The Paradox of Coenzyme Q10 in Aging




 and
and 
Abstract
1. Introduction
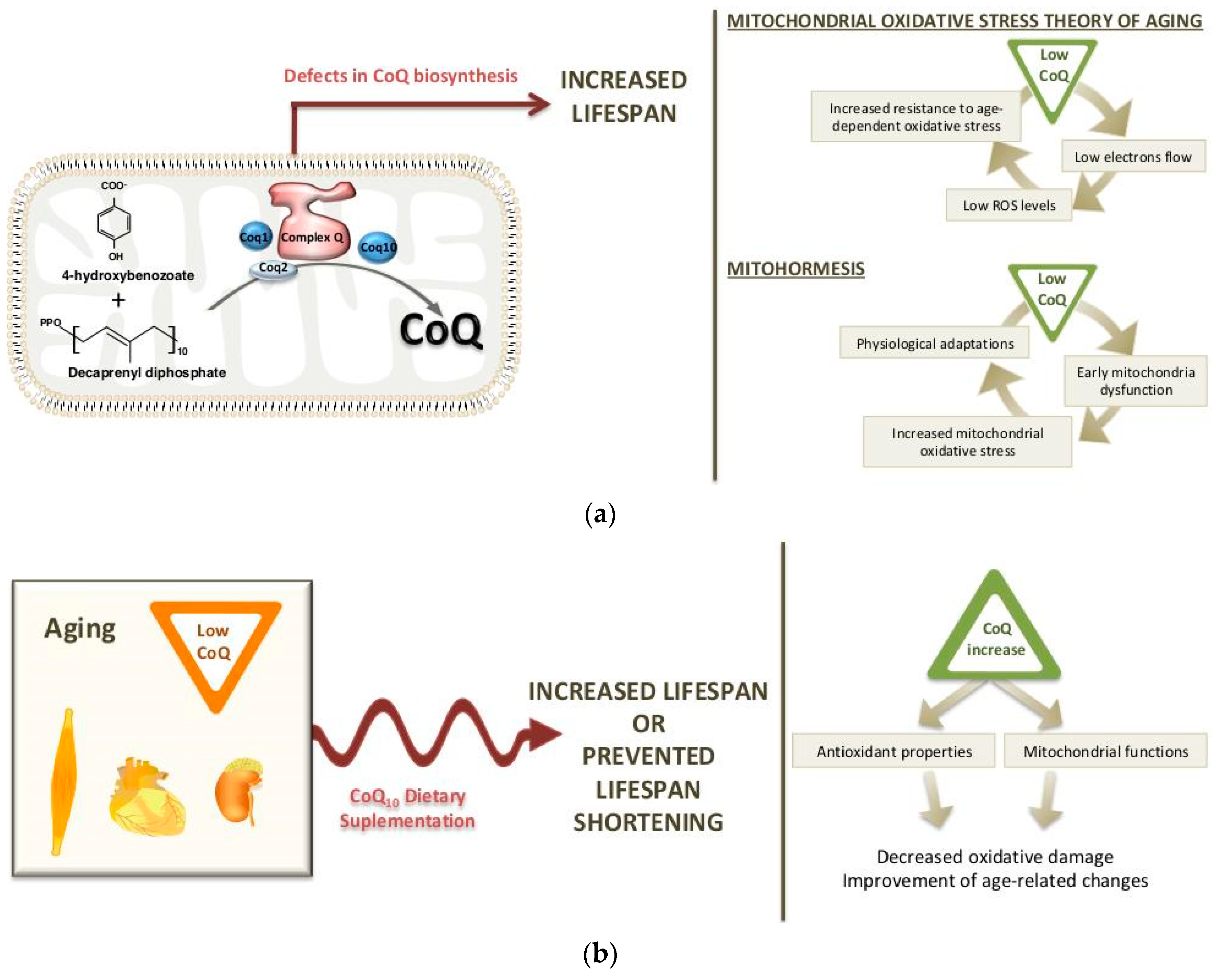
1.1. CoQ Biosynthesis
1.1.1. Quinone Synthesis
1.1.2. Polyisoprenyl Tail Synthesis
1.1.3. Attachment of the Ring/Chain
1.1.4. Next Steps of CoQ Biosynthesis-Ring Modifications
1.1.5. Other Mitochondrial Proteins Involved in CoQ Biosynthesis
1.1.6. Complex Q
1.2. Functions of CoQ
1.2.1. The Role of CoQ in the Mitochondrial Respiratory Chain
1.2.2. The Antioxidant Capacity
1.2.3. Other Controversial Functions
1.3. Pathologic Conditions with Decreased Levels of CoQ
1.3.1. Primary CoQ10 Deficiencies
1.3.2. Secondary CoQ10 Deficiencies
2. Long-Life Animal Models with CoQ Deficiency: Potential Mechanisms
2.1. Worm Models
2.2. Mouse Models
3. Dietary Supplementation of CoQ10 during Aging
3.1. Changes in CoQ Biosynthesis during Aging
3.2. CoQ10 Supplementation in Aging: Effects on Life span and Longevity
3.3. Reversal of Age-Related Changes by CoQ10 Supplementation
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kalén, A.; Appelkvist, E.L.; Chojnacki, T.; Dallner, G. Nonaprenyl-4-hydroxybenzoate transferase, an enzyme involved in ubiquinone biosynthesis, in the endoplasmic reticulum-Golgi system of rat liver. J. Biol. Chem. 1990, 265, 1158–1164. [Google Scholar] [PubMed]
- Kalén, A.; Norling, B.; Appelkvist, E.; Dallner, G. Ubiquinone biosynthesis by the microsomal fraction from rat liver. Biochim. Biophys. Acta 1987, 926, 70–78. [Google Scholar] [CrossRef]
- Eisenberg-Bord, M.; Tsui, H.S.; Antunes, D.; Fernandez-Del-Rio, L.; Bradley, M.C.; Dunn, C.D.; Nguyen, T.P.T.; Rapaport, D.; Clarke, C.F.; Schuldiner, M. The Endoplasmic Reticulum-Mitochondria Encounter Structure Complex Coordinates Coenzyme Q Biosynthesis. Contact 2019, 2. [Google Scholar] [CrossRef] [PubMed]
- Turunen, M.; Olsson, J.; Dallner, G. Metabolism and function of coenzyme Q. Biochim. Biophys. Acta 2004, 1660, 171–199. [Google Scholar] [CrossRef] [PubMed]
- Tzagoloff, A.; Akai, A.; Needleman, R.B.; Zulch, G. Assembly of the mitochondrial membrane system. Cytoplasmic mutants of Saccharomyces cerevisiae with lesions in enzymes of the respiratory chain and in the mitochondrial ATPase. J. Biol. Chem. 1975, 250, 8236–8242. [Google Scholar] [PubMed]
- Tzagoloff, A.; Dieckmann, C.L. PET genes of Saccharomyces cerevisiae. Microbiol. Rev. 1990, 54, 211–225. [Google Scholar] [PubMed]
- Meganathan, R. Ubiquinone biosynthesis in microorganisms. FEMS Microbiol. Lett. 2001, 203, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Hajj Chehade, M.; Loiseau, L.; Lombard, M.; Pecqueur, L.; Ismail, A.; Smadja, M.; Golinelli-Pimpaneau, B.; Mellot-Draznieks, C.; Hamelin, O.; Aussel, L.; et al. ubiI, a new gene in Escherichia coli coenzyme Q biosynthesis, is involved in aerobic C5-hydroxylation. J. Biol. Chem. 2013, 288, 20085–20092. [Google Scholar] [CrossRef]
- Aussel, L.; Loiseau, L.; Hajj Chehade, M.; Pocachard, B.; Fontecave, M.; Pierrel, F.; Barras, F. ubiJ, a new gene required for aerobic growth and proliferation in macrophage, is involved in coenzyme Q biosynthesis in Escherichia coli and Salmonella enterica serovar Typhimurium. J. Bacteriol. 2014, 196, 70–79. [Google Scholar] [CrossRef]
- Awad, A.M.; Bradley, M.C.; Fernández-Del-Río, L.; Nag, A.; Tsui, H.S.; Clarke, C.F. Coenzyme Q10 deficiencies: Pathways in yeast and humans. Essays Biochem. 2018, 62, 361–376. [Google Scholar] [CrossRef]
- Saiki, R.; Nagata, A.; Kainou, T.; Matsuda, H.; Kawamukai, M. Characterization of solanesyl and decaprenyl diphosphate synthases in mice and humans. FEBS J. 2005, 272, 5606–5622. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, K.; Ogiyama, Y.; Yokomi, K.; Nakagawa, T.; Kaino, T.; Kawamukai, M. Functional Conservation of Coenzyme Q Biosynthetic Genes among Yeasts, Plants, and Humans. PLoS ONE 2014, 9, e99038. [Google Scholar] [CrossRef] [PubMed]
- Kawamukai, M. Biosynthesis of coenzyme Q in eukaryotes. Biosci. Biotechnol. Biochem. 2016, 80, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Marbois, B.; Xie, L.X.; Choi, S.; Hirano, K.; Hyman, K.; Clarke, C.F. para-Aminobenzoic Acid Is a Precursor in Coenzyme Q6 Biosynthesis in Saccharomyces cerevisiae. J. Biol. Chem. 2010, 285, 27827–27838. [Google Scholar] [CrossRef] [PubMed]
- Payet, L.-A.; Leroux, M.; Willison, J.C.; Kihara, A.; Pelosi, L.; Pierrel, F. Mechanistic Details of Early Steps in Coenzyme Q Biosynthesis Pathway in Yeast. Cell Chem. Biol. 2016, 23, 1241–1250. [Google Scholar] [CrossRef] [PubMed]
- Stefely, J.A.; Kwiecien, N.W.; Freiberger, E.C.; Richards, A.L.; Jochem, A.; Rush, M.J.P.; Ulbrich, A.; Robinson, K.P.; Hutchins, P.D.; Veling, M.T.; et al. Mitochondrial protein functions elucidated by multi-omic mass spectrometry profiling. Nat. Biotechnol. 2016, 34, 1191–1197. [Google Scholar] [CrossRef] [PubMed]
- Stefely, J.A.; Pagliarini, D.J. Biochemistry of Mitochondrial Coenzyme Q Biosynthesis. Trends Biochem. Sci. 2017, 42, 824–843. [Google Scholar] [CrossRef]
- Doimo, M.; Trevisson, E.; Airik, R.; Bergdoll, M.; Santos-Ocaña, C.; Hildebrandt, F.; Navas, P.; Pierrel, F.; Salviati, L. Effect of vanillic acid on COQ6 mutants identified in patients with coenzyme Q10 deficiency. Biochim. Biophys. Acta 2014, 1842, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo-Gutierrez, A.; Barriocanal-Casado, E.; Bakkali, M.; Diaz-Casado, M.E.; Sanchez-Maldonado, L.; Romero, M.; Sayed, R.K.; Prehn, C.; Escames, G.; Duarte, J.; et al. beta-RA reduces DMQ/CoQ ratio and rescues the encephalopathic phenotype in Coq9 (R239X) mice. EMBO Mol. Med. 2019, 11, e9466. [Google Scholar] [CrossRef]
- Pierrel, F. Impact of Chemical Analogs of 4-Hydroxybenzoic Acid on Coenzyme Q Biosynthesis: From Inhibition to Bypass of Coenzyme Q Deficiency. Front. Physiol. 2017, 8, 436. [Google Scholar] [CrossRef]
- Wang, Y.; Oxer, D.; Hekimi, S. Mitochondrial function and lifespan of mice with controlled ubiquinone biosynthesis. Nat. Commun. 2015, 6, 6393. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Smith, C.; Parboosingh, J.S.; Khan, A.; Innes, M.; Hekimi, S. Pathogenicity of two COQ7 mutations and responses to 2,4-dihydroxybenzoate bypass treatment. J. Cell. Mol. Med. 2017, 21, 2329–2343. [Google Scholar] [CrossRef] [PubMed]
- Luna-Sánchez, M.; Díaz-Casado, E.; Barca, E.; Ángel, T.M.; Ángeles, M.-G.; Cobos, E.J.; Escames, G.; Acuña-Castroviejo, D.; Quinzii, C.M.; López, L.C. The clinical heterogeneity of coenzyme Q10 deficiency results from genotypic differences in the Coq9 gene. EMBO Mol. Med. 2015, 7, 670–687. [Google Scholar]
- Xie, L.X.; Williams, K.J.; He, C.H.; Weng, E.; Khong, S.; Rose, T.E.; Kwon, O.; Bensinger, S.J.; Marbois, B.N.; Clarke, C.F. Resveratrol and para-coumarate serve as ring precursors for coenzyme Q biosynthesis[S]. J. Lipid Res. 2015, 56, 909–919. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Del-Rio, L.; Nag, A.; Gutierrez Casado, E.; Ariza, J.; Awad, A.M.; Joseph, A.I.; Kwon, O.; Verdin, E.; de Cabo, R.; Schneider, C.; et al. Kaempferol increases levels of coenzyme Q in kidney cells and serves as a biosynthetic ring precursor. Free Radic. Biol. Med. 2017, 110, 176–187. [Google Scholar] [CrossRef] [PubMed]
- Gin, P.; Hsu, A.Y.; Rothman, S.C.; Jonassen, T.; Tzagoloff, A.; Clarke, C.F.; Lee, P.T. TheSaccharomyces cerevisiae COQ6Gene Encodes a Mitochondrial Flavin-dependent Monooxygenase Required for Coenzyme Q Biosynthesis. J. Biol. Chem. 2003, 278, 25308–25316. [Google Scholar] [CrossRef] [PubMed]
- Ozeir, M.; Mühlenhoff, U.; Webert, H.; Lill, R.; Fontecave, M.; Pierrel, F. Coenzyme Q Biosynthesis: Coq6 Is Required for the C5-Hydroxylation Reaction and Substrate Analogs Rescue Coq6 Deficiency. Chem. Biol. 2011, 18, 1134–1142. [Google Scholar] [CrossRef] [PubMed]
- Marbois, B.; Gin, P.; Faull, K.F.; Poon, W.W.; Strahan, J.; Clarke, C.F.; Lee, P.T.; Shepherd, J.N. Coq3 and Coq4 Define a Polypeptide Complex in Yeast Mitochondria for the Biosynthesis of Coenzyme Q. J. Biol. Chem. 2005, 280, 20231–20238. [Google Scholar] [CrossRef]
- Zhu, Y.; Wu, B.; Zhang, X.; Fan, X.; Niu, L.; Li, X.; Wang, J.; Teng, M. Structural and biochemical studies reveal UbiG/Coq3 as a class of novel membrane-binding proteins. Biochem. J. 2015, 470, 105–114. [Google Scholar] [CrossRef]
- Cox, G.B.; Young, I.G.; McCann, L.M.; Gibson, F. Biosynthesis of Ubiquinone in Escherichia coli K-12: Location of Genes Affecting the Metabolism of 3-Octaprenyl-4-hydroxybenzoic Acid and 2-Octaprenylphenol. J. Bacteriol. 1969, 99, 450–458. [Google Scholar]
- Young, I.G.; Stroobant, P.; Macdonald, C.G.; Gibson, F. Pathway for Ubiquinone Biosynthesis in Escherichia coli K-12: Gene-Enzyme Relationships and Intermediates. J. Bacteriol. 1973, 114, 42–52. [Google Scholar] [PubMed]
- White, M.D.; Payne, K.A.P.; Fisher, K.; Marshall, S.A.; Parker, D.; Rattray, N.J.W.; Trivedi, D.K.; Goodacre, R.; Rigby, S.E.J.; Scrutton, N.S.; et al. UbiX is a flavin prenyltransferase required for bacterial ubiquinone biosynthesis. Nature 2015, 522, 502–506. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.N.; Zhou, K.; Cao, D.D.; Jiang, Y.L.; Meng, F.; Chi, C.B.; Ren, Y.M.; Chen, Y.; Zhou, C.Z. Crystal structures and catalytic mechanism of the C-methyltransferase Coq5 provide insights into a key step of the yeast coenzyme Q synthesis pathway. Acta Crystallogr. D Biol. Crystallogr. 2014, 70, 2085–2092. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.P.; Casarin, A.; Desbats, M.A.; Doimo, M.; Trevisson, E.; Santos-Ocaña, C.; Navas, P.; Clarke, C.F.; Salviati, L. Molecular characterization of the human COQ5 C-methyltransferase in coenzyme Q10 biosynthesis. Biochim. Biophys. Acta 2014, 1841, 1628–1638. [Google Scholar] [CrossRef] [PubMed]
- Stenmark, P.; Grunler, J.; Mattsson, J.; Sindelar, P.J.; Nordlund, P.; Berthold, D.A. A new member of the family of di-iron carboxylate proteins. Coq7 (clk-1), a membrane-bound hydroxylase involved in ubiquinone biosynthesis. J. Biol. Chem. 2001, 276, 33297–33300. [Google Scholar] [CrossRef] [PubMed]
- Gerards, M.; Bosch, B.V.D.; Calis, C.; Schoonderwoerd, K.; Van Engelen, K.; Tijssen, M.; De Coo, R.; Van Der Kooi, A.; Smeets, H. Nonsense mutations in CABC1/ADCK3 cause progressive cerebellar ataxia and atrophy. Mitochondrion 2010, 10, 510–515. [Google Scholar] [CrossRef] [PubMed]
- Horváth, P.O.; Czermin, B.; Gulati, S.; Pyle, A.; Hassani, A.; Foley, C.; Taylor, R.W.; Chinnery, P.F. 003 Adult-onset cerebellar ataxia due to mutations in the CABC1/ADCK3 gene. J. Neurol. Neurosurg. Psychiatry 2012, 83. [Google Scholar] [CrossRef]
- Lagier-Tourenne, C.; Tazir, M.; López, L.C.; Quinzii, C.M.; Assoum, M.; Drouot, N.; Busso, C.; Makri, S.; Ali-Pacha, L.; Benhassine, T.; et al. ADCK3, an Ancestral Kinase, Is Mutated in a Form of Recessive Ataxia Associated with Coenzyme Q10 Deficiency. Am. J. Hum. Genet. 2008, 82, 661–672. [Google Scholar] [CrossRef]
- Vazquez Fonseca, L.; Doimo, M.; Calderan, C.; Desbats, M.A.; Acosta, M.J.; Cerqua, C.; Cassina, M.; Ashraf, S.; Hildebrandt, F.; Sartori, G.; et al. Mutations in COQ8B (ADCK4) found in patients with steroid-resistant nephrotic syndrome alter COQ8B function. Hum. Mutat. 2018, 39, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.X.; Ozeir, M.; Tang, J.Y.; Chen, J.Y.; Jaquinod, S.-K.; Fontecave, M.; Clarke, C.F.; Pierrel, F. Overexpression of the Coq8 Kinase in Saccharomyces cerevisiae coq Null Mutants Allows for Accumulation of Diagnostic Intermediates of the Coenzyme Q6 Biosynthetic Pathway. J. Biol. Chem. 2012, 287, 23571–23581. [Google Scholar] [CrossRef]
- Reidenbach, A.G.; Kemmerer, Z.A.; Aydin, D.; Jochem, A.; McDevitt, M.T.; Hutchins, P.D.; Stark, J.L.; Stefely, J.A.; Reddy, T.; Hebert, A.S.; et al. Conserved Lipid and Small-Molecule Modulation of COQ8 Reveals Regulation of the Ancient Kinase-like UbiB Family. Cell Chem. Biol. 2018, 25, 154–165 e111. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, E.J.; Gin, P.; Gulmezian, M.; Tran, U.C.; Saiki, R.; Marbois, B.N.; Clarke, C.F. Saccharomyces cerevisiae Coq9 Polypeptide is a Subunit of the Mitochondrial Coenzyme Q Biosynthetic Complex. Arch. Biochem. Biophys. 2007, 463, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Lohman, D.C.; Aydin, D.; Von Bank, H.C.; Smith, R.W.; Linke, V.; Weisenhorn, E.; McDevitt, M.T.; Hutchins, P.; Wilkerson, E.M.; Wancewicz, B.; et al. An Isoprene Lipid-Binding Protein Promotes Eukaryotic Coenzyme Q Biosynthesis. Mol. Cell 2019, 73, 763–774.e10. [Google Scholar] [CrossRef] [PubMed]
- He, C.W.; Black, D.S.; Nguyen, T.P.T.; Wang, C.; Srinivasan, C.; Clarke, C.F. Yeast Coq9 controls deamination of coenzyme Q intermediates that derive from para-aminobenzoic acid. Biochim. Biophys. Acta 2015, 1851, 1227–1239. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lohman, D.C.; Forouhar, F.; Beebe, E.T.; Stefely, M.S.; Minogue, C.E.; Ulbrich, A.; Stefely, J.A.; Sukumar, S.; Luna-Sánchez, M.; Jochem, A.; et al. Mitochondrial COQ9 is a lipid-binding protein that associates with COQ7 to enable coenzyme Q biosynthesis. Proc. Natl. Acad. Sci. USA 2014, 111, E4697–E4705. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Corzo, L.; Luna-Sanchez, M.; Doerrier, C.; Garcia, J.A.; Guaras, A.; Acin-Perez, R.; Bullejos-Peregrin, J.; Lopez, A.; Escames, G.; Enriquez, J.A.; et al. Dysfunctional Coq9 protein causes predominant encephalomyopathy associated with CoQ deficiency. Hum. Mol. Genet. 2013, 22, 1233–1248. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hekimi, S. Molecular genetics of ubiquinone biosynthesis in animals. Crit. Rev. Biochem. Mol. Biol. 2013, 48, 69–88. [Google Scholar] [CrossRef]
- Allan, C.M.; Hill, S.; Morvaridi, S.; Saiki, R.; Johnson, J.S.; Liau, W.S.; Hirano, K.; Kawashima, T.; Ji, Z.; Loo, J.A.; et al. A conserved START domain coenzyme Q-binding polypeptide is required for efficient Q biosynthesis, respiratory electron transport, and antioxidant function in Saccharomyces cerevisiae. Biochim. Biophys. Acta 2013, 1831, 776–791. [Google Scholar] [CrossRef]
- Cui, T.Z.; Kawamukai, M. Coq10, a mitochondrial coenzyme Q binding protein, is required for proper respiration in Schizosaccharomyces pombe. FEBS J. 2009, 276, 748–759. [Google Scholar] [CrossRef]
- Tsui, H.S.; Pham, N.V.B.; Amer, B.R.; Bradley, M.C.; Gosschalk, J.E.; Gallagher-Jones, M.; Ibarra, H.; Clubb, R.T.; Blaby-Haas, C.E.; Clarke, C.F. Human COQ10A and COQ10B are distinct lipid-binding START domain proteins required for coenzyme Q function. J. Lipid Res. 2019, 60, 1293–1310. [Google Scholar] [CrossRef]
- Marbois, B.; Gin, P.; Gulmezian, M.; Clarke, C.F. The yeast Coq4 polypeptide organizes a mitochondrial protein complex essential for coenzyme Q biosynthesis. Biochim. Biophys. Acta 2009, 1791, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Belogrudov, G.I.; Lee, P.T.; Jonassen, T.; Hsu, A.Y.; Gin, P.; Clarke, C.F. Yeast COQ4 Encodes a Mitochondrial Protein Required for Coenzyme Q Synthesis. Arch. Biochem. Biophys. 2001, 392, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Casarin, A.; Jimenez-Ortega, J.C.; Trevisson, E.; Pertegato, V.; Doimo, M.; Ferrero-Gomez, M.L.; Abbadi, S.; Artuch, R.; Quinzii, C.; Hirano, M.; et al. Functional characterization of human COQ4, a gene required for Coenzyme Q10 biosynthesis. Biochem. Biophys. Res. Commun. 2008, 372, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Allan, C.M.; Awad, A.M.; Johnson, J.S.; Shirasaki, D.I.; Wang, C.; Blaby-Haas, C.E.; Merchant, S.S.; Loo, J.A.; Clarke, C.F. Identification of Coq11, a New Coenzyme Q Biosynthetic Protein in the CoQ-Synthome in Saccharomyces cerevisiae. J. Biol. Chem. 2015, 290, 7517–7534. [Google Scholar] [CrossRef] [PubMed]
- He, C.H.; Xie, L.X.; Allan, C.M.; Tran, U.C.; Clarke, C.F. Coenzyme Q supplementation or over-expression of the yeast Coq8 putative kinase stabilizes multi-subunit Coq polypeptide complexes in yeast coq null mutants. Biochim. Biophys. Acta 2014, 1841, 630–644. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Stefely, J.A.; Licitra, F.; Laredj, L.; Reidenbach, A.G.; Kemmerer, Z.A.; Grangeray, A.; Jaeg-Ehret, T.; Minogue, C.E.; Ulbrich, A.; Hutchins, P.D.; et al. Cerebellar Ataxia and Coenzyme Q Deficiency through Loss of Unorthodox Kinase Activity. Mol. Cell 2016, 63, 608–620. [Google Scholar] [CrossRef] [PubMed]
- Crane, F.; Hatefi, Y.; Lester, R.; Widmer, C. Isolation of a quinone from beef heart mitochondria. Biochim. Biophys. Acta 1957, 25, 220–221. [Google Scholar] [CrossRef]
- Quinzii, C.M.; Luna-Sanchez, M.; Ziosi, M.; Hidalgo-Gutierrez, A.; Kleiner, G.; Lopez, L.C. The Role of Sulfide Oxidation Impairment in the Pathogenesis of Primary CoQ Deficiency. Front. Physiol. 2017, 8, 525. [Google Scholar] [CrossRef]
- Ziosi, M.; Di Meo, I.; Kleiner, G.; Gao, X.H.; Barca, E.; Sanchez-Quintero, M.J.; Tadesse, S.; Jiang, H.; Qiao, C.; Rodenburg, R.J.; et al. Coenzyme Q deficiency causes impairment of the sulfide oxidation pathway. EMBO Mol. Med. 2017, 9, 96–111. [Google Scholar] [CrossRef] [PubMed]
- Luna-Sanchez, M.; Hidalgo-Gutierrez, A.; Hildebrandt, T.M.; Chaves-Serrano, J.; Barriocanal-Casado, E.; Santos-Fandila, A.; Romero, M.; Sayed, R.K.; Duarte, J.; Prokisch, H.; et al. CoQ deficiency causes disruption of mitochondrial sulfide oxidation, a new pathomechanism associated with this syndrome. EMBO Mol. Med. 2017, 9, 78–95. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Wakitani, S.; Hayashi, K.; Miki, R.; Kawamukai, M. High production of sulfide in coenzyme Q deficient fission yeast. BioFactors 2008, 32, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Blake, R.L.; Hall, J.G.; Russell, E.S. Mitochondrial proline dehydrogenase deficiency in hyperprolinemic PRO/Re mice: Genetic and enzymatic analyses. Biochem. Genet. 1976, 14, 739–757. [Google Scholar] [CrossRef] [PubMed]
- Battino, M.; Fato, R.; Lenaz, G.; Drahota, Z. Coenzyme Q-pool function in glycerol-3-phosphate oxidation in hamster brown adipose tissue mitochondria. J. Bioenerg. Biomembr. 1992, 24, 235–241. [Google Scholar]
- Evans, D.R.; Guy, H.I. Mammalian Pyrimidine Biosynthesis: Fresh Insights into an Ancient Pathway. J. Biol. Chem. 2004, 279, 33035–33038. [Google Scholar] [CrossRef]
- Jones, E.M. Pyrimidine Nucleotide Biosynthesis in Animals: Genes, Enzymes, and Regulation of UMP Biosynthesis. Annu. Rev. Biochem. 1980, 49, 253–279. [Google Scholar] [CrossRef] [PubMed]
- López-Martín, J.M.; Salviati, L.; Trevisson, E.; Montini, G.; DiMauro, S.; Quinzii, C.; Hirano, M.; Rodriguez-Hernandez, A.; Cordero, M.D.; Sánchez-Alcázar, J.A.; et al. Missense mutation of the COQ2 gene causes defects of bioenergetics and de novo pyrimidine synthesis. Hum. Mol. Genet. 2007, 16, 1091–1097. [Google Scholar] [CrossRef] [PubMed]
- Watmough, N.J.; Frerman, F.E. The electron transfer flavoprotein: Ubiquinone oxidoreductases. Biochim. Biophys. Acta 2010, 1797, 1910–1916. [Google Scholar] [CrossRef] [PubMed]
- Summitt, C.B.; Johnson, L.C.; Jönsson, T.J.; Parsonage, D.; Holmes, R.P.; Lowther, W.T. Proline dehydrogenase 2 (PRODH2) is a hydroxyproline dehydrogenase (HYPDH) and molecular target for treating primary hyperoxaluria. Biochem. J. 2015, 466, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Forsmark-Andrée, P.; Dallner, G.; Ernster, L. Endogenous ubiquinol prevents protein modification accompanying lipid peroxidation in beef heart submitochondrial particles. Free Radic. Biol. Med. 1995, 19, 749–757. [Google Scholar] [CrossRef]
- Godic, A.; Poljsak, B.; Adamič, M.; Dahmane, R. The Role of Antioxidants in Skin Cancer Prevention and Treatment. Oxidative Med. Cell. Longev. 2014, 2014, 1–6. [Google Scholar] [CrossRef]
- Mukai, K.; Kikuchi, S.; Urano, S. Stopped-flow kinetic study of the regeneration reaction of tocopheroxyl radical by reduced ubiquinone-10 in solution. Biochim. Biophys. Acta 1990, 1035, 77–82. [Google Scholar] [CrossRef]
- Frei, B.; Kim, M.C.; Ames, B.N. Ubiquinol-10 is an effective lipid-soluble antioxidant at physiological concentrations. Proc. Natl. Acad. Sci. USA 1990, 87, 4879–4883. [Google Scholar] [CrossRef] [PubMed]
- Forsmark-Andree, P.; Ernster, L. Evidence for a protective effect of endogenous ubiquinol against oxidative damage to mitochondrial protein and DNA during lipid peroxidation. Mol. Asp. Med. 1994, 15, S73–S81. [Google Scholar] [CrossRef]
- Tomasetti, M.; Littarru, G.; Stocker, R.; Alleva, R. Coenzyme Q10 enrichment decreases oxidative DNA damage in human lymphocytes. Free Radic. Biol. Med. 1999, 27, 1027–1032. [Google Scholar] [CrossRef]
- Tomasetti, M.; Alleva, R.; Collins, A.R. In vivo supplementation with coenzyme Q10 enhances the recovery of human lymphocytes from oxidative DNA damage. FASEB J. 2001, 15, 1425–1427. [Google Scholar] [CrossRef] [PubMed]
- Bentinger, M.; Brismar, K.; Dallner, G. The antioxidant role of coenzyme Q. Mitochondrion 2007, 7, S41–S50. [Google Scholar] [CrossRef] [PubMed]
- Bentinger, M.; Dallner, G.; Chojnacki, T.; Swiezewska, E. Distribution and breakdown of labeled coenzyme Q10 in rat. Free Radic. Biol. Med. 2003, 34, 563–575. [Google Scholar] [CrossRef]
- Fontaine, E.; Ichas, F.; Bernardi, P. A Ubiquinone-binding Site Regulates the Mitochondrial Permeability Transition Pore. J. Biol. Chem. 1998, 273, 25734–25740. [Google Scholar] [CrossRef]
- Echtay, K.S.; Winkler, E.; Klingenberg, M.; Echtay, K.S.; Winkler, E.; Echtay, K.S.; Winkler, E.; Klingenberg, M. Coenzyme Q is an obligatory cofactor for uncoupling protein function. Nature 2000, 408, 609–613. [Google Scholar] [CrossRef]
- Echtay, K.S.; Winkler, E.; Frischmuth, K.; Klingenberg, M. Uncoupling proteins 2 and 3 are highly active H(+) transporters and highly nucleotide sensitive when activated by coenzyme Q (ubiquinone). Proc. Natl. Acad. Sci. USA 2001, 98, 1416–1421. [Google Scholar] [CrossRef]
- Jaburek, M.; Garlid, K.D. Reconstitution of recombinant uncoupling proteins: UCP1, -2, and -3 have similar affinities for ATP and are unaffected by coenzyme Q10. J. Biol. Chem. 2003, 278, 25825–25831. [Google Scholar] [CrossRef] [PubMed]
- Esteves, T.C.; Echtay, K.S.; Jonassen, T.; Clarke, C.F.; Brand, M.D. Ubiquinone is not required for proton conductance by uncoupling protein 1 in yeast mitochondria. Biochem. J. 2004, 379, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Sluse, F.E.; Jarmuszkiewicz, W.; Navet, R.; Douette, P.; Mathy, G.; Sluse-Goffart, C.M. Mitochondrial UCPs: New insights into regulation and impact. Biochim. Biophys. Acta 2006, 1757, 480–485. [Google Scholar] [CrossRef] [PubMed]
- Quinzii, C.M.; Lopez, L.C.; Von-Moltke, J.; Naini, A.; Krishna, S.; Schuelke, M.; Salviati, L.; Navas, P.; DiMauro, S.; Hirano, M. Respiratory chain dysfunction and oxidative stress correlate with severity of primary CoQ10 deficiency. FASEB J. 2008, 22, 1874–1885. [Google Scholar] [CrossRef] [PubMed]
- Quinzii, C.M.; Lopez, L.C.; Gilkerson, R.W.; Dorado, B.; Coku, J.; Naini, A.B.; Lagier-Tourenne, C.; Schuelke, M.; Salviati, L.; Carrozzo, R.; et al. Reactive oxygen species, oxidative stress, and cell death correlate with level of CoQ10 deficiency. FASEB J. 2010, 24, 3733–3743. [Google Scholar] [CrossRef] [PubMed]
- Quinzii, C.M.; Garone, C.; Emmanuele, V.; Tadesse, S.; Krishna, S.; Dorado, B.; Hirano, M. Tissue-specific oxidative stress and loss of mitochondria in CoQ-deficient Pdss2 mutant mice. FASEB J. 2013, 27, 612–621. [Google Scholar] [CrossRef] [PubMed]
- Ogasahara, S.; Engel, A.G.; Frens, D.; Mack, D. Muscle coenzyme Q deficiency in familial mitochondrial encephalomyopathy. Proc. Natl. Acad. Sci. USA 1989, 86, 2379–2382. [Google Scholar] [CrossRef] [PubMed]
- Mollet, J.; Giurgea, I.; Schlemmer, D.; Dallner, G.; Chrétien, D.; Delahodde, A.; Bacq, D.; De Lonlay, P.; Munnich, A.; Rötig, A. Prenyldiphosphate synthase, subunit 1 (PDSS1) and OH-benzoate polyprenyltransferase (COQ2) mutations in ubiquinone deficiency and oxidative phosphorylation disorders. J. Clin. Investig. 2007, 117, 765–772. [Google Scholar] [CrossRef]
- Sadowski, C.E.; Lovric, S.; Ashraf, S.; Pabst, W.L.; Gee, H.Y.; Kohl, S.; Engelmann, S.; Vega-Warner, V.; Fang, H.; Halbritter, J.; et al. A single-gene cause in 29.5% of cases of steroid-resistant nephrotic syndrome. J. Am. Soc. Nephrol. 2015, 26, 1279–1289. [Google Scholar]
- López, L.C.; Schuelke, M.; Quinzii, C.M.; Kanki, T.; Rodenburg, R.J.T.; Naini, A.; DiMauro, S.; Hirano, M. Leigh Syndrome with Nephropathy and CoQ10 Deficiency Due to decaprenyl diphosphate synthase subunit 2 (PDSS2) Mutations. Am. J. Hum. Genet. 2006, 79, 1125–1129. [Google Scholar] [CrossRef] [PubMed]
- Quinzii, C.; Naini, A.; Salviati, L.; Trevisson, E.; Navas, P.; Dimauro, S.; Hirano, M. A mutation in para-hydroxybenzoate-polyprenyl transferase (COQ2) causes primary coenzyme Q10 deficiency. Am. J. Hum. Genet. 2006, 78, 345–349. [Google Scholar] [CrossRef] [PubMed]
- Heeringa, S.F.; Chernin, G.; Chaki, M.; Zhou, W.; Sloan, A.J.; Ji, Z.; Xie, L.X.; Salviati, L.; Hurd, T.W.; Vega-Warner, V.; et al. COQ6 mutations in human patients produce nephrotic syndrome with sensorineural deafness. J. Clin. Investig. 2011, 121, 2013–2024. [Google Scholar] [CrossRef] [PubMed]
- Sondheimer, N.; Hewson, S.; Cameron, J.M.; Somers, G.R.; Broadbent, J.D.; Ziosi, M.; Quinzii, C.M.; Naini, A.B. Novel recessive mutations in COQ4 cause severe infantile cardiomyopathy and encephalopathy associated with CoQ10 deficiency. Mol. Genet. Metab. Rep. 2017, 12, 23–27. [Google Scholar] [CrossRef] [PubMed]
- Chung, W.K.; Martin, K.; Jalas, C.; Braddock, S.R.; Juusola, J.; Monaghan, K.G.; Warner, B.; Franks, S.; Yudkoff, M.; Lulis, L.; et al. Mutations in COQ4, an essential component of coenzyme Q biosynthesis, cause lethal neonatal mitochondrial encephalomyopathy. J. Med. Genet. 2015, 52, 627–635. [Google Scholar] [CrossRef] [PubMed]
- Brea-Calvo, G.; Haack, T.B.; Karall, D.; Ohtake, A.; Invernizzi, F.; Carrozzo, R.; Kremer, L.; Dusi, S.; Fauth, C.; Scholl-Burgi, S.; et al. COQ4 mutations cause a broad spectrum of mitochondrial disorders associated with CoQ10 deficiency. Am. J. Hum. Genet. 2015, 96, 309–317. [Google Scholar] [CrossRef]
- Malicdan, M.C.V.; Vilboux, T.; Ben-Zeev, B.; Guo, J.; Eliyahu, A.; Pode-Shakked, B.; Dori, A.; Kakani, S.; Chandrasekharappa, S.C.; Ferreira, C.R.; et al. A novel inborn error of the coenzyme Q10 biosynthesis pathway: Cerebellar ataxia and static encephalomyopathy due to COQ5 C-methyltransferase deficiency. Hum. Mutat. 2018, 39, 69–79. [Google Scholar] [CrossRef]
- Ashraf, S.; Gee, H.Y.; Woerner, S.; Xie, L.X.; Vega-Warner, V.; Lovric, S.; Fang, H.; Song, X.; Cattran, D.C.; Avila-Casado, C.; et al. ADCK4 mutations promote steroid-resistant nephrotic syndrome through CoQ10 biosynthesis disruption. J. Clin. Investig. 2013, 123, 5179–5189. [Google Scholar] [CrossRef]
- Duncan, A.J.; Bitner-Glindzicz, M.; Meunier, B.; Costello, H.; Hargreaves, I.P.; López, L.C.; Hirano, M.; Quinzii, C.M.; Sadowski, M.I.; Hardy, J.; et al. A Nonsense Mutation in COQ9 Causes Autosomal-Recessive Neonatal-Onset Primary Coenzyme Q10 Deficiency: A Potentially Treatable Form of Mitochondrial Disease. Am. J. Hum. Genet. 2009, 84, 558–566. [Google Scholar] [CrossRef]
- Alcázar-Fabra, M.; Trevisson, E.; Brea-Calvo, G. Clinical syndromes associated with Coenzyme Q10 deficiency. Essays Biochem. 2018, 62, 377–398. [Google Scholar] [CrossRef]
- Padilla, S.; González-Mariscal, I.; Martín-Montalvo, A.; Gonzalez-Mariscal, I.; García-Testón, E.; Martin-Montalvo, A.; Pomares-Viciana, T.; Vazquez-Fonseca, L.; Gandolfo-Domínguez, P.; Santos-Ocaña, C.; et al. Regulation of coenzyme Q biosynthesis in yeast: A new complex in the block. IUBMB Life 2014, 66, 63–70. [Google Scholar]
- Yubero, D.; Montero, R.; Martin, M.A.; Montoya, J.; Ribes, A.; Grazina, M.; Trevisson, E.; Rodriguez-Aguilera, J.C.; Hargreaves, I.P.; Salviati, L.; et al. Secondary coenzyme Q10 deficiencies in oxidative phosphorylation (OXPHOS) and non-OXPHOS disorders. Mitochondrion 2016, 30, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Montero, R.; Grazina, M.; López-Gallardo, E.; Montoya, J.; Briones, P.; Navarro-Sastre, A.; Land, J.M.; Hargreaves, I.P.; Artuch, R.; O’Callaghan, M.D.M.; et al. Coenzyme Q10 deficiency in mitochondrial DNA depletion syndromes. Mitochondrion 2013, 13, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Quinzii, C.M.; Kattah, A.G.; Naini, A.; Akman, H.O.; Mootha, V.K.; DiMauro, S.; Hirano, M. Coenzyme Q deficiency and cerebellar ataxia associated with an aprataxin mutation. Neurology 2005, 64, 539–541. [Google Scholar] [CrossRef] [PubMed]
- Gempel, K.; Topaloglu, H.; Talim, B.; Schneiderat, P.; Schoser, B.G.H.; Hans, V.H.; Pálmafy, B.; Kale, G.; Tokatli, A.; Quinzii, C.; et al. The myopathic form of coenzyme Q10 deficiency is caused by mutations in the electron-transferring-flavoprotein dehydrogenase (ETFDH) gene. Brain 2007, 130, 2037–2044. [Google Scholar] [CrossRef] [PubMed]
- Aeby, A.; Sznajer, Y.; Cave, H.; Rebuffat, E.; Van Coster, R.; Rigal, O.; Van Bogaert, P.; Coster, R.; Bogaert, P. Cardiofaciocutaneous (CFC) syndrome associated with muscular coenzyme Q10 deficiency. J. Inherit. Metab. Dis. 2007, 30, 827. [Google Scholar] [CrossRef] [PubMed]
- Balreira, A.; Boczonadi, V.; Barca, E.; Pyle, A.; Bánsági, B.; Appleton, M.; Graham, C.; Hargreaves, I.P.; Rasic, V.M.; Lochmüller, H.; et al. ANO10 mutations cause ataxia and coenzyme Q10 deficiency. J. Neurol. 2014, 261, 2192–2198. [Google Scholar] [CrossRef] [PubMed]
- Miranda, S.; Foncea, R.; Guerrero, J.; Leighton, F. Oxidative Stress and Upregulation of Mitochondrial Biogenesis Genes in Mitochondrial DNA-Depleted HeLa Cells. Biochem. Biophys. Res. Commun. 1999, 258, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Desbats, M.A.; Lunardi, G.; Doimo, M.; Trevisson, E.; Salviati, L. Genetic bases and clinical manifestations of coenzyme Q10 (CoQ10) deficiency. J. Inherit. Metab. Dis. 2015, 38, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Rahman, S.; Clarke, C.F.; Hirano, M. 176th ENMC International Workshop: Diagnosis and treatment of coenzyme Q10 deficiency. Neuromuscul. Disord. 2012, 22, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Marcoff, L.; Thompson, P.D. The role of coenzyme Q10 in statin-associated myopathy: A systematic review. J. Am. Coll. Cardiol. 2007, 49, 2231–2237. [Google Scholar] [CrossRef] [PubMed]
- Kühl, I.; Miranda, M.; Atanassov, I.; Kuznetsova, I.; Hinze, Y.; Mourier, A.; Filipovska, A.; Larsson, N.-G. Transcriptomic and proteomic landscape of mitochondrial dysfunction reveals secondary coenzyme Q deficiency in mammals. eLife 2017, 6, 6. [Google Scholar] [CrossRef] [PubMed]
- Fazakerley, D.J.; Chaudhuri, R.; Yang, P.; Maghzal, G.J.; Thomas, K.C.; Krycer, J.R.; Humphrey, S.J.; Parker, B.L.; Fisher-Wellman, K.H.; Meoli, C.C.; et al. Mitochondrial CoQ deficiency is a common driver of mitochondrial oxidants and insulin resistance. eLife 2018, 7, 7. [Google Scholar] [CrossRef] [PubMed]
- Harman, D. Aging: A Theory Based on Free Radical and Radiation Chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef] [PubMed]
- Harman, D. The Biologic Clock: The Mitochondria? J. Am. Geriatr. Soc. 1972, 20, 145–147. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.; Finkel, T. Mitohormesis. Cell Metab. 2014, 19, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.; Boutis, P.; Hekimi, S. Mutations in the Clk-1 Gene of Caenorhabditis Elegans Affect Developmental and Behavioral Timing. Genetics 1995, 139, 1247–1259. [Google Scholar] [PubMed]
- Cristina, D.; Cary, M.; Lunceford, A.; Clarke, C.; Kenyon, C. A Regulated Response to Impaired Respiration Slows Behavioral Rates and Increases Lifespan in Caenorhabditis elegans. PLoS Genet. 2009, 5, e1000450. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.-Y.; Gangoiti, J.A.; Sedensky, M.M.; Morgan, P.G. The effect of different ubiquinones on lifespan in Caenorhabditis elegans. Mech. Ageing Dev. 2009, 130, 370–376. [Google Scholar] [CrossRef]
- Jonassen, T.; Larsen, P.L.; Clarke, C.F. A dietary source of coenzyme Q is essential for growth of long-lived Caenorhabditis elegans clk-1 mutants. Proc. Natl. Acad. Sci. USA 2001, 98, 421–426. [Google Scholar] [CrossRef]
- Lakowski, B.; Hekimi, S. Determination of Life-Span in Caenorhabditis elegans by Four Clock Genes. Science 1996, 272, 1010–1013. [Google Scholar] [CrossRef]
- Barnes, T.M.; Lakowski, B.; Ewbank, J.J.; Lussier, M.; Bussey, H.; Hekimi, S. Structural and Functional Conservation of the Caenorhabditis elegans Timing Gene clk-1. Science 1997, 275, 980–983. [Google Scholar]
- Shibata, Y.; Branicky, R.; Landaverde, I.O.; Hekimi, S. Redox Regulation of Germline and Vulval Development in Caenorhabditis elegans. Science 2003, 302, 1779–1782. [Google Scholar] [CrossRef] [PubMed]
- Braeckman, B.P.; Houthoofd, K.; De Vreese, A.; Vanfleteren, J.R. Apparent uncoupling of energy production and consumption in long-lived Clk mutants of Caenorhabditis elegans. Curr. Biol. 1999, 9, 493–497. [Google Scholar] [CrossRef]
- Braeckman, B.P.; Houthoofd, K.; Brys, K.; Lenaerts, I.; De Vreese, A.; Van Eygen, S.; Raes, H.; Vanfleteren, J.R. No reduction of energy metabolism in Clk mutants. Mech. Ageing Dev. 2002, 123, 1447–1456. [Google Scholar] [CrossRef]
- Van Voorhies, W.A. The influence of metabolic rate on longevity in the nematode Caenorhabditis elegans. Aging Cell 2002, 1, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Braeckman, B.P.; Houthoofd, K.; Vanfleteren, J.R. Assessing metabolic activity in aging Caenorhabditis elegans: Concepts and controversies. Aging Cell 2002, 1, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Larsen, P.L. Extension of Life-Span in Caenorhabditis elegans by a Diet Lacking Coenzyme Q. Science 2002, 295, 120–123. [Google Scholar] [CrossRef] [PubMed]
- Asencio, C.; Rodríguez-Aguilera, J.C.; Ruiz-Ferrer, M.; Vela, J.; Navas, P. Silencing of ubiquinone biosynthesis genes extends life span in Caenorhabditis elegans. FASEB J. 2003, 17, 1135–1137. [Google Scholar] [CrossRef] [PubMed]
- Kayser, E.B.; Sedensky, M.M.; Morgan, P.G.; Hoppel, C.L. Mitochondrial oxidative phosphorylation is defective in the long-lived mutant clk-1. J. Biol. Chem. 2004, 279, 54479–54486. [Google Scholar] [CrossRef] [PubMed]
- Arroyo, A.; Santos-Ocaña, C.; Ruiz-Ferrer, M.; Padilla, S.; Gavilán, A.; Rodríguez-Aguilera, J.C.; Navas, P. Coenzyme Q is irreplaceable by demethoxy-coenzyme Q in plasma membrane ofCaenorhabditis elegans. FEBS Lett. 2006, 580, 1740–1746. [Google Scholar] [CrossRef] [PubMed]
- Gomez, F.; Saiki, R.; Chin, R.; Srinivasan, C.; Clarke, C.F. Restoring de novo Coenzyme Q biosynthesis in Caenorhabditis elegans coq-3 mutants yields profound rescue compared to exogenous Coenzyme Q supplementation. Gene 2012, 506, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Ishii, N.; Senoo-Matsuda, N.; Miyake, K.; Yasuda, K.; Ishii, T.; Hartman, P.S.; Furukawa, S. Coenzyme Q10 can prolong C. elegans lifespan by lowering oxidative stress. Mech. Ageing Dev. 2004, 125, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.; Niklowitz, P.; Menke, T.; Döring, F. Promotion of growth by Coenzyme Q10 is linked to gene expression in C. elegans. Biochem. Biophys. Res. Commun. 2014, 452, 920–927. [Google Scholar] [CrossRef] [PubMed]
- Hihi, A.K.; Gao, Y.; Hekimi, S. Ubiquinone is necessary for Caenorhabditis elegans development at mitochondrial and non-mitochondrial sites. J. Biol. Chem. 2002, 277, 2202–2206. [Google Scholar] [CrossRef] [PubMed]
- Padilla, S.; Jonassen, T.; Jiménez-Hidalgo, M.A.; Fernández-Ayala, D.J.M.; López-Lluch, G.; Marbois, B.; Navas, P.; Clarke, C.F.; Santos-Ocaña, C.; Fernández-Ayala, D.J.M. Demethoxy-Q, An Intermediate of Coenzyme Q Biosynthesis, Fails to Support Respiration in Saccharomyces cerevisiae and Lacks Antioxidant Activity. J. Biol. Chem. 2004, 279, 25995–26004. [Google Scholar] [CrossRef] [PubMed]
- Miyadera, H.; Kano, K.; Miyoshi, H.; Ishii, N.; Hekimi, S.; Kita, K. Quinones in long-lived clk-1 mutants of Caenorhabditis elegans. FEBS Lett. 2002, 512, 33–37. [Google Scholar] [CrossRef]
- Liu, X.; Jiang, N.; Hughes, B.; Bigras, E.; Shoubridge, E.; Hekimi, S. Evolutionary conservation of the clk-1-dependent mechanism of longevity: Loss of mclk1 increases cellular fitness and lifespan in mice. Genome Res. 2005, 19, 2424–2434. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Hidalgo, M.; Luna-Sánchez, M.; Hidalgo-Gutiérrez, A.; Barriocanal-Casado, E.; Mascaraque, C.; Acuña-Castroviejo, D.; Rivera, M.; Escames, G.; López, L.C. Reduction in the levels of CoQ biosynthetic proteins is related to an increase in lifespan without evidence of hepatic mitohormesis. Sci. Rep. 2018, 8, 14013. [Google Scholar] [CrossRef]
- Takahashi, M.; Shimizu, T.; Moriizumi, E.; Shirasawa, T. Clk-1 deficiency induces apoptosis associated with mitochondrial dysfunction in mouse embryos. Mech. Ageing Dev. 2008, 129, 291–298. [Google Scholar] [CrossRef]
- Levavasseur, F.; Miyadera, H.; Sirois, J.; Tremblay, M.L.; Kita, K.; Shoubridge, E.; Hekimi, S. Ubiquinone Is Necessary for Mouse Embryonic Development but Is Not Essential for Mitochondrial Respiration. J. Biol. Chem. 2001, 276, 46160–46164. [Google Scholar] [CrossRef]
- Holzenberger, M.; Dupont, J.; Ducos, B.; Leneuve, P.; Geloen, A.; Even, P.C.; Cervera, P.; Le Bouc, Y. IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature 2003, 421, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Baba, T.; Shimizu, T.; Suzuki, Y.-I.; Ogawara, M.; Isono, K.-I.; Koseki, H.; Kurosawa, H.; Shirasawa, T. Estrogen, Insulin, and Dietary Signals Cooperatively Regulate Longevity Signals to Enhance Resistance to Oxidative Stress in Mice. J. Biol. Chem. 2005, 280, 16417–16426. [Google Scholar] [CrossRef] [PubMed]
- Napoli, C.; Martin-Padura, I.; De Nigris, F.; Giorgio, M.; Mansueto, G.; Somma, P.; Condorelli, M.; Sica, G.; De Rosa, G.; Pelicci, P. Deletion of the p66Shc longevity gene reduces systemic and tissue oxidative stress, vascular cell apoptosis, and early atherogenesis in mice fed a high-fat diet. Proc. Natl. Acad. Sci. USA 2003, 100, 2112–2116. [Google Scholar] [CrossRef] [PubMed]
- Hauck, S.J.; Aaron, J.M.; Wright, C.; Kopchick, J.J.; Bartke, A. Antioxidant Enzymes, Free-Radical Damage, and Response to Paraquat in Liver and Kidney of Long-Living Growth Hormone Receptor/Binding Protein Gene-Disrupted Mice. Horm. Metab. Res. 2002, 34, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Lapointe, J.; Hekimi, S. Early mitochondrial dysfunction in long-lived Mclk1+/- mice. J. Biol. Chem. 2008, 283, 26217–26227. [Google Scholar] [CrossRef] [PubMed]
- Schulz, T.J.; Zarse, K.; Voigt, A.; Urban, N.; Birringer, M.; Ristow, M. Glucose Restriction Extends Caenorhabditis elegans Life Span by Inducing Mitochondrial Respiration and Increasing Oxidative Stress. Cell Metab. 2007, 6, 280–293. [Google Scholar] [CrossRef] [PubMed]
- Ballard, J.W.O.; Katewa, S.D.; Melvin, R.G.; Chan, G.; Ballard, J.W.O. Comparative Analysis of Mitochondrial Genotype and Aging. Ann. N. Y. Acad. Sci. 2007, 1114, 93–106. [Google Scholar] [CrossRef]
- Brys, K.; Vanfleteren, J.R.; Braeckman, B.P. Testing the rate-of-living/oxidative damage theory of aging in the nematode model Caenorhabditis elegans. Exp. Gerontol. 2007, 42, 845–851. [Google Scholar] [CrossRef]
- Miles, M.V. The uptake and distribution of coenzyme Q(10). Mitochondrion 2007, 7, S72–S77. [Google Scholar] [CrossRef]
- Beyer, R.E.; Burnett, B.-A.; Cartwright, K.J.; Edington, D.W.; Falzon, M.J.; Kreitman, K.R.; Kuhn, T.W.; Ramp, B.J.; Rhee, S.Y.S.; Rosenwasser, M.J.; et al. Tissue coenzyme Q (ubiquinone) and protein concentrations over the life span of the laboratory rat. Mech. Ageing Dev. 1985, 32, 267–281. [Google Scholar] [CrossRef]
- Kalén, A.; Appelkvist, E.-L.; Dallner, G. Age-related changes in the lipid compositions of rat and human tissues. Lipids 1989, 24, 579–584. [Google Scholar] [CrossRef] [PubMed]
- Battino, M.; Gorini, A.; Villa, R.; Genova, M.; Bovina, C.; Sassi, S.; Littarru, G.; Lenaz, G. Coenzyme Q content in synaptic and non-synaptic mitochondria from different brain regions in the ageing rat. Mech. Ageing Dev. 1995, 78, 173–187. [Google Scholar] [CrossRef]
- Åberg, F.; Zhang, Y.; Teclebrhan, H.; Appelkvist, E.-L.; Dallner, G. Increases in tissue levels of ubiquinone in association with peroxisome proliferation. Chem. Interact. 1996, 99, 205–218. [Google Scholar] [CrossRef]
- Lass, A.; Forster, M.J.; Sohal, R.S. Effects of coenzyme Q10 and α-tocopherol administration on their tissue levels in the mouse: Elevation of mitochondrial α-tocopherol by coenzyme Q10. Free Radic. Biol. Med. 1999, 26, 1375–1382. [Google Scholar] [CrossRef]
- Lass, A.; Sohal, R.S. Effect of coenzyme Q(10) and alpha-tocopherol content of mitochondria on the production of superoxide anion radicals. FASEB J. 2000, 14, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Kwong, L.K.; Kamzalov, S.; Rebrin, I.; Bayne, A.-C.V.; Jana, C.K.; Morris, P.; Forster, M.J.; Sohal, R.S. Effects of coenzyme Q10 administration on its tissue concentrations, mitochondrial oxidant generation, and oxidative stress in the rat. Free Radic. Biol. Med. 2002, 33, 627–638. [Google Scholar] [CrossRef]
- Sohal, R.S.; Kamzalov, S.; Sumien, N.; Ferguson, M.; Rebrin, I.; Heinrich, K.R.; Forster, M.J. Effect of coenzyme Q10 intake on endogenous coenzyme Q content, mitochondrial electron transport chain, antioxidative defenses, and life span of mice. Free Radic. Biol. Med. 2006, 40, 480–487. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.-Y.; Yum, K.-S. Effect of Coenzyme Q10 on Insulin Resistance in Korean Patients with Prediabetes: A Pilot Single-Center, Randomized, Double-Blind, Placebo-Controlled Study. BioMed Res. Int. 2018, 2018, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Dahri, M.; Tarighat-Esfanjani, A.; Asghari-Jafarabadi, M.; Hashemilar, M. Oral coenzyme Q10 supplementation in patients with migraine: Effects on clinical features and inflammatory markers. Nutr. Neurosci. 2019, 22, 607–615. [Google Scholar] [CrossRef] [PubMed]
- McGarry, A.; McDermott, M.; Kieburtz, K.; de Blieck, E.A.; Beal, F.; Marder, K.; Ross, C.; Shoulson, I.; Gilbert, P.; Mallonee, W.M.; et al. A randomized, double-blind, placebo-controlled trial of coenzyme Q10 in Huntington disease. Neurology 2017, 88, 152–159. [Google Scholar] [CrossRef]
- Varela-López, A.; Giampieri, F.; Battino, M.; Quiles, J.L. Coenzyme Q and Its Role in the Dietary Therapy against Aging. Molecules 2016, 21, 373. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, J.; Chen, X.-Q.; Chen, C.-Y.O. Ubiquinol is superior to ubiquinone to enhance Coenzyme Q10 status in older men. Food Funct. 2018, 9, 5653–5659. [Google Scholar] [CrossRef] [PubMed]
- Hidaka, T.; Fujii, K.; Funahashi, I.; Fukutomi, N.; Hosoe, K. Safety assessment of coenzyme Q10 (CoQ10). BioFactors 2008, 32, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Hathcock, J.N.; Shao, A. Risk assessment for coenzyme Q10 (Ubiquinone). Regul. Toxicol. Pharmacol. 2006, 45, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Shetty, R.A.; Forster, M.J.; Sumien, N. Coenzyme Q10 supplementation reverses age-related impairments in spatial learning and lowers protein oxidation. AGE 2013, 35, 1821–1834. [Google Scholar] [CrossRef] [PubMed]
- Rivara, M.B.; Yeung, C.K.; Robinson-Cohen, C.; Phillips, B.R.; Ruzinski, J.; Rock, D.; Linke, L.; Shen, D.D.; Ikizler, T.A.; Himmelfarb, J. Effect of Coenzyme Q10 on Biomarkers of Oxidative Stress and Cardiac Function in Hemodialysis Patients: The CoQ10 Biomarker Trial. Am. J. Kidney Dis. 2017, 69, 389–399. [Google Scholar] [CrossRef] [PubMed]
- García-Corzo, L.; Luna-Sánchez, M.; Doerrier, C.; Ortiz, F.; Escames, G.; Acuña-Castroviejo, D.; López, L.C. Ubiquinol-10 ameliorates mitochondrial encephalopathy associated with CoQ deficiency. Biochim. Biophys. Acta 2014, 1842, 893–901. [Google Scholar] [CrossRef] [PubMed]
- Asencio, C.; Navas, P.; Cabello, J.; Schnabel, R.; Cypser, J.R.; Johnson, T.E.; Rodríguez-Aguilera, J.C. Coenzyme Q supports distinct developmental processes in Caenorhabditis elegans. Mech. Ageing Dev. 2009, 130, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-K.; Pugh, T.D.; Klopp, R.G.; Edwards, J.; Allison, D.B.; Weindruch, R.; Prolla, A.T. The impact of α-lipoic acid, coenzyme Q10 and caloric restriction on life span and gene expression patterns in mice. Free Radic. Biol. Med. 2004, 36, 1043–1057. [Google Scholar] [CrossRef] [PubMed]
- Lönnrot, K.; Alho, H.; Holm, P.; Lagerstedt, A.; Huhtala, H. The effects of lifelong ubiquinone Q10 supplementation on the Q9 and Q10 tissue concentrations and life span of male rats and mice. IUBMB Life 1998, 44, 727–737. [Google Scholar] [CrossRef]
- Ramirez-Tortosa, C.L.; Varela-López, A.; Navarro-Hortal, M.D.; Ramos-Pleguezuelos, F.M.; Márquez-Lobo, B.; Ramirez-Tortosa, M.C.; Ochoa, J.J.; Battino, M.; Quiles, J.L. Longevity and cause of death in male Wistar rats fed lifelong diets based on virgin olive oil, sunflower oil or fish oil. J. Gerontol. Ser. A 2019. [Google Scholar] [CrossRef] [PubMed]
- Reeves, P.G. Components of the AIN-93 Diets as Improvements in the AIN-76A Diet. J. Nutr. 1997, 127, 838S–841S. [Google Scholar] [CrossRef] [PubMed]
- Varela-Lopez, A.; Bullon, P.; Battino, M.; Ramirez-Tortosa, M.; Ochoa, J.J.; Cordero, M.D.; Ramirez-Tortosa, C.L.; Rubini, C.; Zizzi, A.; Quiles, J.L. Coenzyme Q Protects Against Age-Related Alveolar Bone Loss Associated to n-6 Polyunsaturated Fatty Acid Rich-Diets by Modulating Mitochondrial Mechanisms. J. Gerontol. Ser. A Biomed. Sci. Med. Sci. 2015, 71, 593–600. [Google Scholar] [CrossRef] [PubMed]
- Varela-Lopez, A.; Ochoa, J.J.; Llamas-Elvira, J.M.; Lopez-Frias, M.; Planells, E.; Speranza, L.; Battino, M.; Quiles, J.L. Loss of Bone Mineral Density Associated with Age in Male Rats Fed on Sunflower Oil Is Avoided by Virgin Olive Oil Intake or Coenzyme Q Supplementation. Int. J. Mol. Sci. 2017, 18, 1397. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Alonso, A.; Ramirez-Tortosa, C.L.; Varela-López, A.; Roche, E.; Arribas, M.I.; Ramirez-Tortosa, M.C.; Giampieri, F.; Ochoa, J.J.; Quiles, J.L. Sunflower Oil but Not Fish Oil Resembles Positive Effects of Virgin Olive Oil on Aged Pancreas after Life-Long Coenzyme Q Addition. Int. J. Mol. Sci. 2015, 16, 23425–23445. [Google Scholar] [CrossRef] [PubMed]
- Huertas, J.R.; Ibáñez, S.; López-Frias, M.; Ochoa, J.J.; Quiles, J.L.; Castelli, G.P.; Mataix, J.; Lenaz, G.; Huertas, J.F.R.; Martinez-Velasco, E.; et al. Virgin olive oil and coenzyme Q10protect heart mitochondria from peroxidative damage during aging. BioFactors 1999, 9, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Ochoa, J.J.; Pamplona, R.; Ramirez-Tortosa, M.C.; Granados-Principal, S.; Perez-Lopez, P.; Naudí, A.; Portero-Otin, M.; López-Frías, M.; Battino, M.; Quiles, J.L. Age-related changes in brain mitochondrial DNA deletion and oxidative stress are differentially modulated by dietary fat type and coenzyme Q10. Free Radic. Biol. Med. 2011, 50, 1053–1064. [Google Scholar] [CrossRef] [PubMed]
- Ochoa, J.J.; Quiles, J.L.; López-Frías, M.; Huertas, J.R.; Mataix, J.; Huertas, J.F.R. Effect of Lifelong Coenzyme Q10 Supplementation on Age-Related Oxidative Stress and Mitochondrial Function in Liver and Skeletal Muscle of Rats Fed on a Polyunsaturated Fatty Acid (PUFA)-Rich Diet. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2007, 62, 1211–1218. [Google Scholar] [CrossRef] [PubMed]
- Quiles, J.L.; Ochoa, J.J.; Battino, M.; Nepomuceno, E.A.; Frías, M.L.; Huertas, J.R.; Mataix, J.; Gutierrez-Rios, P.; Huertas, J.F.R.; Gutierrez-Rios, P. Life-long supplementation with a low dosage of coenzyme Q10 in the rat: Effects on antioxidant status and DNA damage. BioFactors 2005, 25, 73–86. [Google Scholar] [CrossRef]
- Quiles, J.L.; Ochoa, J.J.; Huertas, J.R.; Mataix, J.; Huertas, J.F.R. Coenzyme Q supplementation protects from age-related DNA double-strand breaks and increases lifespan in rats fed on a PUFA-rich diet. Exp. Gerontol. 2004, 39, 189–194. [Google Scholar] [CrossRef]
- Quiles, J.L.; Pamplona, R.; Ramirez-Tortosa, M.C.; Naudí, A.; Portero-Otin, M.; Araujo-Nepomuceno, E.; López-Frías, M.; Battino, M.; Ochoa, J.J. Coenzyme Q addition to an n-6 PUFA-rich diet resembles benefits on age-related mitochondrial DNA deletion and oxidative stress of a MUFA-rich diet in rat heart. Mech. Ageing Dev. 2010, 131, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Varela-Lopez, A.; Ochoa, J.J.; Llamas-Elvira, J.M.; Lopez-Frias, M.; Planells, E.; Ramirez-Tortosa, M.; Ramirez-Tortosa, C.L.; Giampieri, F.; Battino, M.; Quiles, J.L. Age-Related Loss in Bone Mineral Density of Rats Fed Lifelong on a Fish Oil-Based Diet Is Avoided by Coenzyme Q10 Addition. Nutrients 2017, 9, 176. [Google Scholar] [CrossRef] [PubMed]
- Bello, R.I.; Gómez-Díaz, C.; Burón, M.I.; Alcain, F.J.; Navas, P.; Villalba, J.M. Enhanced anti-oxidant protection of liver membranes in long-lived rats fed on a coenzyme Q10-supplemented diet. Exp. Gerontol. 2005, 40, 694–706. [Google Scholar] [CrossRef] [PubMed]
- Bello, R.I.; Herman, M.D.; Navas, P.; Villalba, J.M.; Gómez-Díaz, C.; Burón, M.I.; Alcain, F.J.; González-Ojeda, R.; González-Reyes, J.A. Effect of dietary coenzyme Q and fatty acids on the antioxidant status of rat tissues. Protoplasma 2003, 221, 11–17. [Google Scholar]
- Dash, S.; Xiao, C.; Morgantini, C.; Lewis, G.F. New Insights into the Regulation of Chylomicron Production. Annu. Rev. Nutr. 2015, 35, 265–294. [Google Scholar] [CrossRef] [PubMed]
- Birru, W.A.; Warren, D.B.; Williams, H.D.; Benameur, H.; Porter, C.J.H.; Chalmers, D.K.; Ibrahim, A.; Pouton, C.W. Digestion of Phospholipids after Secretion of Bile into the Duodenum Changes the Phase Behavior of Bile Components. Mol. Pharm. 2014, 11, 2825–2834. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Ogawara, M.; Shimizu, T.; Shirasawa, T. Restoration of the behavioral rates and lifespan in clk-1 mutant nematodes in response to exogenous coenzyme Q10. Exp. Gerontol. 2012, 47, 276–279. [Google Scholar] [CrossRef] [PubMed]
- Gavilán, A.; Asencio, C.; Cabello, J.; Rodríguez-Aguilera, J.C.; Schnabel, R.; Navas, P.C. elegans knockouts in ubiquinone biosynthesis genes result in different phenotypes during larval development. BioFactors 2005, 25, 21–29. [Google Scholar] [CrossRef]
- Moradi, M.; Haghighatdoost, F.; Feizi, A.; Larijani, B.; Azadbakht, L. Effect of Coenzyme Q10 Supplementation on Diabetes Biomarkers: A Systematic Review and Meta-analysis of Randomized Controlled Clinical Trials. Arch. Iran. Med. 2016, 19, 588–596. [Google Scholar] [PubMed]
- Raygan, F.; Rezavandi, Z.; Dadkhah Tehrani, S.; Farrokhian, A.; Asemi, Z. The effects of coenzyme Q10 administration on glucose homeostasis parameters, lipid profiles, biomarkers of inflammation and oxidative stress in patients with metabolic syndrome. Eur. J. Nutr. 2016, 55, 2357–2364. [Google Scholar] [CrossRef]
- Suksomboon, N.; Poolsup, N.; Juanak, N. Effects of coenzyme Q10supplementation on metabolic profile in diabetes: A systematic review and meta-analysis. J. Clin. Pharm. Ther. 2015, 40, 413–418. [Google Scholar] [CrossRef]
- Xu, Z.; Huo, J.; Ding, X.; Yang, M.; Li, L.; Dai, J.; Hosoe, K.; Kubo, H.; Mori, M.; Higuchi, K.; et al. Coenzyme Q10 Improves Lipid Metabolism and Ameliorates Obesity by Regulating CaMKII-Mediated PDE4 Inhibition. Sci. Rep. 2017, 7, 8253. [Google Scholar] [CrossRef]
- Witting, P.K.; Pettersson, K.; Letters, J.; Stocker, R. Anti-atherogenic effect of coenzyme Q10 in apolipoprotein E gene knockout mice. Free Radic. Biol. Med. 2000, 29, 295–305. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, H.; Hao, Y.; Xu, L.; Zhang, T.; Liu, Y.; Guo, L.; Zhu, L.; Pei, Z. Coenzyme Q10 protects against hyperlipidemia-induced cardiac damage in apolipoprotein E-deficient mice. Lipids Heal. Dis. 2018, 17, 279. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Moreno, J.; Quintana-Navarro, G.M.; Delgado-Lista, J.; Garcia-Rios, A.; Alcala-Diaz, J.F.; Gomez-Delgado, F.; Camargo, A.; Perez-Martinez, P.; Tinahones, F.J.; Striker, G.E.; et al. Mediterranean Diet Supplemented With Coenzyme Q10 Modulates the Postprandial Metabolism of Advanced Glycation End Products in Elderly Men and Women. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2018, 73, 340–346. [Google Scholar]
- Mirhashemi, S.M.; Najafi, V.; Raygan, F.; Asemi, Z. The effects of coenzyme Q10 supplementation on cardiometabolic markers in overweight type 2 diabetic patients with stable myocardial infarction: A randomized, double-blind, placebo-controlled trial. ARYA Atheroscler 2016, 12, 158–165. [Google Scholar] [PubMed]
- Ayaz, M.; Tuncer, S.; Okudan, N.; Gokbel, H. Coenzyme Q(10) and alpha-lipoic acid supplementation in diabetic rats: Conduction velocity distributions. Methods Find. Exp. Clin. Pharmacol. 2008, 30, 367. [Google Scholar] [CrossRef] [PubMed]
- Monsef, A.; Shahidi, S.; Komaki, A. Influence of Chronic Coenzyme Q10 Supplementation on Cognitive Function, Learning, and Memory in Healthy and Diabetic Middle-Aged Rats. Neuropsychobiology 2019, 77, 92–100. [Google Scholar] [CrossRef]
- Schmelzer, C.; Kubo, H.; Mori, M.; Sawashita, J.; Kitano, M.; Hosoe, K.; Boomgaarden, I.; Doring, F.; Higuchi, K. Supplementation with the reduced form of Coenzyme Q10 decelerates phenotypic characteristics of senescence and induces a peroxisome proliferator-activated receptor-alpha gene expression signature in SAMP1 mice. Mol. Nutr. Food Res. 2010, 54, 805–815. [Google Scholar] [CrossRef]
- Flowers, N.; Hartley, L.; Todkill, D.; Stranges, S.; Rees, K. Co-enzyme Q10 supplementation for the primary prevention of cardiovascular disease. Cochrane Database Syst. Rev. 2014. [Google Scholar] [CrossRef]
- Yubero-Serrano, E.M.; Gonzalez-Guardia, L.; Rangel-Zuñiga, O.; Delgado-Lista, J.; Gutierrez-Mariscal, F.M.; Perez-Martinez, P.; Delgado-Casado, N.; Cruz-Teno, C.; Tinahones, F.J.; Villalba, J.M.; et al. Mediterranean Diet Supplemented With Coenzyme Q10 Modifies the Expression of Proinflammatory and Endoplasmic Reticulum Stress–Related Genes in Elderly Men and Women. J. Gerontol. Ser. A 2012, 67A, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Fotino, A.D.; Thompson-Paul, A.M.; Bazzano, L.A. Effect of coenzyme Q10 supplementation on heart failure: A meta-analysis. Am. J. Clin. Nutr. 2013, 97, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Madmani, M.E.; Yusuf Solaiman, A.; Tamr Agha, K.; Madmani, Y.; Shahrour, Y.; Essali, A.; Kadro, W. Coenzyme Q10 for heart failure. Cochrane Database Syst. Rev. 2014. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, A.; Kawarazaki, H.; Ando, K.; Fujita, M.; Fujita, T.; Homma, Y. Renal preservation effect of ubiquinol, the reduced form of coenzyme Q10. Clin. Exp. Nephrol. 2011, 15, 30–33. [Google Scholar] [CrossRef] [PubMed]
- Bolignano, D.; Cernaro, V.; Gembillo, G.; Baggetta, R.; Buemi, M.; D’Arrigo, G. Antioxidant agents for delaying diabetic kidney disease progression: A systematic review and meta-analysis. PLoS ONE 2017, 12, 0178699. [Google Scholar] [CrossRef] [PubMed]
- žmitek, J.; Šmidovnik, A.; Fir, M.; Prošek, M.; Zmitek, K.; Walczak, J.; Pravst, I. Relative Bioavailability of Two Forms of a Novel Water-Soluble Coenzyme Q10. Ann. Nutr. Metab. 2008, 52, 281–287. [Google Scholar]
- Galasko, D.R.; Peskind, E.; Clark, C.M.; Quinn, J.F.; Ringman, J.M.; Jicha, G.A.; Cotman, C.; Cottrell, B.; Montine, T.J.; Thomas, R.G.; et al. Antioxidants for Alzheimer disease: A randomized clinical trial with cerebrospinal fluid biomarker measures. Arch. Neurol. 2012, 69, 836–841. [Google Scholar] [CrossRef]
- Fan, L.; Feng, Y.; Chen, G.-C.; Qin, L.-Q.; Fu, C.-L.; Chen, L.-H. Effects of coenzyme Q10 supplementation on inflammatory markers: A systematic review and meta-analysis of randomized controlled trials. Pharmacol. Res. 2017, 119, 128–136. [Google Scholar] [CrossRef]
- Mazidi, M.; Kengne, A.P.; Banach, M. Effects of coenzyme Q10 supplementation on plasma C-reactive protein concentrations: A systematic review and meta-analysis of randomized controlled trials. Pharmacol. Res. 2018, 128, 130–136. [Google Scholar] [CrossRef]
- Tian, G.; Sawashita, J.; Kubo, H.; Nishio, S.-Y.; Hashimoto, S.; Suzuki, N.; Yoshimura, H.; Tsuruoka, M.; Wang, Y.; Liu, Y.; et al. Ubiquinol-10 Supplementation Activates Mitochondria Functions to Decelerate Senescence in Senescence-Accelerated Mice. Antioxid. Redox Signal. 2014, 20, 2606–2620. [Google Scholar] [CrossRef]
- Yan, J.; Fujii, K.; Yao, J.; Kishida, H.; Hosoe, K.; Sawashita, J.; Takeda, T.; Mori, M.; Higuchi, K. Reduced coenzyme Q10 supplementation decelerates senescence in SAMP1 mice. Exp. Gerontol. 2006, 41, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Bullon, P.; Battino, M.; Varela-Lopez, A.; Perez-Lopez, P.; Granados-Principal, S.; Ramirez-Tortosa, M.C.; Ochoa, J.J.; Cordero, M.D.; Gonzalez-Alonso, A.; Ramirez-Tortosa, C.L.; et al. Diets based on virgin olive oil or fish oil but not on sunflower oil prevent age-related alveolar bone resorption by mitochondrial-related mechanisms. PLoS ONE 2013, 8, e74234. [Google Scholar] [CrossRef] [PubMed]
- Ochoa, J.J.; Quiles, J.L.; Huertas, J.R.; Mataix, J.; Huertas, J.F.R. Coenzyme Q10 Protects From Aging-Related Oxidative Stress and Improves Mitochondrial Function in Heart of Rats Fed a Polyunsaturated Fatty Acid (PUFA)-Rich Diet. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2005, 60, 970–975. [Google Scholar] [CrossRef] [PubMed]
- Ochoa, J.J.; Quiles, J.L.; Ibáñez, S.; Martínez, E.; López-Frías, M.; Huertas, J.F.R.; Mataix, J. Aging-Related Oxidative Stress Depends on Dietary Lipid Source in Rat Postmitotic Tissues. J. Bioenerg. Biomembr. 2003, 35, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Quiles, J.L.; Martínez, E.; Ibáñez, S.; Ochoa, J.J.; Martín, Y.; López-Frías, M.; Huertas, J.F.R.; Mataix, J. Ageing-Related Tissue-Specific Alterations in Mitochondrial Composition and Function Are Modulated by Dietary Fat Type in the Rat. J. Bioenerg. Biomembr. 2002, 34, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Orlando, P.; Silvestri, S.; Brugè, F.; Tiano, L.; Klöting, I.; Falcioni, G.; Polidori, C.; Sonia, S. High-fat diet-induced met-hemoglobin formation in rats prone (WOKW) or resistant (DA) to the metabolic syndrome: Effect of CoQ10supplementation. BioFactors 2014, 40, 603–609. [Google Scholar] [CrossRef] [PubMed]
- Sohet, F.M.; Neyrinck, A.M.; Pachikian, B.D.; De Backer, F.C.; Bindels, L.B.; Niklowitz, P.; Menke, T.; Cani, P.D.; Delzenne, N.M. Coenzyme Q10 supplementation lowers hepatic oxidative stress and inflammation associated with diet-induced obesity in mice. Biochem. Pharmacol. 2009, 78, 1391–1400. [Google Scholar] [CrossRef] [PubMed]
- Salehpour, F.; Farajdokht, F.; Mahmoudi, J.; Erfani, M.; Farhoudi, M.; Karimi, P.; Rasta, S.H.; Sadigh-Eteghad, S.; Hamblin, M.R.; Gjedde, A. Photobiomodulation and Coenzyme Q10 Treatments Attenuate Cognitive Impairment Associated With Model of Transient Global Brain Ischemia in Artificially Aged Mice. Front. Cell. Neurosci. 2019, 13, 74. [Google Scholar] [CrossRef] [PubMed]
- Safwat, G.M.; Pisanò, S.; D’Amore, E.; Borioni, G.; Napolitano, M.; Kamal, A.A.; Ballanti, P.; Botham, K.M.; Bravo, E. Induction of non-alcoholic fatty liver disease and insulin resistance by feeding a high-fat diet in rats: Does coenzyme Q monomethyl ether have a modulatory effect? Nutrition 2009, 25, 1157–1168. [Google Scholar] [CrossRef] [PubMed]
- Cano, A.; Ciaffoni, F.; Safwat, G.M.; Aspichueta, P.; Ochoa, B.; Bravo, E.; Botham, K.M.; Olascoaga, B.O. Hepatic VLDL assembly is disturbed in a rat model of nonalcoholic fatty liver disease: Is there a role for dietary coenzyme Q? J. Appl. Physiol. 2009, 107, 707–717. [Google Scholar] [CrossRef] [PubMed]
- Heidari, A.; Hamidi, G.; Soleimani, A.; Aghadavod, E.; Asemi, Z. Effects of Coenzyme Q10 Supplementation on Gene Expressions Related to Insulin, Lipid, and Inflammation Pathways in Patients With Diabetic Nephropathy. Iran. J. Kidney Dis. 2018, 12, 14–21. [Google Scholar] [PubMed]
- Kujjo, L.L.; Acton, B.M.; Perkins, G.A.; Ellisman, M.H.; D’Estaing, S.G.; Casper, R.F.; Jurisicova, A.; Perez, G.I. Ceramide and its transport protein (CERT) contribute to deterioration of mitochondrial structure and function in aging oocytes. Mech. Ageing Dev. 2013, 134, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Ben-Meir, A.; Burstein, E.; Borrego-Alvarez, A.; Chong, J.; Wong, E.; Yavorska, T.; Naranian, T.; Chi, M.; Wang, Y.; Bentov, Y.; et al. Coenzyme Q10 restores oocyte mitochondrial function and fertility during reproductive aging. Aging Cell 2015, 14, 887–895. [Google Scholar] [CrossRef] [PubMed]
- Bentov, Y.; Yavorska, T.; Esfandiari, N.; Jurisicova, A.; Casper, R.F. The contribution of mitochondrial function to reproductive aging. J. Assist. Reprod. Genet. 2011, 28, 773–783. [Google Scholar] [CrossRef] [PubMed]
- Ben-Meir, A.; Kim, K.; McQuaid, R.; Esfandiari, N.; Bentov, Y.; Casper, R.F.; Jurisicova, A. Co-Enzyme Q10 Supplementation Rescues Cumulus Cells Dysfunction in a Maternal Aging Model. Antioxidants 2019, 8, 58. [Google Scholar] [CrossRef] [PubMed]
- Gvozdjakova, A.; Kucharská, J.; Dubravicky, J.; Mojto, V.; Singh, R.B. Coenzyme Q10, α-Tocopherol, and Oxidative Stress Could Be Important Metabolic Biomarkers of Male Infertility. Dis. Markers 2015, 2015, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Salas-Huetos, A.; Rosique-Esteban, N.; Becerra-Tomás, N.; Vizmanos, B.; Bulló, M.; Salas-Salvadó, J. The Effect of Nutrients and Dietary Supplements on Sperm Quality Parameters: A Systematic Review and Meta-Analysis of Randomized Clinical Trials. Adv. Nutr. 2018, 9, 833–848. [Google Scholar] [CrossRef] [PubMed]
- Yang, E.J.; Kim, S.; Kim, J.S.; Choi, I.H. Inflammasome formation and IL-1beta release by human blood monocytes in response to silver nanoparticles. Biomaterials 2012, 33, 6858–6867. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Ohsawa, I.; Shirasawa, T.; Takahashi, M. Early-onset motor impairment and increased accumulation of phosphorylated alpha-synuclein in the motor cortex of normal aging mice are ameliorated by coenzyme Q. Exp. Gerontol. 2016, 81, 65–75. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Model | Strain | Age, Gender, n | CoQ form (Daily Dose/Conc), Treatment Duration | Diet/Food | Effect on Longevity | Ref |
|---|---|---|---|---|---|---|
| Caenorhabditis elegans | N2 Bristol wild-type | Egg, n = 88–105 | Water-soluble CoQ10 1 (dose) | Nematode growth medium (NGM) with E. coli OP50 | No effect | [187] |
| L1, n = 96–98 | CoQ10 (50g/mL) | NGM with E. coli OP50 | Average life span extended by 6% | [188] | ||
| CoQ10 (150g/mL) | NGM with E. coli OP50 | Average life span extended by 18% | [188] | |||
| Clk-1 mutant | Egg, n = 88–105 | Water-soluble CoQ10 1 (dose), 24 h | NGM with E. coli OP50 | No effect | [187] | |
| Eggs, n = 100 | Different engineered Escherichia coli strains producing either CoQ6 to CoQ10 | NGM with different engineered E. coli strains or E. coli OP50 (which produce CoQ8) | Median adult life span increased by 19% but only with CoQ10-producing bacteria | [118] | ||
| Mev-1 (kn1) mutant | L1, n = 96–98 | CoQ10 (50g/mL) | NGM with E. coli OP50 | Average life span increased by 13% | [132] | |
| CoQ10 (150g/mL) | NGM with E. coli OP50 | Average life span increased by 19% | ||||
| Mouse (Mus musculus) | C57BL/6 | 3.5 months old, n = 50 | CoQ10 (93 mg/kg of bw) | ad libitum Purina diet 5001 | No effect | [157] |
| CoQ10 (371 mg/kg of bw) | ad libitum Purina diet 5001 | No effect | [157] | |||
| C57/B17 | 2 m old, male, n = 43 | CoQ10 (10 mg/kg of bw) | normal animal diet | No effect | [170] | |
| C57BL/6 C3H (B6C3F1) | 14 m old, male n = 60 | CoQ10 (100 mg/kg) | AIN93 diet | No effect | [169] | |
| Rat (Rattus norvegicus) | Sprague–Dawley | From pregnancy, male, n = 75 | CoQ10 (10 mg/kg) | normal animal diet | No effect | [170] |
| Wistar | 28 d old (weaning), male, n = 43 | CoQ10 (2.5 mg/kg of bw) | AIN93 diet but with 8% sunflower oil or virgin olive oil as unique dietary fats | Median life span increased by 11.7%. | [180] | |
| 28 d old (weaning), male, n = 22–25 | CoQ10 (2.5 mg/kg of bw) | AIN93 diet with sunflower oil as unique dietary fat (4%) | Increased median life span by 25.5% | [171] | ||
| AIN93 diet with fish oil as unique dietary fat (4%) | No effect | [171] | ||||
| AIN93 diet with virgin olive oil as unique dietary fat (4%) | No effect | [171] |
| Model | Strain, Age, Sex | CoQ form (Dose), Duration | Diet/Food | Tissues, Organs or Systems (Sample Size Per Group) | Consequences in Age-Related Changes | Ref |
|---|---|---|---|---|---|---|
| C. elegans | N2 Bristol wild-type, Egg | Water-soluble CoQ10 1 | NGM containing 200 μg/mL streptomycin with E. coli OP50, 24 h | Nervous system | No effect on pharyngeal contraction and defecation rate. | [187] |
| N2 Bristol wild-type, L1 | CoQ10 (50 or 150 g/mL) | With a lawn of E. coli OP50 | - | Reduced O2− in the presence of succinate, although in a slight manner. | [131] | |
| Clk-1 mutant, Egg | Water-soluble CoQ10 1 (0.1, 1, or 10 μM) | NGM containing 200 μg/mL streptomycin with E. coli OP50, 24 h | Nervous system | Increased pharyngeal pumping rate and defecation rate slowed in a dose-dependent manner, with comparable values found in wild-type strains. | [187] | |
| Different engineered E. coli strains producing CoQ6 to CoQ10 | NGM with different engineered strains or the OP50 strain of E. coli | - (100) | Bacteria containing CoQ6, CoQ7 or CoQ10 decreased complex I-dependent respiration rates compared to those containing CoQ8 or CoQ9. Bacteria containing CoQ7 or CoQ10 decreased complex II-dependent respiration rates compared to those fed on bacteria containing vitamin K12, CoQ6 or CoQ8. | [228] | ||
| Mouse (Mus musculus) | C57BL/6, 3.5 m old, male, | CoQ10 (0.072 or 0.281%, w/w), 16/22 m of age | ad libitum Purina diet 5001 | Liver, heart, skeletal muscle and brain (2–6) | No effect on enzymatic antioxidant defenses (SOD, catalase and GPX activities). No effect on protein oxidative damage (carbonyl content). No effect on mitochondrial Reactive Oxygen Species (ROS) production. No effect on glutathione redox state. No effect on mitochondrial function (mtETC complexes activities). | [157] |
| C57BL/6NCr, 15/6 m old, male, | Water-soluble CoQ10 1 (150 μM) via drinking water | Standard chow diet | Brain (motor cortex) (6–9) | Restored aging-associated decreases in mitochondrial function (OCR). Restored aging-associated motor function, phosphorylated α-synuclein and glutamate transporter 1 levels. | [229] | |
| SAMP1, 4 wk old, male and female | CoQ10 (0.2%, w/w), 10/12/14/16 m | Standard laboratory mouse diet | General (9–11) | No effect on senescence evaluated by the grading system by Hosokawa et al., 1984. | [211] | |
| Urine (9–11) | No effect on oxidative damage (acrolein-lysine adduct and OhdG). | |||||
| Brain (9–11) | No effect on senile amyloid deposition rate. | [211] | ||||
| CoQ10 H2 (0.2%, w/w), 10/12/14/16 m | General (10) | Slowed senescence evaluated by the grading system. | [211] | |||
| Urine (10) | No effect on oxidative damage (acrolein-lysine adduct and OhdG). | [211] | ||||
| Brain (10) | No effect on senile amyloid deposition rate. | [211] | ||||
| SAMP1/Sku Slc, 4 wk old, female | CoQ10 H2 (0.3%, w/w), 2/7/13/ and 19 m of age | CE-2 | General (11–20) | Slowed degree of senescence 2. Slowed the rate of age-related hearing loss. | [210] | |
| Liver (11–20) | Prevented age-related decreases in the expression of sirtuin gene family members and increased intracellular cyclic AMP (cAMP) levels. Maintained mitochondrial biogenesis and oxidative metabolism by maintaining PPARγ coactivator (PGC)-1α activated. Maintained enzymatic antioxidant defenses (SOD and isocitrate dehydrogenase [IDH]2). Increased mitochondrial function (complex I activity). Decreased protein, lipid and DNA oxidative damage (protein carbonyls, MDA and apurinic/apyrimidinic sites). Increased the GSH:GSSG ratio. | [210] | ||||
| Rat (Rattus novergicus) | Sprague–Dawley, 14 m old, male | CoQ10 (0.324%, w/w), 13 wk | NIH-31 diet | Blood (8) | Increased the GSH:GSSG ratio. | [156] |
| Liver (8) | No effect on protein oxidative damage (protein carbonyls). No effect on enzymatic antioxidant defense (catalase, SOD and GPX). | [156] | ||||
| Heart and Brain | No effect on lipid oxidative damage (hydroperoxides). No effect on mitochondrial ROS (H2O2) production. | [156] | ||||
| Skeletal muscle | No effect on lipid oxidative damage (hydroperoxides). Decreased protein oxidative damage (protein carbonyls) at mitochondria. No effect on mitochondrial ROS (H2O2) production. No effect on enzymatic antioxidant defense (catalase, SOD and GPX). | [156] | ||||
| Wistar, 28 d old (weaning) male | CoQ10 (0.005%, w/w), 6/24 m of age | AIN93 diet with sunflower oil as unique fat source (4%) | Urine (6) | Reduced aging-associated increase in urinary F2-isoprostanes. | [174] | |
| Pancreas (6) | Improved endocrine pancreas structure, in particular β-cell mass. | [175] | ||||
| Bone (6) | Prevented aging-associated bone mass loss decline. | [174] | ||||
| Alveolar bone (6) | Attenuated aging-associated alveolar bone loss. | [173] | ||||
| Gingivae (6) | Increased antioxidant enzymatic defenses (antioxidant enzyme gene expression). Increased mitochondrial biogenesis markers. | [173] | ||||
| AIN93 diet with fish oil as unique fat source (4%) | PBMCs | Reduced DNA oxidative damage markers (DNA strand breaks) in 24-m-old rats. | [182] | |||
| Urine | Reduced lipid oxidative damage markers (F2-isoprostanes) in 6-m-old rats. | [182] | ||||
| Pancreas | No effect on structural alterations in exocrine compartment. | [175] | ||||
| Bone | Increased bone mass density in 24-m-old rats. | [182] | ||||
| Alveolar bone | No effect on aging-associated alveolar bone loss. | [173] | ||||
| Gingivae | No effect on mitochondrial biogenesis markers. | [173] | ||||
| AIN93 diet with virgin olive oil as unique fat source (4%) | Urine | No effect on lipid oxidative damage (F2-isoprostanes) markers. | [174] | |||
| Pancreas | No effect on histopathological alterations. | [175] | ||||
| Bone | No effect on aging-associated bone mass density loss. | [174] | ||||
| Alveolar bone | No effect on aging-associated alveolar bone loss. | [173] | ||||
| Gingivae | No effect on mitochondrial biogenesis markers. No effect on enzymatic antioxidant defense | [173] | ||||
| CoQ10 (0.005%, w/w), 6/12 m | AIN93 diet but with 8% of sunflower oil as fat source | Heart (8) | Attenuated an aging-associated increase in lipid oxidative damage (hydroperoxides). | [176] | ||
| CoQ10 (0.062%, w/w), 6/12 m of age | AIN93 diet but with 8% of sunflower oil as fat source | Liver (8) | Decreased cytosolic and membrane-bound NQO1 activity. | [184] | ||
| Brain (8) | Decreased cytosolic and membrane-bound NQO1 activity | [184] | ||||
| AIN93 diet but with 8% of virgin olive oil as fat source | Liver (8) | Decreased cytosolic and membrane-bound NQO1 activity. | [184] | |||
| Brain (8) | Decreased cytosolic and membrane-bound NQO1 activity. | [184] | ||||
| CoQ10 (0.005%, w/w), 6/12/18/24 m of age | AIN93 diet but with 8% of sunflower oil as fat source | Blood (8) | Decreased DNA oxidative damage markers (DNA strand breaks) in PBMCs in 18- and 24-m-old rats. | [180] | ||
| CoQ10 (0.005%, w/w), 6/12/24 m of age | AIN93 diet but with 8% of sunflower oil as fat source | Heart (20) | Decreased lipid oxidative damage (hydroperoxides) in 12- and 14-m-old rats. | [213] | ||
| Liver (8) | Prevented an aging-associated decrease in glutathione-S-transferase (GST) activity but Se-dep GPX was not clearly affected. No effect on basal lipid oxidative damage markers (hydroperoxides) but attenuated formation against AAPH in old rats. | [183] | ||||
| CoQ10 (0.005%, w/w) 6/24 m of age | AIN93 diet with 8% of sunflower oil as fat source | Blood (20) | Increased non-enzymatic antioxidant defenses (α-tocopherol and retinol) and total antioxidant capacity in aged rats. Decreased DNA oxidative damage markers (DNA strand breaks) in PBMCs in young rats. | [179] | ||
| Liver | Prevented an aging-associated increase in lipid oxidative damage markers (hydroperoxides). Increased non-enzymatic antioxidant defenses (α-Tocopherol). Prevented an aging-associated decrease in enzymatic antioxidant defenses (catalase activity). | [178] | ||||
| Skeletal muscle | Increased non-enzymatic antioxidant defenses (α-Tocopherol) in young rats but attenuated its aging-associated increase. Reduced lipid oxidative damage markers (hydroperoxides) at any age. Prevented an aging-associated increase in enzymatic antioxidant defenses (catalase activity). | [178] | ||||
| Heart | Increased non-enzymatic antioxidant defenses. Partially prevented an age-associated mitochondrial function (mtETC II and III and COX were decreased). Prevented an age-associated increase in lipid and DNA oxidative damage in mitochondria (deleted mtDNA and hydroperoxides). Improved mitochondrial ultrastructure (area, perimeter, cristae density) in aged rats. | [181] | ||||
| Brain | Increased non-enzymatic antioxidants (α-tocopherol) at mitochondria. Decreased mitochondrial ROS production. Decreased enzymatic antioxidant defenses (GPX content) in cytosol. Increased mitochondrial function (mtETC complex I, IV and III activities) in young rats, but this decreased (complex I activity) in aged rats. Decreased oxidative DNA and lipid damage markers at mitochondria (hydroperoxides and deleted mtDNA). | [178] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Díaz-Casado, M.E.; Quiles, J.L.; Barriocanal-Casado, E.; González-García, P.; Battino, M.; López, L.C.; Varela-López, A. The Paradox of Coenzyme Q10 in Aging. Nutrients 2019, 11, 2221. https://doi.org/10.3390/nu11092221
Díaz-Casado ME, Quiles JL, Barriocanal-Casado E, González-García P, Battino M, López LC, Varela-López A. The Paradox of Coenzyme Q10 in Aging. Nutrients. 2019; 11(9):2221. https://doi.org/10.3390/nu11092221
Chicago/Turabian StyleDíaz-Casado, M. Elena, José L. Quiles, Eliana Barriocanal-Casado, Pilar González-García, Maurizio Battino, Luis C. López, and Alfonso Varela-López. 2019. "The Paradox of Coenzyme Q10 in Aging" Nutrients 11, no. 9: 2221. https://doi.org/10.3390/nu11092221
APA StyleDíaz-Casado, M. E., Quiles, J. L., Barriocanal-Casado, E., González-García, P., Battino, M., López, L. C., & Varela-López, A. (2019). The Paradox of Coenzyme Q10 in Aging. Nutrients, 11(9), 2221. https://doi.org/10.3390/nu11092221