, E.; Załęski, P.; Waszkiel, D.; Ładny, J.R.; Żukowski, P.; et al. The Redox Balance in Erythrocytes, Plasma, and Periosteum of Patients with Titanium Fixation of the Jaw. Front. Physiol. 2017, 8, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Borys, J.; Maciejczyk, M.; Antonowicz, B.; Kretowski, A.; Sidun, J.; Domel, E.; Dąbrowski, J.R.; Ładny, J.R.; Morawska, K.; Zalewska, A. Glutathione Metabolism, Mitochondria Activity, and Nitrosative Stress in Patients Treated for Mandible Fractures. J. Clin. Med. 2019, 8, 127. [Google Scholar] [CrossRef] [PubMed]
- Grisham, M.B.; Johnson, G.G.; Lancaster, J.R. Quantitation of nitrate and nitrite in extracellular fluids. Methods Enzymol. 1996, 268, 237–246. [Google Scholar] [CrossRef]
- Meki, A.-R.M.; Esmail, E.E.-D.F.; Hussein, A.A.; Hassanein, H.M. Caspase-3 and heat shock protein-70 in rat liver treated with aflatoxin B1: Effect of melatonin. Toxicon 2004, 43, 93–100. [Google Scholar] [CrossRef]
- Chung, A.P.Y.S.; Gurtu, S.; Chakravarthi, S.; Moorthy, M.; Palanisamy, U.D. Geraniin Protects High-Fat Diet-Induced Oxidative Stress in Sprague Dawley Rats. Front. Nutr. 2018, 5, 17. [Google Scholar] [CrossRef]
- Raza, H.; John, A.; Howarth, F.C. Increased Oxidative Stress and Mitochondrial Dysfunction in Zucker Diabetic Rat Liver and Brain. Cell. Physiol. Biochem. 2015, 35, 1241–1251. [Google Scholar] [CrossRef]
- Maciejczyk, M.; Żebrowska, E.; Zalewska, A.; Chabowski, A. Redox Balance, Antioxidant Defense, and Oxidative Damage in the Hypothalamus and Cerebral Cortex of Rats with High Fat Diet-Induced Insulin Resistance. Oxidative Med. Cell. Longev. 2018, 2018, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kjaergaard, M.; Nilsson, C.; Nielsen, M.O.; Grove, K.; Raun, K. Hypothalamic oxidative stress and inflammation, and peripheral glucose homeostasis in Sprague-Dawley rat offspring exposed to maternal and postnatal chocolate and soft drink. Nutr. Diabetes 2018, 8, 44. [Google Scholar] [CrossRef]
- Codella, R.; Terruzzi, I.; Luzi, L. Sugars, exercise and health. J. Affect. Disord. 2017, 224, 76–86. [Google Scholar] [CrossRef]
- Wang, G.-J. Impact of sugar on the body brain and behavior. Front. Biosci. 2018, 23, 2255–2266. [Google Scholar] [CrossRef]
- Peng, Y.; Liu, J.; Shi, L.; Tang, Y.; Gao, D.; Long, J.; Liu, J. Mitochondrial dysfunction precedes depression of AMPK/AKT signaling in insulin resistance induced by high glucose in primary cortical neurons. J. Neurochem. 2016, 137, 701–713. [Google Scholar] [CrossRef] [PubMed]
- Lushchak, V.I. Classification of oxidative stress based on its intensity. EXCLI J. 2014, 13, 922–937. [Google Scholar]
- Zińczuk, J.; Maciejczyk, M.; Zaręba, K.; Pryczynicz, A.; Dymicka-Piekarska, V.; Kamińska, J.; Koper-Lenkiewicz, O.; Matowicka-Karna, J.; Kędra, B.; Zalewska, A.; et al. Pro-Oxidant Enzymes, Redox Balance and Oxidative Damage to Proteins, Lipids and DNA in Colorectal Cancer Tissue. Is Oxidative Stress Dependent on Tumour Budding and Inflammatory Infiltration? Cancers 2020, 12, 1636. [Google Scholar] [CrossRef]
- Day, B.J. Catalase and glutathione peroxidase mimics. Biochem. Pharmacol. 2009, 77, 285–296. [Google Scholar] [CrossRef]
- Lushchak, V.I. Free radicals, reactive oxygen species, oxidative stress and its classification. Chem. Interact. 2014, 224, 164–175. [Google Scholar] [CrossRef]
- Morawska, K.; Zalewska, A.; Popławski, Ł.; Popławska-Kita, A.; Krętowski, A.J.; Maciejczyk, M. Enhanced Salivary and General Oxidative Stress in Hashimoto’s Thyroiditis Women in Euthyreosis. J. Clin. Med. 2020, 9, 2102. [Google Scholar] [CrossRef] [PubMed]
