Distinct TP53 Mutation Subtypes Differentially Influence Cellular Iron Metabolism

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture Conditions and Creation of Inducible Cell Lines
2.2. Western Blots
2.3. Intracellular Iron Measurement
2.4. RNA Isolation and Real-Time qPCR
2.5. Aconitase Assays
2.6. IRP RNA Binding Assays
2.7. MTT Assays
2.8. Statistical Analyses
3. Results
3.1. Induction of Mutant TP53 Expression Alone is Sufficient to Alter Cellular Iron Levels
3.2. Iron-Related mRNA Expresion Differs among Cells Harboring Distinct TP53 Mutation Subtypes
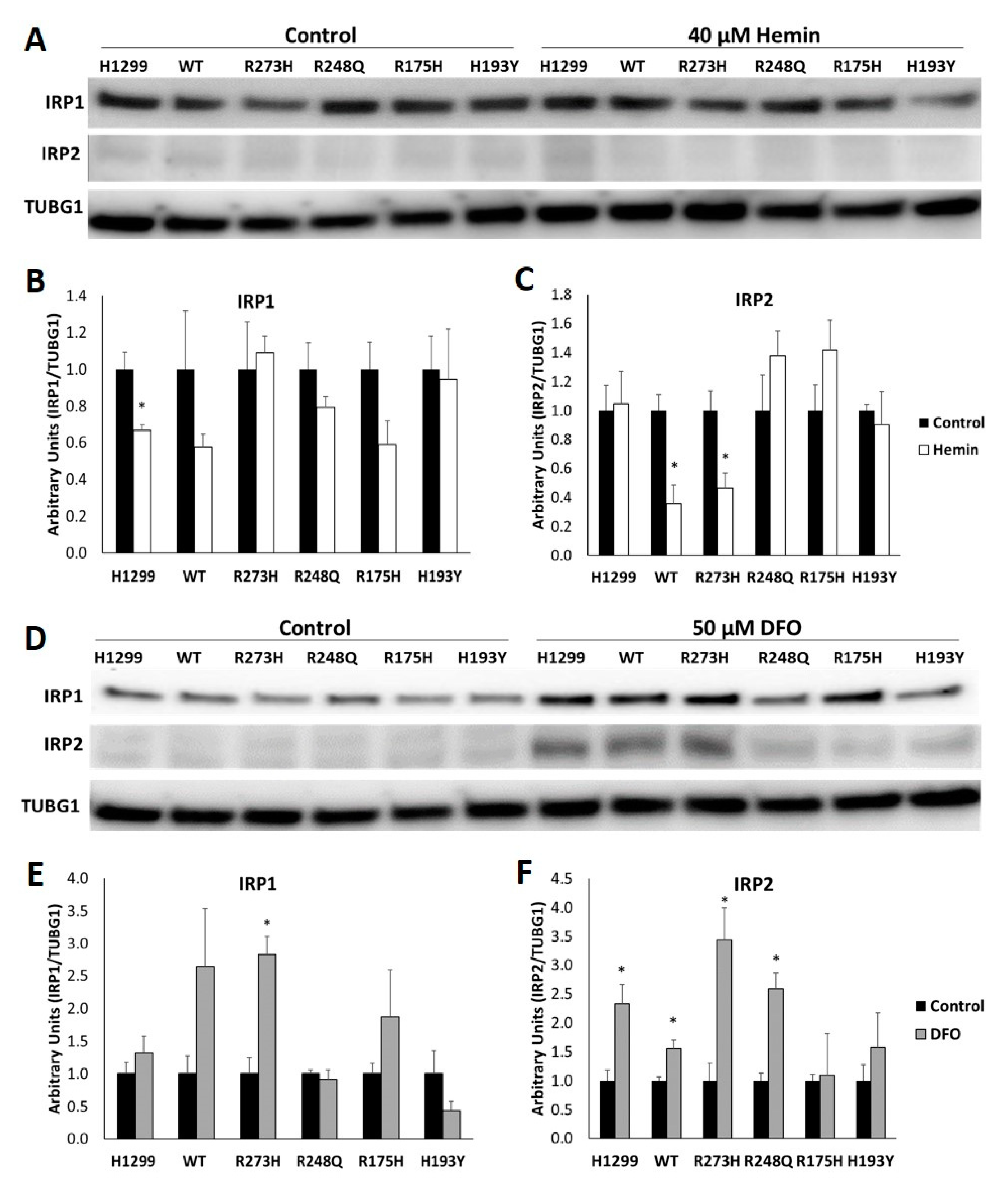
3.3. Cells with Distinct TP53 Mutations Exhibit Differential Changes in IRP RNA Binding Activity in Response to Modifications in Iron Availability
3.4. Reduced FDXR Expression is Associated with Decreased Mitochondrial Aconitase Activity in R175H Mutants
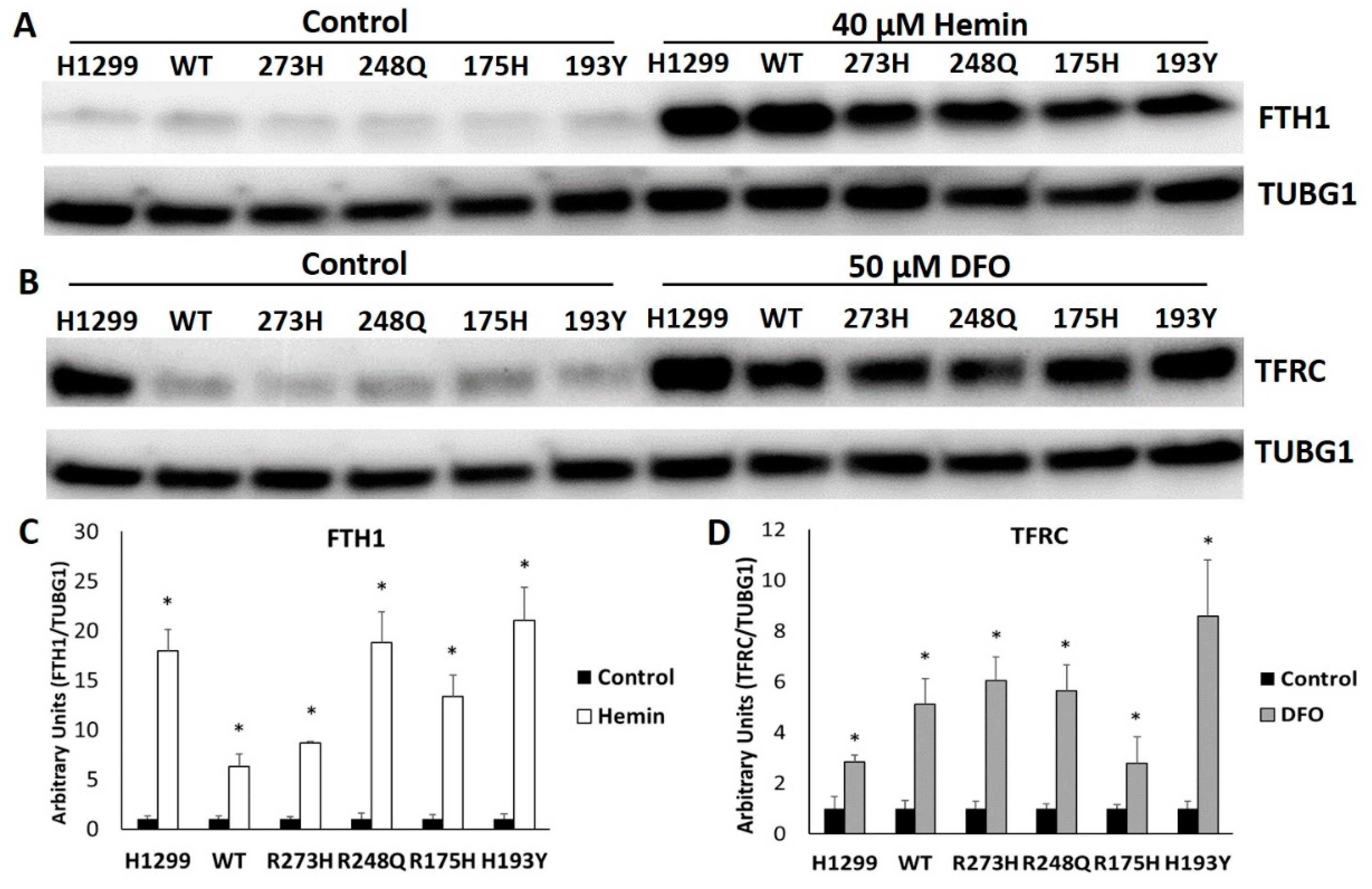
3.5. Mutant TP53 Expressing Cells Display Robust FTH1 and TFRC Responses to Changes in Iron Availability
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lee, H.J.; Lee, E.K.; Lee, K.J.; Hong, S.W.; Yoon, Y.; Kim, J.S. Ectopic expression of neutrophil gelatinase-associated lipocalin suppresses the invasion and liver metastasis of colon cancer cells. Int. J. Cancer 2006, 118, 2490–2497. [Google Scholar] [CrossRef] [PubMed]
- Miller, L.D.; Coffman, L.G.; Chou, J.W.; Black, M.A.; Bergh, J.; D’Agostino, R., Jr.; Torti, S.V.; Torti, F.M. An iron regulatory gene signature predicts outcome in breast cancer. Cancer Res. 2011, 71, 6728–6737. [Google Scholar] [CrossRef] [PubMed]
- Pinnix, Z.K.; Miller, L.D.; Wang, W.; D’Agostino, R., Jr.; Kute, T.; Willingham, M.C.; Hatcher, H.; Tesfay, L.; Sui, G.; Di, X.; et al. Ferroportin and iron regulation in breast cancer progression and prognosis. Sci. Transl. Med. 2010, 2, 43ra56. [Google Scholar] [CrossRef] [PubMed]
