Diet-Induced Rodent Models of Diabetic Peripheral Neuropathy, Retinopathy and Nephropathy

 , , , ,
, , , , 


Abstract
1. Introduction
2. Diet-Induced Models of Diabetic Peripheral Neuropathy
3. Diet-Induced Models of Diabetic Retinopathy
4. Diet-Induced Models of Diabetic Nephropathy
5. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- IDF. IDF Diabetes Atlas; The International Diabetes Federation (IDF): Brussels, Belgium, 2019. [Google Scholar]
- Kasmauski, K. Type 2 diabetes: The urgent need to protect young people. Lancet 2018, 392, 2325. [Google Scholar] [CrossRef]
- Stehouwer, C.D. Microvascular Dysfunction and Hyperglycemia: A Vicious Cycle With Widespread Consequences. Diabetes 2018, 67, 1729–1741. [Google Scholar] [CrossRef] [PubMed]
- Spijkerman, A.M.; Dekker, J.M.; Nijpels, G.; Adriaanse, M.C.; Kostense, P.J.; Ruwaard, D.; Stehouwer, C.D.; Bouter, L.M.; Heine, R.J. Microvascular complications at time of diagnosis of type 2 diabetes are similar among diabetic patients detected by targeted screening and patients newly diagnosed in general practice: The hoorn screening study. Diabetes Care 2003, 26, 2604–2608. [Google Scholar] [CrossRef] [PubMed]
- Valencia, W.M.; Florez, H. How to prevent the microvascular complications of type 2 diabetes beyond glucose control. BMJ 2017, 356, i6505. [Google Scholar] [CrossRef]
- Fernandes, R.; Viana, S.D.; Nunes, S.; Reis, F. Diabetic gut microbiota dysbiosis as an inflammaging and immunosenescence condition that fosters progression of retinopathy and nephropathy. Biochim. Biophys Acta Mol. Basis Dis. 2019, 1865, 1876–1897. [Google Scholar] [CrossRef]
- Barrett, E.J.; Liu, Z.; Khamaisi, M.; King, G.L.; Klein, R.; Klein, B.E.K.; Hughes, T.M.; Craft, S.; Freedman, B.I.; Bowden, D.W.; et al. Diabetic Microvascular Disease: An Endocrine Society Scientific Statement. J. Clin. Endocrinol. Metab. 2017, 102, 4343–4410. [Google Scholar] [CrossRef]
- Barriere, D.A.; Noll, C.; Roussy, G.; Lizotte, F.; Kessai, A.; Kirby, K.; Belleville, K.; Beaudet, N.; Longpre, J.M.; Carpentier, A.C.; et al. Combination of high-fat/high-fructose diet and low-dose streptozotocin to model long-term type-2 diabetes complications. Sci. Rep. 2018, 8, 424. [Google Scholar] [CrossRef]
- Srinivasan, K.; Ramarao, P. Animal models in type 2 diabetes research: An overview. Indian J. Med. Res. 2007, 125, 451–472. [Google Scholar]
- Beigrezaei, S.; Ghiasvand, R.; Feizi, A.; Iraj, B. Relationship between Dietary Patterns and Incidence of Type 2 Diabetes. Int. J. Prev. Med. 2019, 10, 122. [Google Scholar] [CrossRef]
- Hintze, K.J.; Benninghoff, A.D.; Cho, C.E.; Ward, R.E. Modeling the Western Diet for Preclinical Investigations. Adv. Nutr. 2018, 9, 263–271. [Google Scholar] [CrossRef]
- Rice Bradley, B.H. Dietary Fat and Risk for Type 2 Diabetes: A Review of Recent Research. Curr. Nutr. Rep. 2018, 7, 214–226. [Google Scholar] [CrossRef] [PubMed]
- Sami, W.; Ansari, T.; Butt, N.S.; Hamid, M.R.A. Effect of diet on type 2 diabetes mellitus: A review. Int. J. Health Sci. 2017, 11, 65–71. [Google Scholar]
- Forouhi, N.G.; Misra, A.; Mohan, V.; Taylor, R.; Yancy, W. Dietary and nutritional approaches for prevention and management of type 2 diabetes. BMJ 2018, 361, k2234. [Google Scholar] [CrossRef] [PubMed]
- Reuter, T.Y. Diet-induced models for obesity and type 2 diabetes. Drug Discov. Today Dis. Models 2007, 4, 3–8. [Google Scholar] [CrossRef]
- Barbosa-Da-Silva, S.; Sarmento, I.B.; Bargut, T.C.L.; Souza-Mello, V.; Aguila, M.B.; Mandarim-De-Lacerda, C.A. Animal Models of Nutritional Induction of Type 2 Diabetes Mellitus. Int. J. Morphol. 2014, 32, 279–293. [Google Scholar] [CrossRef]
- Speakman, J.R. Use of high-fat diets to study rodent obesity as a model of human obesity. Int. J. Obes. 2019, 43, 1491–1492. [Google Scholar] [CrossRef]
- Hicks, C.W.; Selvin, E. Epidemiology of Peripheral Neuropathy and Lower Extremity Disease in Diabetes. Curr. Diabetes Rep. 2019, 19, 86. [Google Scholar] [CrossRef]