- Stadtman, E.R.; Levine, R.L. Free radical-mediated oxidation of free amino acids and amino acid residues in proteins. Amino Acids 2003, 25, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Yan, L.-J. Protein Oxidative Modifications: Beneficial Roles in Disease and Health. J. Biochem. Pharmacol. Res. 2013, 1, 15–26. [Google Scholar]
- Ledesma-Osuna, A.I.; Ramos-Clamont, G.; Vázquez-Moreno, L. Characterization of bovine serum albumin glycated with glucose, galactose and lactose. Acta Biochim. Pol. 2008, 55, 491–497. [Google Scholar] [CrossRef] [PubMed]
- Pawlukianiec, C.; Gryciuk, M.E.; Mil, K.M.; Żendzian-Piotrowska, M.; Zalewska, A.; Maciejczyk, M. A New Insight into Meloxicam: Assessment of Antioxidant and Anti-Glycating Activity in In Vitro Studies. Pharmaceuticals 2020, 13, 240. [Google Scholar] [CrossRef]
- Griffiths, H.R.; Dias, I.H.; Willetts, R.S.; Devitt, A. Redox regulation of protein damage in plasma. Redox Biol. 2014, 2, 430–435. [Google Scholar] [CrossRef]
- Maciejczyk, M.; Szulimowska, J.; Taranta-Janusz, K.; Wasilewska, A.; Zalewska, A. Salivary Gland Dysfunction, Protein Glycooxidation and Nitrosative Stress in Children with Chronic Kidney Disease. J. Clin. Med. 2020, 9, 1285. [Google Scholar] [CrossRef]
- De La Monte, S.M.; Re, E.; Longato, L.; Tong, M. Dysfunctional Pro-Ceramide, ER Stress, and Insulin/IGF Signaling Networks with Progression of Alzheimer’s Disease. J. Alzheimer’s Dis. 2012, 30, S217–S229. [Google Scholar] [CrossRef]
- Srikanth, V.; Maczurek, A.; Phan, T.; Steele, M.; Westcott, B.; Juskiw, D.; Münch, G. Advanced glycation endproducts and their receptor RAGE in Alzheimer’s disease. Neurobiol. Aging 2011, 32, 763–777. [Google Scholar] [CrossRef]
- Klimiuk, A.; Maciejczyk, M.; Choromańska, M.; Fejfer, K.; Waszkiewicz, N.; Zalewska, A. Salivary Redox Biomarkers in Different Stages of Dementia Severity. J. Clin. Med. 2019, 8, 840. [Google Scholar] [CrossRef]
- Qin, L.; Liu, Y.; Hong, J.-S.; Crews, F.T. NADPH oxidase and aging drive microglial activation, oxidative stress, and dopaminergic neurodegeneration following systemic LPS administration. Glia 2013, 61, 855–868. [Google Scholar] [CrossRef]
- Zhang, X.; Dong, F.; Ren, J.; Driscoll, M.J.; Culver, B. High dietary fat induces NADPH oxidase-associated oxidative stress and inflammation in rat cerebral cortex. Exp. Neurol. 2005, 191, 318–325. [Google Scholar] [CrossRef]
- Adams, L.; Franco, M.C.; Estevez, A.G. Reactive nitrogen species in cellular signaling. Exp. Biol. Med. 2015, 240, 711–717. [Google Scholar] [CrossRef] [PubMed]
- Borys, J.; Maciejczyk, M.; Antonowicz, B.; Sidun, J.; Świderska, M.; Zalewska, A. Free Radical Production, Inflammation and Apoptosis in Patients Treated With Titanium Mandibular Fixations—An Observational Study. Front. Immunol. 2019, 10, 2662. [Google Scholar] [CrossRef] [PubMed]
- Cavaliere, G.; Viggiano, E.; Trinchese, G.; De Filippo, C.; Messina, A.; Monda, V.; Valenzano, A.; Cincione, R.I.; Zammit, C.; Cimmino, F.; et al. Long Feeding High-Fat Diet Induces Hypothalamic Oxidative Stress and Inflammation, and Prolonged Hypothalamic AMPK Activation in Rat Animal Model. Front. Physiol. 2018, 9. [Google Scholar] [CrossRef]
- Zhao, W.-Q.; Alkon, D.L. Role of insulin and insulin receptor in learning and memory. Mol. Cell. Endocrinol. 2001, 177, 125–134. [Google Scholar] [CrossRef]
- Kleinridders, A.; Cai, W.; Cappellucci, L.; Ghazarian, A.; Collins, W.R.; Vienberg, S.G.; Pothos, E.N.; Kahn, C.R. Insulin resistance in brain alters dopamine turnover and causes behavioral disorders. Proc. Natl. Acad. Sci. USA 2015, 112, 3463–3468. [Google Scholar] [CrossRef]