- Wenners, A.S.; Mehta, K.; Loibl, S.; Park, H.; Mueller, B.; Arnold, N.; Hamann, S.; Weimer, J.; Ataseven, B.; Darb-Esfahani, S.; et al. Neutrophil gelatinase-associated lipocalin (NGAL) predicts response to neoadjuvant chemotherapy and clinical outcome in primary human breast cancer. PLoS ONE 2012, 7, e45826. [Google Scholar] [CrossRef] [PubMed]
- Funauchi, Y.; Tanikawa, C.; Yi Lo, P.H.; Mori, J.; Daigo, Y.; Takano, A.; Miyagi, Y.; Okawa, A.; Nakamura, Y.; Matsuda, K. Regulation of iron homeostasis by the p53-ISCU pathway. Sci. Rep. 2015, 5, 16497. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Sheng, X.; Chang, Z.; Wu, Q.; Wang, S.; Xuan, Z.; Li, D.; Wu, Y.; Shang, Y.; Kong, X.; et al. Iron metabolism regulates p53 signaling through direct heme-p53 interaction and modulation of p53 localization, stability, and function. Cell Rep. 2014, 7, 180–193. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Wang, W.; Tsuji, Y.; Torti, S.V.; Torti, F.M. Post-transcriptional modulation of iron homeostasis during p53-dependent growth arrest. J. Biol. Chem. 2008, 283, 33911–33918. [Google Scholar] [CrossRef] [PubMed]
- Eisenstein, R.S. Iron regulatory proteins and the molecular control of mammalian iron metabolism. Annu. Rev. Nutr. 2000, 20, 627–662. [Google Scholar] [CrossRef]
- Johnson, N.B.; Deck, K.M.; Nizzi, C.P.; Eisenstein, R.S. A synergistic role of IRP1 and FBXL5 proteins in coordinating iron metabolism during cell proliferation. J. Biol. Chem. 2017, 292, 15976–15989. [Google Scholar] [CrossRef]
- Freed-Pastor, W.A.; Prives, C. Mutant p53: One name, many proteins. Genes Dev. 2012, 26, 1268–1286. [Google Scholar] [CrossRef]
- Di Agostino, S.; Strano, S.; Emiliozzi, V.; Zerbini, V.; Mottolese, M.; Sacchi, A.; Blandino, G.; Piaggio, G. Gain of function of mutant p53: The mutant p53/NF-Y protein complex reveals an aberrant transcriptional mechanism of cell cycle regulation. Cancer Cell 2006, 10, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Ling, S.; Lin, W.C. TopBP1 mediates mutant p53 gain of function through NF-Y and p63/p73. Mol. Cell. Biol. 2011, 31, 4464–4481. [Google Scholar] [CrossRef] [PubMed]