- Dyck, P.J.; Albers, J.W.; Andersen, H.; Arezzo, J.C.; Biessels, G.J.; Bril, V.; Feldman, E.L.; Litchy, W.J.; O’Brien, P.C.; Russell, J.W.; et al. Diabetic polyneuropathies: Update on research definition, diagnostic criteria and estimation of severity. Diabetes Metab. Res. Rev. 2011, 27, 620–628. [Google Scholar] [CrossRef]
- Sumner, C.J.; Sheth, S.; Griffin, J.W.; Cornblath, D.R.; Polydefkis, M. The spectrum of neuropathy in diabetes and impaired glucose tolerance. Neurology 2003, 60, 108–111. [Google Scholar] [CrossRef]
- O’Brien, P.D.; Sakowski, S.A.; Feldman, E.L. Mouse models of diabetic neuropathy. ILAR J. 2014, 54, 259–272. [Google Scholar] [CrossRef]
- Tesfaye, S.; Boulton, A.J.; Dyck, P.J.; Freeman, R.; Horowitz, M.; Kempler, P.; Lauria, G.; Malik, R.A.; Spallone, V.; Vinik, A.; et al. Diabetic neuropathies: Update on definitions, diagnostic criteria, estimation of severity, and treatments. Diabetes Care 2010, 33, 2285–2293. [Google Scholar] [CrossRef] [PubMed]
- Salvage, J. King’s Fund nursing development. Nurs. Stand. 1989, 3, 25. [Google Scholar] [CrossRef] [PubMed]
- Richner, M.; Ferreira, N.; Dudele, A.; Jensen, T.S.; Vaegter, C.B.; Goncalves, N.P. Functional and Structural Changes of the Blood-Nerve-Barrier in Diabetic Neuropathy. Front. Neurosci. 2018, 12, 1038. [Google Scholar] [CrossRef] [PubMed]
- Eid, S.; Sas, K.M.; Abcouwer, S.F.; Feldman, E.L.; Gardner, T.W.; Pennathur, S.; Fort, P.E. New insights into the mechanisms of diabetic complications: Role of lipids and lipid metabolism. Diabetologia 2019, 62, 1539–1549. [Google Scholar] [CrossRef]
- Van Dam, P.S.; Cotter, M.A.; Bravenboer, B.; Cameron, N.E. Pathogenesis of diabetic neuropathy: Focus on neurovascular mechanisms. Eur. J. Pharmacol. 2013, 719, 180–186. [Google Scholar] [CrossRef]
- Boulton, A.J.; Vinik, A.I.; Arezzo, J.C.; Bril, V.; Feldman, E.L.; Freeman, R.; Malik, R.A.; Maser, R.E.; Sosenko, J.M.; Ziegler, D.; et al. Diabetic neuropathies: A statement by the American Diabetes Association. Diabetes Care 2005, 28, 956–962. [Google Scholar] [CrossRef]
- Petropoulos, I.N.; Ponirakis, G.; Khan, A.; Almuhannadi, H.; Gad, H.; Malik, R.A. Diagnosing Diabetic Neuropathy: Something Old, Something New. Diabetes Metab. J. 2018, 42, 255–269. [Google Scholar] [CrossRef]
- Biessels, G.J.; Bril, V.; Calcutt, N.A.; Cameron, N.E.; Cotter, M.A.; Dobrowsky, R.; Feldman, E.L.; Fernyhough, P.; Jakobsen, J.; Malik, R.A.; et al. Phenotyping animal models of diabetic neuropathy: A consensus statement of the diabetic neuropathy study group of the EASD (Neurodiab). J. Peripher. Nerv. Syst. 2014, 19, 77–87. [Google Scholar] [CrossRef]
- Sima, A.A.; Zhang, W.; Xu, G.; Sugimoto, K.; Guberski, D.; Yorek, M.A. A comparison of diabetic polyneuropathy in type II diabetic BBZDR/Wor rats and in type I diabetic BB/Wor rats. Diabetologia 2000, 43, 786–793. [Google Scholar] [CrossRef]
- Feng, H.; Lu, G.; Li, Q.; Liu, Z. Inhibition of Adenylyl Cyclase in the Spinal Cord Alleviates Painful Diabetic Neuropathy in Zucker Diabetic Fatty Rats. Can. J. Diabetes 2017, 41, 177–183. [Google Scholar] [CrossRef]
- Xie, J.; Rao, N.; Zhai, Y.; Li, J.; Zhao, Y.; Ge, L.; Wang, Y. Therapeutic effects of stem cells from human exfoliated deciduous teeth on diabetic peripheral neuropathy. Diabetol. Metab. Syndr. 2019, 11, 38. [Google Scholar] [CrossRef] [PubMed]
- Fan, B.; Li, C.; Szalad, A.; Wang, L.; Pan, W.; Zhang, R.; Chopp, M.; Zhang, Z.G.; Liu, X.S. Mesenchymal stromal cell-derived exosomes ameliorate peripheral neuropathy in a mouse model of diabetes. Diabetologia 2019. [Google Scholar] [CrossRef] [PubMed]
- McGregor, B.A.; Eid, S.; Rumora, A.E.; Murdock, B.; Guo, K.; de Anda-Jauregui, G.; Porter, J.E.; Feldman, E.L.; Hur, J. Conserved Transcriptional Signatures in Human and Murine Diabetic Peripheral Neuropathy. Sci. Rep. 2018, 8, 17678. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, P.D.; Hur, J.; Hayes, J.M.; Backus, C.; Sakowski, S.A.; Feldman, E.L. BTBR ob/ob mice as a novel diabetic neuropathy model: Neurological characterization and gene expression analyses. Neurobiol. Dis. 2015, 73, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Stino, A.M.; Smith, A.G. Peripheral neuropathy in prediabetes and the metabolic syndrome. J. Diabetes Investig. 2017, 8, 646–655. [Google Scholar] [CrossRef]