- De La Monte, S.M. Brain Insulin Resistance and Deficiency as Therapeutic Targets in Alzheimers Disease. Curr. Alzheimer Res. 2012, 9, 35–66. [Google Scholar] [CrossRef]
- De La Monte, S.M.; Wands, J.R. Alzheimer’s Disease is Type 3 Diabetes—Evidence Reviewed. J. Diabetes Sci. Technol. 2008, 2, 1101–1113. [Google Scholar] [CrossRef]
- Bosco, D.; Fava, A.; Plastino, M.; Montalcini, T.; Pujia, A. Possible implications of insulin resistance and glucose metabolism in Alzheimer’s disease pathogenesis. J. Cell. Mol. Med. 2011, 15, 1807–1821. [Google Scholar] [CrossRef]
- Villa, R.F.; Ferrari, F.; Gorini, A. Energy metabolism of rat cerebral cortex, hypothalamus and hypophysis during ageing. Neuroscience 2012, 227, 55–66. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | CD | HSD |
|---|---|---|
| Body weight (g) | 356 ± 11.47 | 411 ± 7.54 *** |
| BMI (g/cm2) | 0.64 ± 0.024 | 0.70 ± 0.016 * |
| Food intake (g/day) | 21.4 ± 1.32 | 21.8 ± 1.42 |
| Energy intake (g/rat/week) | 82.4 ± 5.07 | 84 ± 5.46 |
| Glucose concentration (mg/dL) | 137 ± 5.54 | 169 ± 5.01 *** |
| Insulin concentration (mU/mL) | 0.18 ± 0.007 | 0.27 ± 0.03 * |
| HOMA-IR | 2.73 ± 0.13 | 19.2 ± 0.95 **** |
| Parameter | CD | HSD |
|---|---|---|
| Enzymatic and non-enzymatic antioxidants | ||
| GPx (mU/mg protein) | 0.88 ± 0.09 | 1.9 ± 0.16 *** |
| CAT (nmol H2O2 min−1 mg protein−1) | 4.52 ± 0.64 | 3.84 ± 0.69 |
| SOD-1 (mU/mg protein) | 43.5 ± 1.21 | 30.9 ± 1.1 **** |
| UA (µg/mg protein) | 0.44 ± 0.05 | 0.41 ± 0.05 |
| Glutathione metabolism | ||
| Total glutathione (nmol/mg protein) | 20.9 ± 1.28 | 16.5 ± 2.72 |
| GSH (nmol/mg protein) | 20.4 ± 1.27 | 16.0 ± 2.71 |
| GSSG (nmol/mg protein) | 0.50 ± 0.03 | 0.51 ± 0.03 |
| [GSH]2/[GSSG] | 882 ± 116 | 634 ± 172 |
| Thiol groups (nmol/mg protein) | 3.22 ± 0.19 | 2.51 ± 0.20 * |
| Redox status | ||
| TAC (Trolox nmol/mg protein) | 0.09 ± 0.002 | 0.12 ± 0.01 * |
| TOS (nmol H2O2 Equiv./mg protein) | 0.79 ± 0.07 | 2.77 ± 0.31 *** |
| OSI (TOS/TAC ratio) | 9.24 ± 0.93 | 21.65 ± 5.25 |
| Oxidative damage | ||
| AGEs (AFU/mg protein) | 297 ± 6.17 | 408 ± 22.7 ** |
| Amadori Products (nmol/mg protein) | 11.1 ± 0.77 | 18.3 ± 1.01 ** |
| AOPPs (μmol/mg protein) | 6.2 ± 1.2 | 14.8 ± 1.3 ** |
| PCs (nmol/mg protein) | 0.70 ± 0.74 | 3.6 ± 0.56 ** |
| Dityrosine (AFU/mg protein) | 507 ± 37.03 | 681 ± 18.44 ** |
| Kynurenine (AFU/mg protein) | 330 ± 22.9 | 424 ± 22.0 * |
| N-formylkynurenine (AFU/mg protein) | 270 ± 15.34 | 383 ± 15.75 ** |
| Tryptophan (AFU/mg protein) | 28.9 ± 1.12 | 19.04 ± 2.55 * |
| Apoptosis markers | ||
| NO (μmol/mg protein) | 21.5 ± 1.42 | 23.2 ± 2.83 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Żebrowska, E.; Chabowski, A.; Zalewska, A.; Maciejczyk, M. High-Sugar Diet Disrupts Hypothalamic but Not Cerebral Cortex Redox Homeostasis. Nutrients 2020, 12, 3181. https://doi.org/10.3390/nu12103181
Żebrowska E, Chabowski A, Zalewska A, Maciejczyk M. High-Sugar Diet Disrupts Hypothalamic but Not Cerebral Cortex Redox Homeostasis. Nutrients. 2020; 12(10):3181. https://doi.org/10.3390/nu12103181
Chicago/Turabian StyleŻebrowska, Ewa, Adrian Chabowski, Anna Zalewska, and Mateusz Maciejczyk. 2020. "High-Sugar Diet Disrupts Hypothalamic but Not Cerebral Cortex Redox Homeostasis" Nutrients 12, no. 10: 3181. https://doi.org/10.3390/nu12103181
APA StyleŻebrowska, E., Chabowski, A., Zalewska, A., & Maciejczyk, M. (2020). High-Sugar Diet Disrupts Hypothalamic but Not Cerebral Cortex Redox Homeostasis. Nutrients, 12(10), 3181. https://doi.org/10.3390/nu12103181