- Stambolsky, P.; Tabach, Y.; Fontemaggi, G.; Weisz, L.; Maor-Aloni, R.; Siegfried, Z.; Shiff, I.; Kogan, I.; Shay, M.; Kalo, E.; et al. Modulation of the vitamin D3 response by cancer-associated mutant p53. Cancer Cell 2010, 17, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Nekanti, U.; Dastidar, S.; Venugopal, P.; Totey, S.; Ta, M. Increased proliferation and analysis of differential gene expression in human Wharton’s jelly-derived mesenchymal stromal cells under hypoxia. Int. J. Biol. Sci. 2010, 6, 499–512. [Google Scholar] [CrossRef]
- Wang, W.; Di, X.; D’Agostino, R.B., Jr.; Torti, S.V.; Torti, F.M. Excess capacity of the iron regulatory protein system. J. Biol. Chem. 2007, 282, 24650–24659. [Google Scholar] [CrossRef]
- Davis, M.R.; Shawron, K.M.; Rendina, E.; Peterson, S.K.; Lucas, E.A.; Smith, B.J.; Clarke, S.L. Hypoxia inducible factor-2 alpha is translationally repressed in response to dietary iron deficiency in Sprague-Dawley rats. J. Nutr. 2011, 141, 1590–1596. [Google Scholar] [CrossRef]
- Eriksson, M.; Ambroise, G.; Ouchida, A.T.; Lima Queiroz, A.; Smith, D.; Gimenez-Cassina, A.; Iwanicki, M.P.; Muller, P.A.; Norberg, E.; Vakifahmetoglu-Norberg, H. Effect of Mutant p53 Proteins on Glycolysis and Mitochondrial Metabolism. Mol. Cell. Biol. 2017, 37. [Google Scholar] [CrossRef]
- Nam, S.Y.; Sabapathy, K. p53 promotes cellular survival in a context-dependent manner by directly inducing the expression of haeme-oxygenase-1. Oncogene 2011, 30, 4476–4486. [Google Scholar] [CrossRef]
- Stein, S.; Thomas, E.K.; Herzog, B.; Westfall, M.D.; Rocheleau, J.V.; Jackson, R.S., 2nd; Wang, M.; Liang, P. NDRG1 is necessary for p53-dependent apoptosis. J. Biol. Chem. 2004, 279, 48930–48940. [Google Scholar] [CrossRef]
- Lymboussaki, A.; Pignatti, E.; Montosi, G.; Garuti, C.; Haile, D.J.; Pietrangelo, A. The role of the iron responsive element in the control of ferroportin1/IREG1/MTP1 gene expression. J. Hepatol. 2003, 39, 710–715. [Google Scholar] [CrossRef]
- Le, N.T.; Richardson, D.R. Iron chelators with high antiproliferative activity up-regulate the expression of a growth inhibitory and metastasis suppressor gene: A link between iron metabolism and proliferation. Blood 2004, 104, 2967–2975. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Qian, Y.; Zhang, J.; Yan, W.; Jung, Y.S.; Chen, M.; Huang, E.; Lloyd, K.; Duan, Y.; Wang, J.; et al. Ferredoxin reductase is critical for p53-dependent tumor suppression via iron regulatory protein 2. Genes Dev. 2017, 31, 1243–1256. [Google Scholar] [CrossRef] [PubMed]
- Hanson, E.S.; Foot, L.M.; Leibold, E.A. Hypoxia post-translationally activates iron-regulatory protein 2. J. Biol. Chem. 1999, 274, 5047–5052. [Google Scholar] [CrossRef] [PubMed]
- Deck, K.M.; Vasanthakumar, A.; Anderson, S.A.; Goforth, J.B.; Kennedy, M.C.; Antholine, W.E.; Eisenstein, R.S. Evidence that phosphorylation of iron regulatory protein 1 at Serine 138 destabilizes the [4Fe-4S] cluster in cytosolic aconitase by enhancing 4Fe-3Fe cycling. J. Biol. Chem. 2009, 284, 12701–12709. [Google Scholar] [CrossRef] [PubMed]
- Salahudeen, A.A.; Thompson, J.W.; Ruiz, J.C.; Ma, H.W.; Kinch, L.N.; Li, Q.; Grishin, N.V.; Bruick, R.K. An E3 ligase possessing an iron-responsive hemerythrin domain is a regulator of iron homeostasis. Science 2009, 326, 722–726. [Google Scholar] [CrossRef] [PubMed]
- Vashisht, A.A.; Zumbrennen, K.B.; Huang, X.; Powers, D.N.; Durazo, A.; Sun, D.; Bhaskaran, N.; Persson, A.; Uhlen, M.; Sangfelt, O.; et al. Control of iron homeostasis by an iron-regulated ubiquitin ligase. Science 2009, 326, 718–721. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Ghosh, M.; Kovtunovych, G.; Crooks, D.R.; Rouault, T.A. Both human ferredoxins 1 and 2 and ferredoxin reductase are important for iron-sulfur cluster biogenesis. Biochim. Biophys. Acta 2012, 1823, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Bogdan, A.R.; Miyazawa, M.; Hashimoto, K.; Tsuji, Y. Regulators of Iron Homeostasis: New Players in Metabolism, Cell Death, and Disease. Trends Biochem. Sci. 2016, 41, 274–286. [Google Scholar] [CrossRef]
- Bykov, V.J.N.; Eriksson, S.E.; Bianchi, J.; Wiman, K.G. Targeting mutant p53 for efficient cancer therapy. Nat. Rev. Cancer 2018, 18, 89–102. [Google Scholar] [CrossRef]
- Yue, X.; Zhao, Y.; Xu, Y.; Zheng, M.; Feng, Z.; Hu, W. Mutant p53 in Cancer: Accumulation, Gain-of-Function, and Therapy. J. Mol. Biol. 2017, 429, 1595–1606. [Google Scholar] [CrossRef] [PubMed]
- Bradbear, R.A.; Bain, C.; Siskind, V.; Schofield, F.D.; Webb, S.; Axelsen, E.M.; Halliday, J.W.; Bassett, M.L.; Powell, L.W. Cohort study of internal malignancy in genetic hemochromatosis and other chronic nonalcoholic liver diseases. J. Natl. Cancer Inst. 1985, 75, 81–84. [Google Scholar] [PubMed]
- Campbell, J.A. Effects of Precipitated Silica and of Iron Oxide on the Incidence of Primary Lung Tumours in Mice. Br. Med. J. 1940, 2, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Richmond, H.G. Induction of sarcoma in the rat by iron-dextran complex. Br. Med. J. 1959, 1, 947–949. [Google Scholar] [CrossRef] [PubMed]
- Stevens, R.G.; Graubard, B.I.; Micozzi, M.S.; Neriishi, K.; Blumberg, B.S. Moderate elevation of body iron level and increased risk of cancer occurrence and death. Int. J. Cancer. 1994, 56, 364–369. [Google Scholar] [CrossRef] [PubMed]
- Eberhard, Y.; McDermott, S.P.; Wang, X.; Gronda, M.; Venugopal, A.; Wood, T.E.; Hurren, R.; Datti, A.; Batey, R.A.; Wrana, J.; et al. Chelation of intracellular iron with the antifungal agent ciclopirox olamine induces cell death in leukemia and myeloma cells. Blood 2009, 114, 3064–3073. [Google Scholar] [CrossRef]
- Fukushima, T.; Kawabata, H.; Nakamura, T.; Iwao, H.; Nakajima, A.; Miki, M.; Sakai, T.; Sawaki, T.; Fujita, Y.; Tanaka, M.; et al. Iron chelation therapy with deferasirox induced complete remission in a patient with chemotherapy-resistant acute monocytic leukemia. Anticancer Res. 2011, 31, 1741–1744. [Google Scholar]
- Martin, L.K.; Grecula, J.; Jia, G.; Wei, L.; Yang, X.; Otterson, G.A.; Wu, X.; Harper, E.; Kefauver, C.; Zhou, B.S.; et al. A dose escalation and pharmacodynamic study of triapine and radiation in patients with locally advanced pancreas cancer. Int. J. Radiat. Oncol. Biol. Phys. 2012, 84, e475–e481. [Google Scholar] [CrossRef]
- Yu, Y.; Gutierrez, E.; Kovacevic, Z.; Saletta, F.; Obeidy, P.; Suryo Rahmanto, Y.; Richardson, D.R. Iron chelators for the treatment of cancer. Curr. Med. Chem. 2012, 19, 2689–2702. [Google Scholar] [CrossRef]
- Chen, Z.; Li, Y.; Zhang, H.; Huang, P.; Luthra, R. Hypoxia-regulated microRNA-210 modulates mitochondrial function and decreases ISCU and COX10 expression. Oncogene 2010, 29, 4362–4368. [Google Scholar] [CrossRef]
- Gammella, E.; Buratti, P.; Cairo, G.; Recalcati, S. The transferrin receptor: The cellular iron gate. Metallomics 2017, 9, 1367–1375. [Google Scholar] [CrossRef] [PubMed]
- Melotte, V.; Qu, X.; Ongenaert, M.; van Criekinge, W.; de Bruine, A.P.; Baldwin, H.S.; van Engeland, M. The N-myc downstream regulated gene (NDRG) family: Diverse functions, multiple applications. FASEB J. 2010, 24, 4153–4166. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Brand, M.; Zenke, Y.; Tashiro, S.; Groudine, M.; Igarashi, K. Heme regulates the dynamic exchange of Bach1 and NF-E2-related factors in the Maf transcription factor network. Proc. Natl. Acad. Sci. USA 2004, 101, 1461–1466. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.P.; Shen, M.; Eisenstein, R.S.; Leibold, E.A. Mammalian iron metabolism and its control by iron regulatory proteins. Biochim. Biophys. Acta 2012, 1823. [Google Scholar] [CrossRef] [PubMed]
- Chen, O.S.; Schalinske, K.L.; Eisenstein, R.S. Dietary iron intake modulates the activity of iron regulatory proteins and the abundance of ferritin and mitochondrial aconitase in rat liver. J. Nutr. 1997, 127, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Clarke, S.L.; Vasanthakumar, A.; Anderson, S.A.; Pondarre, C.; Koh, C.M.; Deck, K.M.; Pitula, J.S.; