Inconsistencies in the Nutrition Management of Glutaric Aciduria Type 1: An International Survey



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
3. Results
3.1. Management of Patients with GA-1 Who Are under 6 Years of Age and Were Identified by Newborn Screening
3.1.1. Breastfeeding
3.1.2. Medical Food
3.1.3. Counting Intact Protein
3.1.4. Arginine Supplementation
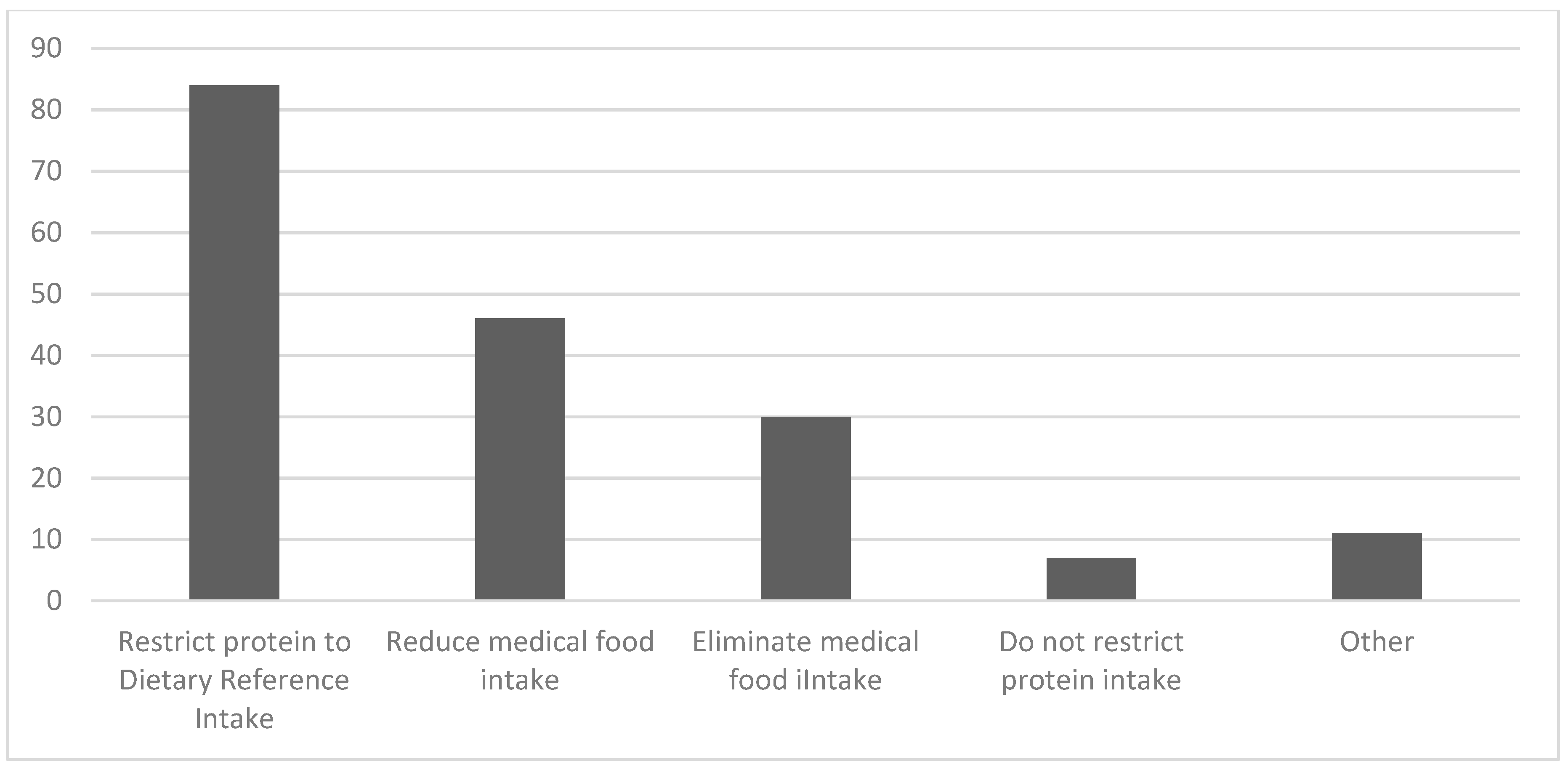
3.2. Diet Liberalization
3.2.1. Definition of Diet Liberalization
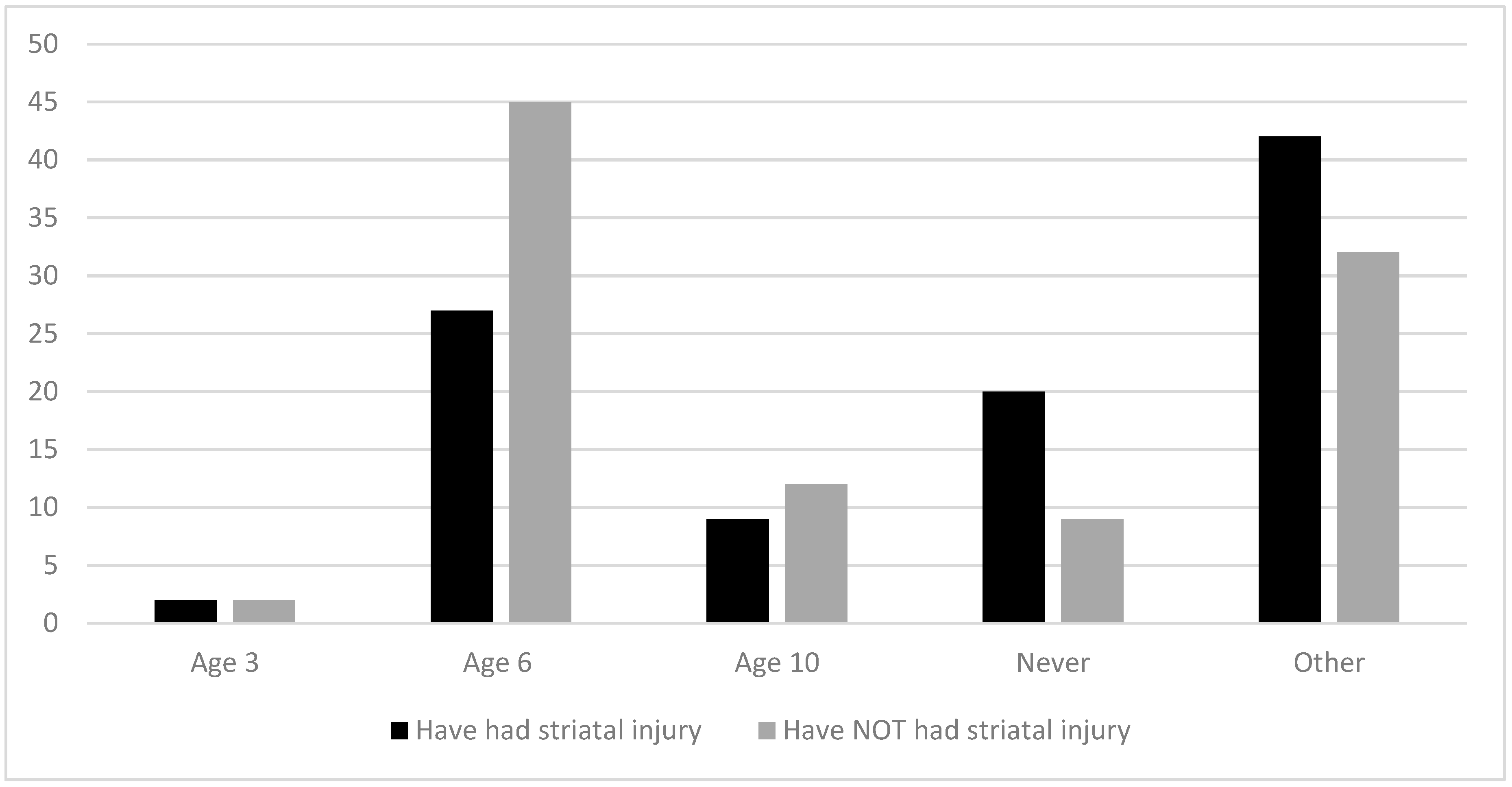
3.2.2. Age of Diet Liberalization
3.2.3. Definition of a Protein-Controlled Diet
3.2.4. Tracking Protein Intake after Diet Liberalization
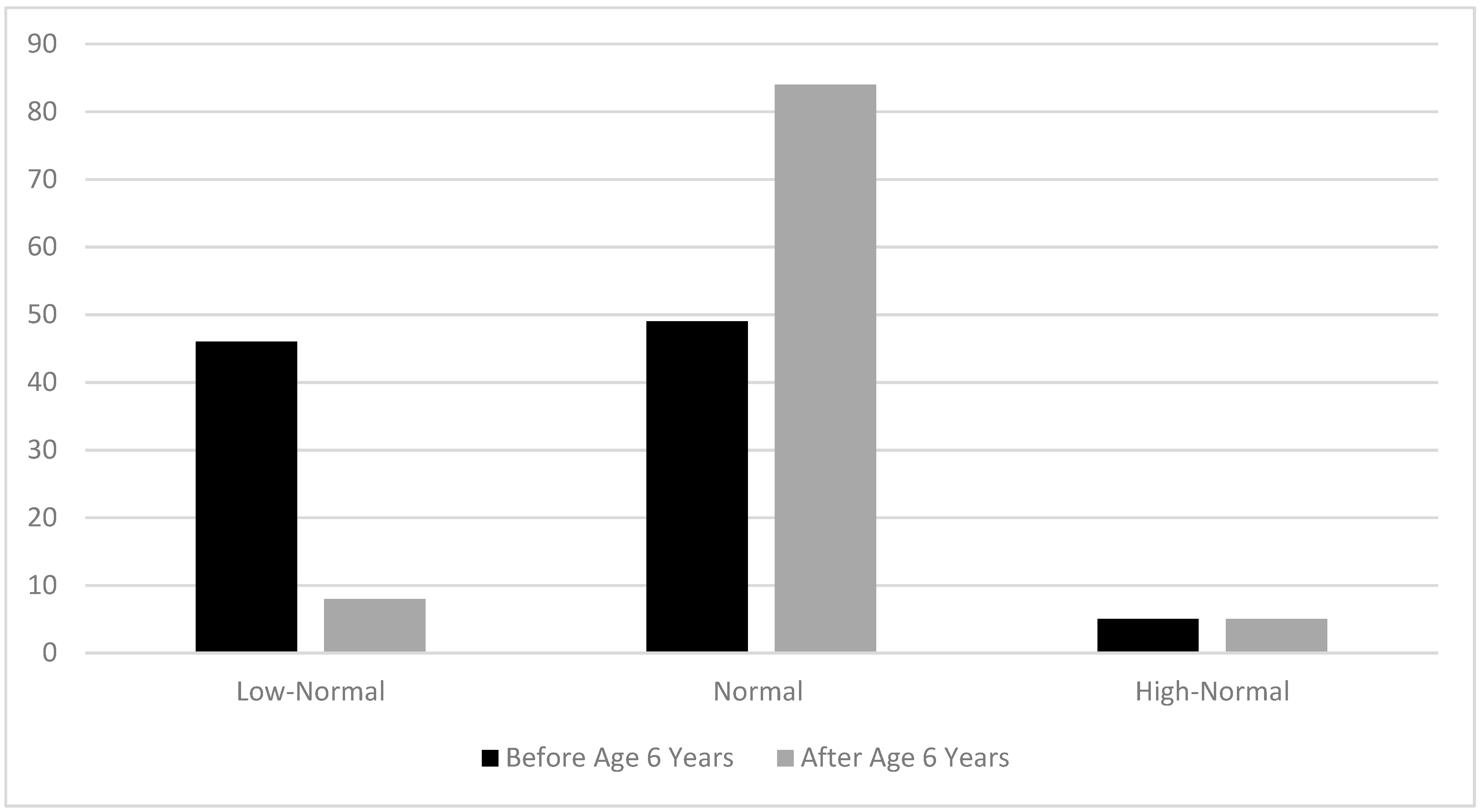
3.2.5. Goals for Plasma Lysine Concentrations
3.2.6. Diet Monitoring
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Goodman, S.I.; Markey, S.P.; Moe, P.G.; Miles, B.S.; Teng, C.C. Glutaric aciduria; A “new” disorder of amino acid metabolism. Biochem. Med. 1975, 12, 12–21. [Google Scholar] [CrossRef]
- Kölker, S.; Garbade, S.F.; Greenberg, C.R.; Leonard, J.V.; Saudubray, J.-M.; Ribees, A.; Kalkanoglu, H.S.; Lund, A.M.; Meeerineroo, B.; Wajner, M.; et al. Natural history, outcome, and treatment efficacy in children and adults with glutaryl-CoA dehydrogenase deficiency. Pediatr. Res. 2006, 59, 840–847. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, G.F.; Athanassopoulos, S.; Burlina, A.B.; Duran, M.; de Klerk, J.B.; Lehnert, W.; Leonard, J.V.; Monavari, A.A.; Müller, E.; Muntau, A.C.; et al. Clinical course, early diagnosis, treatment, and prevention of disease in glutaryl-CoA dehydrogenase deficiency. Neuropediatrics 1996, 27, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Busquets, C.; Merinero, B.; Christensen, E.; Gelpí, J.L.; Campistol, J.; Pineda, M.; Fernández-Alvarez, E.; Prats, J.M.; Sans, A.; Arteaga, R.; et al. Glutaryl-CoA dehydrogenase deficiency in Spain: Evidence of two groups of patients, genetically, and biochemically distinct. Pediatr. Res. 2000, 48, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Strauss, K.A.; Puffenberger, E.G.; Robinson, D.L.; Morton, D.H. Type I glutaric aciduria, part 1: Natural history of 77 patients. Am. J. Med. Genet. C Semin. Med. Genet. 2003, 121, 38–52. [Google Scholar] [CrossRef]
- Boy, N.; Mengler, K.; Thimm, E.; Schiergens, K.A.; Marquardt, T.; Weinhold, N.; Marquardt, I.; Das, A.M.; Freisinger, P.; Grünert, S.C.; et al. Newborn screening: A disease-changing intervention for glutaric aciduria type 1. Ann. Neurol. 2018, 83, 970–979. [Google Scholar] [CrossRef] [PubMed]
- Lenich, A.C.; Goodman, S.I. The purification and characterization of glutaryl-coenzyme. A dehydrogenase from porcine and human liver. J. Biol. Chem. 1986, 261, 4090–4096. [Google Scholar]
- Goodman, S.I. Development of pathogenic concepts in glutaryl-CoA dehydrogenase deficiency: The challenge. J. Inherit. Meta. Dis. 2004, 27, 801–803. [Google Scholar] [CrossRef]
- Kölker, S.; Ahlemeyer, B.; Krieglstein, J.; Hoffmann, G.F. Maturation-dependent neurotoxicity of 3-hydroxyglutaric and glutaric acids in vitro: A new pathophysiologic approach to glutaryl-CoA dehydrogenase deficiency. Pediatr. Res. 2000, 47, 495–503. [Google Scholar] [CrossRef] [PubMed]
- Kölker, S.; Ahlemeyer, B.; Krieglstein, J.; Hoffmann, G.F. 3-Hydroxyglutaric and glutaric acids are neurotoxic through NMDA receptors in vitro. J. Inherit. Metab. Dis. 1999, 22, 259–262. [Google Scholar] [CrossRef]
- Boy, N.; Mühlhausen, C.; Maier, E.M.; Heringer, J.; Assmann, B.; Burgard, P.; Dixon, M.; Fleissner, S.; Greenberg, C.R.; Harting, I.; et al. Additional individual contributors, proposed recommendations for diagnosing and managing individuals with glutaric aciduria type I: Second revision. J. Inherit. Metab. Dis. 2017, 40, 75–101. [Google Scholar] [CrossRef] [PubMed]
- Seccombe, D.W.; James, L.; Booth, F.L. Carnitine treatment in glutaric aciduria type I. Neurology 1986, 36, 264–267. [Google Scholar] [CrossRef] [PubMed]
- Kyllerman, M.; Steen, G. Intermittently progressive dyskinetic syndrome in glutaric aciduria. Neuropadiatrie 1977, 8, 397–404. [Google Scholar] [CrossRef]
- Brandt, N.J.; Gregersen, N.; Christensen, E.; Grøn, I.H.; Rasmussen, K. Treatment of glutaryl-CoA dehydrogenase deficiency (glutaric aciduria). Experience with diet, riboflavin, and GABA analogue. J. Pediatr. 1979, 94, 669–673. [Google Scholar] [CrossRef]
- Collins, J.; Leonard, J.V. Natural history of glutaric aciduria type 1. Arch. Dis. Child. 2000, 83, 87. [Google Scholar] [CrossRef] [PubMed]
- Mühlhausen, C.; Hoffmann, G.F.; Strauss, K.A.; Kölker, S.; Okun, J.G.; Greenberg, C.R.; Naughten, E.R.; Ullrich, K. Maintenance treatment of glutaryl-CoA dehydrogenase deficiency. J. Inherit. Metab. Dis. 2004, 27, 885–892. [Google Scholar] [CrossRef]
- Kölker, S.; Christensen, E.; Leonard, J.V.; Greenberg, C.R.; Burlina, A.B.; Burlina, A.P.; Dixon, M.; Duran, M.; Goodman, S.I.; Koeller, D.M.; et al. Guideline for the diagnosis and management of glutaryl-CoA dehydrogenase deficiency (glutaric aciduria type I). J. Inherit. Metab. Dis. 2007, 30, 5–22. [Google Scholar] [CrossRef]
- Kölker, S.; Christensen, E.; Leonard, J.V.; Greenberg, C.R.; Boneh, A.; Burlina, A.B.; Burlina, A.P.; Dixon, M.; Duran, M.; García Cazorla, A.; et al. Diagnosis and management of glutaric aciduria type I-revised recommendations. J. Inherit. Metab. Dis. 2011, 34, 677–694. [Google Scholar] [CrossRef]
- Yannicelli, S.; Rohr, F.; Warman, M.L. Nutrition support for glutaric acidemia type I. J. Am. Diet. Assoc. 1994, 94, 183–188. [Google Scholar] [CrossRef]
- Müller, E.; Kölker, S. Reduction of lysine intake while avoiding malnutrition—Major goals and major problems in dietary treatment of glutaryl-CoA dehydrogenase deficiency. J. Inherit. Metab. Dis. 2004, 27, 903–910. [Google Scholar] [CrossRef]
- Gokmen-Ozel, H.; MacDonald, A.; Daly, A.; Ashmore, C.; Preece, M.A.; Hendriksz, C.; Vijay, S.; Chakrapaniv, A. Dietary practices in glutaric aciduria type 1 over 16 years. J. Hum. Nut. Diet. 2012, 25, 514–519. [Google Scholar] [CrossRef] [PubMed]
- Pierson, T.M.; Nezhad, M.; Tremblay, M.A.; Lewis, R.; Wong, D.; Salamon, N.; Sicotte, N. Adult-onset glutaric aciduria type I presenting with white matter abnormalities and subependymal nodules. Neurogenetics 2015, 16, 325–328. [Google Scholar] [CrossRef] [PubMed]
- Sonmez, G.; Mutlu, H.; Ozturk, E.; Sildiroglu, H.O.; Keskin, A.T.; Basekim, C.C.; Kizilkaya, E. Magnetic resonance imaging findings of adult-onset glutaric aciduria type I. Acta Radiologica 2007, 48, 557–559. [Google Scholar] [CrossRef] [PubMed]
- Bähr, O.; Mader, I.; Zschocke, J.; Dichgans, J.; Schulz, J.B. Adult onset glutaric aciduria type I presenting with a leukoencephalopathy. Neurology 2002, 59, 1802–1804. [Google Scholar] [CrossRef] [PubMed]
- Herskovitz, M.; Goldsher, D.; Sela, B.-A.; Mandel, H. Subependymal mass lesions and peripheral polyneuropathy in adult-onset glutaric aciduria type I. Neurology 2013, 81, 849–850. [Google Scholar] [CrossRef]
- Külkens, S.; Harting, I.; Sauer, S.; Zschocke, J.; Hoffmann, G.F.; Gruber, S.; Bodamer, O.A.; Kölker, S. Late-onset neurologic disease in glutaryl-CoA dehydrogenase deficiency. Neurology 2005, 64, 2142–2144. [Google Scholar] [CrossRef]
- Heringer, J.; Boy, S.P.N.; Ensenauer, R.; Assmann, B.; Zschocke, J.; Harting, I.; Lücke, T.; Maier, E.M.; Mühlhausen, C.; Haege, G.; et al. Use of guidelines improves the neurological outcome in glutaric aciduria type I. Ann. Neurol. 2010, 68, 743–752. [Google Scholar] [CrossRef]
- Höliner, I.; Simma, B.; Reiter, A.; Sass, J.O.; Zschocke, J.; Huemer, M. Compliance to clinical guidelines determines outcome in glutaric aciduria type I in the era of newborn screening. Klin. Padiatr. 2010, 222, 35–37. [Google Scholar] [CrossRef]
- Afroze, B.; Yunis, Z.M. Glutaric aciduria type 1—Importance of early diagnosis and treatment. J. Pak. Med. Assoc. 2014, 64, 593–595. [Google Scholar]
- Viau, K.; Ernst, S.L.; Vanzo, R.J.; Botto, L.D.; Pasquali, M.; Longo, N. Glutaric acidemia type 1: Outcomes before and after expanded newborn screening. Mol. Genet. Metab. 2012, 106, 430–438. [Google Scholar] [CrossRef]
- Iwashima, S.; Ishikawa, T.; Ohzeki, T.; Endou, Y. Institute of Medicine, Protein and Amino Acids, Dietary Reference Intakes for Energy, Carbohydrate, Fiber, Fat, Fatty Acids, Cholesterol, Protein, and Amino Acids; The National Academies Press: Washington, DC, USA, 2005; pp. 589–768. [Google Scholar]
- Pasiakos, S.M.; Agarwal, S.; Lieberman, H.R.; Fulgoni, V. Sources and amounts of animal, dairy, and plant protein intake of US adults in 2007–2010. Nutrients 2015, 7, 7058–7069. [Google Scholar] [CrossRef] [PubMed]
- Berryman, C.E.; Liberman, H.R.; Fulgoni, V.L.; Pasiakos, S.M. Protein intake trends and conformity with the dietary reference intakes in the USA: Analysis of the national health and nutrition examination survey, 2001–2014. Am. J. Clin. Nutr. 2018, 108, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Strauss, K.A.; Brumbaugh, J.; Duffy, A.; Wardley, B.; Robinson, D.; Hendrickson, C.; Tortorelli, S.; Moser, A.B.; Puffenberger, E.G.; Rider, N.L.; et al. Safety, efficacy and physiological actions of a lysine-free, arginine-rich formula to treat glutaryl-CoA dehydrogenase deficiency: Focus on cerebral amino acid influx. Mol. Genet. Metab. 2011, 104, 93–106. [Google Scholar] [CrossRef] [PubMed]
- Boneh, A.; Beauchamp, M.; Humphrey, M.; Watkins, J.; Peters, H.; Yaplito-Lee, J. Newborn screening for glutaric aciduria type I in Victoria: Treatment and outcome. J. Mol. Genet. Metab. 2008, 94, 287–291. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bernstein, L.; Coughlin, C.R.; Drumm, M.; Yannicelli, S.; Rohr, F. Inconsistencies in the Nutrition Management of Glutaric Aciduria Type 1: An International Survey. Nutrients 2020, 12, 3162. https://doi.org/10.3390/nu12103162
Bernstein L, Coughlin CR, Drumm M, Yannicelli S, Rohr F. Inconsistencies in the Nutrition Management of Glutaric Aciduria Type 1: An International Survey. Nutrients. 2020; 12(10):3162. https://doi.org/10.3390/nu12103162
Chicago/Turabian StyleBernstein, Laurie, Curtis R. Coughlin, Morgan Drumm, Steven Yannicelli, and Fran Rohr. 2020. "Inconsistencies in the Nutrition Management of Glutaric Aciduria Type 1: An International Survey" Nutrients 12, no. 10: 3162. https://doi.org/10.3390/nu12103162
APA StyleBernstein, L., Coughlin, C. R., Drumm, M., Yannicelli, S., & Rohr, F. (2020). Inconsistencies in the Nutrition Management of Glutaric Aciduria Type 1: An International Survey. Nutrients, 12(10), 3162. https://doi.org/10.3390/nu12103162