- Kopf, S.; Groener, J.B.; Kender, Z.; Fleming, T.; Bischoff, S.; Jende, J.; Schumann, C.; Ries, S.; Bendszus, M.; Schuh-Hofer, S.; et al. Deep phenotyping neuropathy: An underestimated complication in patients with pre-diabetes and type 2 diabetes associated with albuminuria. Diabetes Res. Clin. Pract. 2018, 146, 191–201. [Google Scholar] [CrossRef]
- Vincent, A.M.; Hinder, L.M.; Pop-Busui, R.; Feldman, E.L. Hyperlipidemia: A new therapeutic target for diabetic neuropathy. J. Peripher. Nerv. Syst. 2009, 14, 257–267. [Google Scholar] [CrossRef]
- Vincent, A.M.; Hayes, J.M.; McLean, L.L.; Vivekanandan-Giri, A.; Pennathur, S.; Feldman, E.L. Dyslipidemia-induced neuropathy in mice: The role of oxLDL/LOX-1. Diabetes 2009, 58, 2376–2385. [Google Scholar] [CrossRef]
- Obrosova, I.G.; Ilnytska, O.; Lyzogubov, V.V.; Pavlov, I.A.; Mashtalir, N.; Nadler, J.L.; Drel, V.R. High-fat diet induced neuropathy of pre-diabetes and obesity: Effects of “healthy” diet and aldose reductase inhibition. Diabetes 2007, 56, 2598–2608. [Google Scholar] [CrossRef]
- Sullivan, K.A.; Hayes, J.M.; Wiggin, T.D.; Backus, C.; Su Oh, S.; Lentz, S.I.; Brosius, F., 3rd; Feldman, E.L. Mouse models of diabetic neuropathy. Neurobiol. Dis. 2007, 28, 276–285. [Google Scholar] [CrossRef]
- Coppey, L.; Davidson, E.; Lu, B.; Gerard, C.; Yorek, M. Vasopeptidase inhibitor ilepatril (AVE7688) prevents obesity- and diabetes-induced neuropathy in C57Bl/6J mice. Neuropharmacology 2011, 60, 259–266. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, P.D.; Hinder, L.M.; Rumora, A.E.; Hayes, J.M.; Dauch, J.R.; Backus, C.; Mendelson, F.E.; Feldman, E.L. Juvenile murine models of prediabetes and type 2 diabetes develop neuropathy. Dis. Model. Mech. 2018, 11. [Google Scholar] [CrossRef] [PubMed]
- Coppey, L.J.; Shevalye, H.; Obrosov, A.; Davidson, E.P.; Yorek, M.A. Determination of peripheral neuropathy in high-fat diet fed low-dose streptozotocin-treated female C57Bl/6J mice and Sprague-Dawley rats. J. Diabetes Investig. 2018, 9, 1033–1040. [Google Scholar] [CrossRef] [PubMed]
- Xie, F.; Fu, H.; Hou, J.F.; Jiao, K.; Costigan, M.; Chen, J. High energy diets-induced metabolic and prediabetic painful polyneuropathy in rats. PLoS ONE 2013, 8, e57427. [Google Scholar] [CrossRef] [PubMed]
- Coppey, L.; Lu, B.; Gerard, C.; Yorek, M.A. Effect of Inhibition of Angiotensin-Converting Enzyme and/or Neutral Endopeptidase on Neuropathy in High-Fat-Fed C57Bl/6J Mice. J. Obes. 2012, 2012, 326806. [Google Scholar] [CrossRef] [PubMed]
- Stavniichuk, R.; Drel, V.R.; Shevalye, H.; Vareniuk, I.; Stevens, M.J.; Nadler, J.L.; Obrosova, I.G. Role of 12/15-lipoxygenase in nitrosative stress and peripheral prediabetic and diabetic neuropathies. Free Radic. Biol. Med. 2010, 49, 1036–1045. [Google Scholar] [CrossRef][Green Version]
- Hinder, L.M.; O’Brien, P.D.; Hayes, J.M.; Backus, C.; Solway, A.P.; Sims-Robinson, C.; Feldman, E.L. Dietary reversal of neuropathy in a murine model of prediabetes and metabolic syndrome. Dis. Model. Mech. 2017, 10, 717–725. [Google Scholar] [CrossRef]
- Cheung, N.; Mitchell, P.; Wong, T.Y. Diabetic retinopathy. Lancet 2010, 376, 124–136. [Google Scholar] [CrossRef]
- Chawla, A.; Chawla, R.; Jaggi, S. Microvasular and macrovascular complications in diabetes mellitus: Distinct or continuum? Indian J Endocrinol Metab 2016, 20, 546–551. [Google Scholar] [CrossRef]
- Jenkins, A.J.; Joglekar, M.V.; Hardikar, A.A.; Keech, A.C.; O’Neal, D.N.; Januszewski, A.S. Biomarkers in Diabetic Retinopathy. Rev. Diabet. Stud. 2015, 12, 159–195. [Google Scholar] [CrossRef]
- Mastropasqua, R.; D’Aloisio, R.; Di Antonio, L.; Erroi, E.; Borrelli, E.; Evangelista, F.; D’Onofrio, G.; Di Nicola, M.; Di Martino, G.; Toto, L. Widefield optical coherence tomography angiography in diabetic retinopathy. Acta Diabetol. 2019, 56, 1293–1303. [Google Scholar] [CrossRef] [PubMed]
- Cunha-Vaz, J.G. Pathophysiology of diabetic retinopathy. Br. J. Ophthalmol. 1978, 62, 351–355. [Google Scholar] [CrossRef] [PubMed]
- Santiago, A.R.; Boia, R.; Aires, I.D.; Ambrosio, A.F.; Fernandes, R. Sweet Stress: Coping With Vascular Dysfunction in Diabetic Retinopathy. Front. Physiol. 2018, 9, 820. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.; Wong, T.Y.; Sabanayagam, C. Epidemiology of diabetic retinopathy, diabetic macular edema and related vision loss. Eye Vis. 2015, 2, 17. [Google Scholar] [CrossRef]
- Xu, G.; Kang, D.; Zhang, C.; Lou, H.; Sun, C.; Yang, Q.; Lu, L.; Xu, G.T.; Zhang, J.; Wang, F. Erythropoietin Protects Retinal Cells in Diabetic Rats Through Upregulating ZnT8 via Activating ERK Pathway and Inhibiting HIF-1alpha Expression. Investig. Ophthalmol. Vis. Sci. 2015, 56, 8166–8178. [Google Scholar] [CrossRef]
- Robinson, R.; Barathi, V.A.; Chaurasia, S.S.; Wong, T.Y.; Kern, T.S. Update on animal models of diabetic retinopathy: From molecular approaches to mice and higher mammals. Dis. Model. Mech. 2012, 5, 444–456. [Google Scholar] [CrossRef]
- Olivares, A.M.; Althoff, K.; Chen, G.F.; Wu, S.; Morrisson, M.A.; DeAngelis, M.M.; Haider, N. Animal Models of Diabetic Retinopathy. Curr. Diabetes Rep. 2017, 17, 93. [Google Scholar] [CrossRef]
- Zhu, H.; Zhang, W.; Zhao, Y.; Shu, X.; Wang, W.; Wang, D.; Yang, Y.; He, Z.; Wang, X.; Ying, Y. GSK3beta-mediated tau hyperphosphorylation triggers diabetic retinal neurodegeneration by disrupting synaptic and mitochondrial functions. Mol. Neurodegener. 2018, 13, 62. [Google Scholar] [CrossRef]
- Rajagopal, R.; Bligard, G.W.; Zhang, S.; Yin, L.; Lukasiewicz, P.; Semenkovich, C.F. Functional Deficits Precede Structural Lesions in Mice With High-Fat Diet-Induced Diabetic Retinopathy. Diabetes 2016, 65, 1072–1084. [Google Scholar] [CrossRef]
- Datilo, M.N.; Sant’Ana, M.R.; Formigari, G.P.; Rodrigues, P.B.; de Moura, L.P.; da Silva, A.S.R.; Ropelle, E.R.; Pauli, J.R.; Cintra, D.E. Omega-3 from Flaxseed Oil Protects Obese Mice Against Diabetic Retinopathy Through GPR120 Receptor. Sci. Rep. 2018, 8, 14318. [Google Scholar] [CrossRef]
- Cheng, Y.; Yu, X.; Zhang, J.; Chang, Y.; Xue, M.; Li, X.; Lu, Y.; Li, T.; Meng, Z.; Su, L.; et al. Pancreatic kallikrein protects against diabetic retinopathy in KK Cg-A(y)/J and high-fat diet/streptozotocin-induced mouse models of type 2 diabetes. Diabetologia 2019, 62, 1074–1086. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Xiao, X.; Zheng, J.; Li, M.; Yu, M.; Ping, F.; Wang, T.; Wang, X. Compound Danshen Dripping Pill Inhibits Retina Cell Apoptosis in Diabetic Rats. Front. Physiol. 2018, 9, 1501. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Li, C.; Liu, J.; Zhang, C.; Fu, Y.; Wang, N.; Ma, H.; Lu, H.; Kong, H.; Kong, L. Thioredoxin plays a key role in retinal neuropathy prior to endothelial damage in diabetic mice. Oncotarget 2017, 8, 61350–61364. [Google Scholar] [CrossRef] [PubMed]
- Tofovic, S.P.; Kusaka, H.; Kost, C.K., Jr.; Bastacky, S. Renal function and structure in diabetic, hypertensive, obese ZDFxSHHF-hybrid rats. Ren. Fail. 2000, 22, 387–406. [Google Scholar] [CrossRef]
- Bilan, V.P.; Salah, E.M.; Bastacky, S.; Jones, H.B.; Mayers, R.M.; Zinker, B.; Poucher, S.M.; Tofovic, S.P. Diabetic nephropathy and long-term treatment effects of rosiglitazone and enalapril in obese ZSF1 rats. J. Endocrinol. 2011, 210, 293–308. [Google Scholar] [CrossRef]
- Caolo, V.; Roblain, Q.; Lecomte, J.; Carai, P.; Peters, L.; Cuijpers, I.; Robinson, E.L.; Derks, K.; Sergeys, J.; Noel, A.; et al. Resistance to retinopathy development in obese, diabetic and hypertensive ZSF1 rats: An exciting model to identify protective genes. Sci. Rep. 2018, 8, 11922. [Google Scholar] [CrossRef]
- Glassock, R.J.; Warnock, D.G.; Delanaye, P. The global burden of chronic kidney disease: Estimates, variability and pitfalls. Nat. Rev. Nephrol. 2017, 13, 104–114. [Google Scholar] [CrossRef]
- Jha, V.; Garcia-Garcia, G.; Iseki, K.; Li, Z.; Naicker, S.; Plattner, B.; Saran, R.; Wang, A.Y.; Yang, C.W. Chronic kidney disease: Global dimension and perspectives. Lancet 2013, 382, 260–272. [Google Scholar] [CrossRef]
- Arora, M.K.; Singh, U.K. Molecular mechanisms in the pathogenesis of diabetic nephropathy: An update. Vascul. Pharmacol. 2013, 58, 259–271. [Google Scholar] [CrossRef]
- Gross, J.L.; de Azevedo, M.J.; Silveiro, S.P.; Canani, L.H.; Caramori, M.L.; Zelmanovitz, T. Diabetic nephropathy: Diagnosis, prevention, and treatment. Diabetes Care 2005, 28, 164–176. [Google Scholar] [CrossRef]
- Nazar, C.M. Diabetic nephropathy; principles of diagnosis and treatment of diabetic kidney disease. J. Nephropharmacol. 2014, 3, 15–20. [Google Scholar] [PubMed]
- Kitada, M.; Ogura, Y.; Koya, D. Rodent models of diabetic nephropathy: Their utility and limitations. Int. J. Nephrol. Renovasc. Dis. 2016, 9, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Tervaert, T.W.; Mooyaart, A.L.; Amann, K.; Cohen, A.H.; Cook, H.T.; Drachenberg, C.B.; Ferrario, F.; Fogo, A.B.; Haas, M.; de Heer, E.; et al. Pathologic classification of diabetic nephropathy. J. Am. Soc. Nephrol. 2010, 21, 556–563. [Google Scholar] [CrossRef] [PubMed]
- Brosius, F.C., 3rd; Alpers, C.E.; Bottinger, E.P.; Breyer, M.D.; Coffman, T.M.; Gurley, S.B.; Harris, R.C.; Kakoki, M.; Kretzler, M.; Leiter, E.H.; et al. Mouse models of diabetic nephropathy. J. Am. Soc. Nephrol. 2009, 20, 2503–2512. [Google Scholar] [CrossRef]
- Soler, M.J.; Riera, M.; Batlle, D. New experimental models of diabetic nephropathy in mice models of type 2 diabetes: Efforts to replicate human nephropathy. Exp. Diabetes Res. 2012, 2012, 616313. [Google Scholar] [CrossRef]
- Kaur, M.; Bedi, O.; Sachdeva, S.; Reddy, B.V.; Kumar, P. Rodent animal models: From mild to advanced stages of diabetic nephropathy. Inflammopharmacology 2014, 22, 279–293. [Google Scholar] [CrossRef]
- Betz, B.; Conway, B.R. An Update on the Use of Animal Models in Diabetic Nephropathy Research. Curr. Diabetes Rep. 2016, 16, 18. [Google Scholar] [CrossRef]
- Apirion, D.; Neil, J.; Watson, N. Consequences of losing ribonuclease III on the Escherichia coli cell. Mol. Gen. Genet. 1976, 144, 185–190. [Google Scholar] [CrossRef]
- Deji, N.; Kume, S.; Araki, S.; Soumura, M.; Sugimoto, T.; Isshiki, K.; Chin-Kanasaki, M.; Sakaguchi, M.; Koya, D.; Haneda, M.; et al. Structural and functional changes in the kidneys of high-fat diet-induced obese mice. Am. J. Physiol. Renal. Physiol. 2009, 296, F118–F126. [Google Scholar] [CrossRef]
- Rangel Silvares, R.; Nunes Goulart da Silva Pereira, E.; Eduardo Ilaquita Flores, E.; Lino Rodrigues, K.; Ribeiro Silva, A.; Goncalves-de-Albuquerque, C.F.; Daliry, A. High-fat diet-induced kidney alterations in rats with metabolic syndrome: Endothelial dysfunction and decreased antioxidant defense. Diabetes Metab. Syndr. Obes. 2019, 12, 1773–1781. [Google Scholar] [CrossRef]
- Danda, R.S.; Habiba, N.M.; Rincon-Choles, H.; Bhandari, B.K.; Barnes, J.L.; Abboud, H.E.; Pergola, P.E. Kidney involvement in a nongenetic rat model of type 2 diabetes. Kidney Int. 2005, 68, 2562–2571. [Google Scholar] [CrossRef] [PubMed]
- Sugano, M.; Yamato, H.; Hayashi, T.; Ochiai, H.; Kakuchi, J.; Goto, S.; Nishijima, F.; Iino, N.; Kazama, J.J.; Takeuchi, T.; et al. High-fat diet in low-dose-streptozotocin-treated heminephrectomized rats induces all features of human type 2 diabetic nephropathy: A new rat model of diabetic nephropathy. Nutr. Metab. Cardiovasc. Dis. 2006, 16, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Prabhakar, S.; Starnes, J.; Shi, S.; Lonis, B.; Tran, R. Diabetic nephropathy is associated with oxidative stress and decreased renal nitric oxide production. J. Am. Soc. Nephrol. 2007, 18, 2945–2952. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Widomski, D.; Ma, J.; Namovic, M.; Nikkel, A.; Leys, L.; Olson, L.; Salte, K.; Donnelly-Roberts, D.; Esbenshade, T.; et al. Longitudinal Changes in Measured Glomerular Filtration Rate, Renal Fibrosis and Biomarkers in a Rat Model of Type 2 Diabetic Nephropathy. Am. J. Nephrol. 2016, 44, 339–353. [Google Scholar] [CrossRef] [PubMed]
- Dower, K.; Zhao, S.; Schlerman, F.J.; Savary, L.; Campanholle, G.; Johnson, B.G.; Xi, L.; Nguyen, V.; Zhan, Y.; Lech, M.P.; et al. High resolution molecular and histological analysis of renal disease progression in ZSF1 fa/faCP rats, a model of type 2 diabetic nephropathy. PLoS ONE 2017, 12, e0181861. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Lozada, L.G.; Tapia, E.; Jimenez, A.; Bautista, P.; Cristobal, M.; Nepomuceno, T.; Soto, V.; Avila-Casado, C.; Nakagawa, T.; Johnson, R.J.; et al. Fructose-induced metabolic syndrome is associated with glomerular hypertension and renal microvascular damage in rats. Am. J. Physiol. Renal. Physiol. 2007, 292, F423–F429. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, T.; Kosugi, T.; Gersch, M.; Connor, T.; Sanchez-Lozada, L.G.; Lanaspa, M.A.; Roncal, C.; Perez-Pozo, S.E.; Johnson, R.J.; Nakagawa, T. Dietary fructose causes tubulointerstitial injury in the normal rat kidney. Am. J. Physiol. Renal. Physiol. 2010, 298, F712–F720. [Google Scholar] [CrossRef]
- Toyoda, K.; Suzuki, Y.; Muta, K.; Masuyama, T.; Kakimoto, K.; Kobayashi, A.; Shoda, T.; Sugai, S. High fructose diet feeding accelerates diabetic nephropathy in Spontaneously Diabetic Torii (SDT) rats. J. Toxicol. Sci. 2018, 43, 45–58. [Google Scholar] [CrossRef]
- Sasase, T.; Ohta, T.; Masuyama, T.; Yokoi, N.; Kakehashi, A.; Shinohara, M. The spontaneously diabetic torii rat: An animal model of nonobese type 2 diabetes with severe diabetic complications. J. Diabetes Res. 2013, 2013, 976209. [Google Scholar] [CrossRef]
- Aoyama, M.; Isshiki, K.; Kume, S.; Chin-Kanasaki, M.; Araki, H.; Araki, S.; Koya, D.; Haneda, M.; Kashiwagi, A.; Maegawa, H.; et al. Fructose induces tubulointerstitial injury in the kidney of mice. Biochem. Biophys. Res. Commun. 2012, 419, 244–249. [Google Scholar] [CrossRef]
- Kelly, D.J.; Zhang, Y.; Hepper, C.; Gow, R.M.; Jaworski, K.; Kemp, B.E.; Wilkinson-Berka, J.L.; Gilbert, R.E. Protein kinase C beta inhibition attenuates the progression of experimental diabetic nephropathy in the presence of continued hypertension. Diabetes 2003, 52, 512–518. [Google Scholar] [CrossRef] [PubMed]
- Marques, C.; Mega, C.; Goncalves, A.; Rodrigues-Santos, P.; Teixeira-Lemos, E.; Teixeira, F.; Fontes-Ribeiro, C.; Reis, F.; Fernandes, R. Sitagliptin prevents inflammation and apoptotic cell death in the kidney of type 2 diabetic animals. Mediat. Inflamm. 2014, 2014, 538737. [Google Scholar] [CrossRef] [PubMed]
- Mega, C.; de Lemos, E.T.; Vala, H.; Fernandes, R.; Oliveira, J.; Mascarenhas-Melo, F.; Teixeira, F.; Reis, F. Diabetic nephropathy amelioration by a low-dose sitagliptin in an animal model of type 2 diabetes (Zucker diabetic fatty rat). Exp. Diabetes Res. 2011, 2011, 162092. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, A.; Leal, E.; Paiva, A.; Teixeira Lemos, E.; Teixeira, F.; Ribeiro, C.F.; Reis, F.; Ambrosio, A.F.; Fernandes, R. Protective effects of the dipeptidyl peptidase IV inhibitor sitagliptin in the blood-retinal barrier in a type 2 diabetes animal model. Diabetes Obes. Metab. 2012, 14, 454–463. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.J.; Segal, M.S.; Sautin, Y.; Nakagawa, T.; Feig, D.I.; Kang, D.H.; Gersch, M.S.; Benner, S.; Sanchez-Lozada, L.G. Potential role of sugar (fructose) in the epidemic of hypertension, obesity and the metabolic syndrome, diabetes, kidney disease, and cardiovascular disease. Am. J. Clin. Nutr. 2007, 86, 899–906. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.J.; Perez-Pozo, S.E.; Sautin, Y.Y.; Manitius, J.; Sanchez-Lozada, L.G.; Feig, D.I.; Shafiu, M.; Segal, M.; Glassock, R.J.; Shimada, M.; et al. Hypothesis: Could excessive fructose intake and uric acid cause type 2 diabetes? Endocr. Rev. 2009, 30, 96–116. [Google Scholar] [CrossRef]
- American Diabetes Association Task Force for Writing Nutrition Principles; Recommendations for the Management of Diabetes and Related Complications. American Diabetes Association position statement: Evidence-based nutrition principles and recommendations for the treatment and prevention of diabetes and related complications. J. Am. Diet. Assoc. 2002, 102, 109–118. [Google Scholar] [CrossRef]
- Shoham, D.A.; Durazo-Arvizu, R.; Kramer, H.; Luke, A.; Vupputuri, S.; Kshirsagar, A.; Cooper, R.S. Sugary soda consumption and albuminuria: Results from the National Health and Nutrition Examination Survey, 1999–2004. PLoS ONE 2008, 3, e3431. [Google Scholar] [CrossRef]
- Bjornstad, P.; Lanaspa, M.A.; Ishimoto, T.; Kosugi, T.; Kume, S.; Jalal, D.; Maahs, D.M.; Snell-Bergeon, J.K.; Johnson, R.J.; Nakagawa, T. Fructose and uric acid in diabetic nephropathy. Diabetologia 2015, 58, 1993–2002. [Google Scholar] [CrossRef]
- Dissard, R.; Klein, J.; Caubet, C.; Breuil, B.; Siwy, J.; Hoffman, J.; Sicard, L.; Ducasse, L.; Rascalou, S.; Payre, B.; et al. Long term metabolic syndrome induced by a high fat high fructose diet leads to minimal renal injury in C57BL/6 mice. PLoS ONE 2013, 8, e76703. [Google Scholar] [CrossRef]
- Calcutt, N.A.; Cooper, M.E.; Kern, T.S.; Schmidt, A.M. Therapies for hyperglycaemia-induced diabetic complications: From animal models to clinical trials. Nat. Rev. Drug Discov. 2009, 8, 417–429. [Google Scholar] [CrossRef] [PubMed]
- Surwit, R.S.; Feinglos, M.N.; Rodin, J.; Sutherland, A.; Petro, A.E.; Opara, E.C.; Kuhn, C.M.; Rebuffe-Scrive, M. Differential effects of fat and sucrose on the development of obesity and diabetes in C57BL/6J and A/J mice. Metabolism 1995, 44, 645–651. [Google Scholar] [CrossRef]


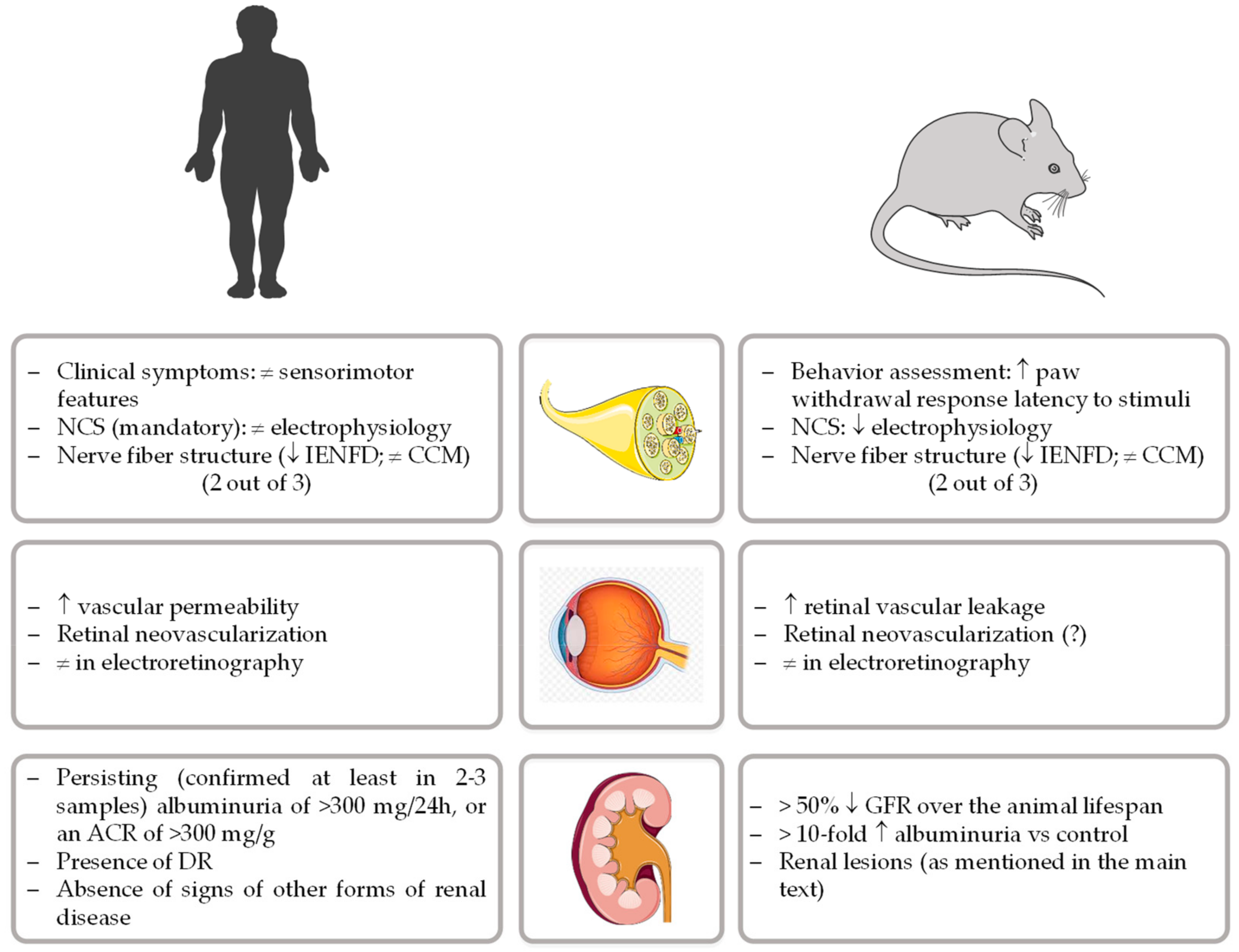{kind=link}
| Diet Type | Composition (%) | Duration (Wks) | Species/Strain | Main Features of DPN Evolution | Ref. |
|---|---|---|---|---|---|
| HFD | 45% calories from fat | 34 | Mice/C57BL/6J | Thermal hypoalgesia (week 12, 34); Motor and sensory nerve conduction abnormalities (week 12, 34); Decreased IENFD (week 34) | [39] |
| 45% calories from fat | 12 | Mice/C57BL/6J | Thermal hypoalgesia (week 6); Sensory nerve conduction abnormalities (week 16, 24 and 36); Decreased IENFD (week 12) | [42] | |
| 60% calories from fat | 31 | Mice/C57BL/6J | Thermal hypoalgesia (week 24 and 36); Motor and sensory nerve conduction abnormalities (week 16, 24 and 36); Decreased IENFD (week 36) | [43] | |
| 60% calories from fat | 12 | Rat/Sprague-Dawley | Thermal hypoalgesia and mechanical allodynia (week 12); Sensory nerve conduction abnormalities (week 12); Decreased IENFD and corneal nerve fiber length (week 12) | [44] | |
| HFD + moderate dose STZ | 60% calories from fat | 31 | Mice/C57BL/6J | Thermal hypoalgesia (week 24 and 36); Motor and sensory nerve conduction abnormalities (week 16, 24 and 36); Decreased IENFD (week 36) | [43] |
| HFD + low dose STZ | 60% calories from fat | 12 | Rat/Sprague-Dawley | Thermal hypoalgesia and mechanical allodynia (week 12); Motor and sensory nerve conduction abnormalities (week 12); Decreased IENFD and corneal nerve fiber length (week 12) | [41] |
| HFSD | 46% calories from fat, 36% calories from CH | 16 | Rat/Sprague-Dawley | Reduced paw withdrawal mechanical threshold and thermal latency (week 8); Motor coordination impairments (week 16); Severe damage of large myelinated fibers along with mild damage to small myelinated and unmyelinated fibers (week 16) | [45] |
| Diet Type | Composition (%) | Duration (Weeks) | Specie/Strain | Main Features of DR Evolution | Ref. |
|---|---|---|---|---|---|
| HFD | 45% calories from fat, 35% calories from CH | 16 to 24 | Mice/C57BL/6 | Electrophysiological impairment of retinal ganglion cells, including delayed and diminished visual responses to patterned light stimuli, precede increased retinal vascular permeability | [59] |
| 42% calories from fat, 43% calories from CH | 24 to 48 | Mice/C57BL/6J | Accelerated weight gain and adiposity, with abnormal glucose metabolism and electroretinographic dysfunction manifested by increased latencies and reduced amplitudes of OP (6 weeks); evidence of microvascular disease (48 weeks) | [60] | |
| 35% calories from fat | 16 | Mice/Swiss | Pro-inflammatory state in the retinal tissue; Increased VEGF protein levels | [61] | |
| HFD + low dose STZ | 17.9% calories from fat | 12 | Mice/C57BL/6J | Pericytes dropout, acellular capillaries formation, increased vascular leakage, increased oxidative stress, chronic retinal inflammation and increased apoptosis | [62] |
| 10% calories from fat | 16 | Rat/Sprague–Dawley | ERG abnormalities (abnormal b-wave amplitudes and OPs); Thinner ONLs and fewer cells in the ONLs of the retinas | [63] | |
| HFD + high fructose | 25.7% calories from fat; 46.5 wt% fructose | 20 | Rat/Wistar | Thickening of the retinal parenchyma and neovascularization | [8] |
| Diet Type | Composition (%) | Duration (Weeks) | Species/Strain | Main Features of DN Evolution | Ref. |
|---|---|---|---|---|---|
| HFD | 30% calories from fat | 20 to 28 | Rat/Wistar | Unable to develop major changes on renal function and basal microvascular blood flow | [81] |
| 60% calories from fat | 12 | Mice/C57BL/6 | Increased UAE and glomerular lesions associated with extracellular matrix protein accumulation, together with impaired sodium handling | [80] | |
| HFD + low dose STZ | 60% calories from fat | 5 | Rat/Sprague–Dawley | Despite lower blood glucose levels and proteinuria, animals will develop more severe kidney lesions | [82] |
| 11.3% calories from fat | 15 to 25 | Rat/Sprague–Dawley | Microalbuminuria and augmented creatinine clearance, followed (25 weeks) by overt proteinuria, mesangial expansion and glomerular sclerosis | [83] | |
| High-CH | 56% calories from CH | 20 | Rat/Obese ZSF1 | Renal lesion resembling early DN, such as arteriolar thickening, tubular dilation and atrophy, thickening of GBM and mesangial expansion, with proteinuria and tubulointerstitial fibrosis. | [65,66,84,85,86] |
| 45–50 | Progressive aggravation of renal disease with death accompanied by ESRD when aged 45–50 weeks | ||||
| High-fructose | 6o% calories from fructose | 6 to 8 | Rat/Sprague–Dawley | Renal hypertrophy, arteriolopathy, glomerular hypertension, and cortical vasoconstriction, together with cell proliferation and hyperplasia in proximal tubules (focal tubulointerstitial injury) | [87,88] |
| 66% calories from fructose | 4 | Rat/SDT | Renal lesions accompanied by increases in urine volume and renal function parameters, which progresses with aging to severe tubular lesions, diffuse and nodular glomerular lesions | [89,90] | |
| 67% calories from fructose | 16 | Mice DBA/2N | Kidney injury was compared in C57Bl/6J, CBA/JN and DBA/2N, and only the DBA/2N mice was able to develop tubulointerstitial fibrosis | [91] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Preguiça, I.; Alves, A.; Nunes, S.; Gomes, P.; Fernandes, R.; Viana, S.D.; Reis, F. Diet-Induced Rodent Models of Diabetic Peripheral Neuropathy, Retinopathy and Nephropathy. Nutrients 2020, 12, 250. https://doi.org/10.3390/nu12010250
Preguiça I, Alves A, Nunes S, Gomes P, Fernandes R, Viana SD, Reis F. Diet-Induced Rodent Models of Diabetic Peripheral Neuropathy, Retinopathy and Nephropathy. Nutrients. 2020; 12(1):250. https://doi.org/10.3390/nu12010250
Chicago/Turabian StylePreguiça, Inês, André Alves, Sara Nunes, Pedro Gomes, Rosa Fernandes, Sofia D. Viana, and Flávio Reis. 2020. "Diet-Induced Rodent Models of Diabetic Peripheral Neuropathy, Retinopathy and Nephropathy" Nutrients 12, no. 1: 250. https://doi.org/10.3390/nu12010250
APA StylePreguiça, I., Alves, A., Nunes, S., Gomes, P., Fernandes, R., Viana, S. D., & Reis, F. (2020). Diet-Induced Rodent Models of Diabetic Peripheral Neuropathy, Retinopathy and Nephropathy. Nutrients, 12(1), 250. https://doi.org/10.3390/nu12010250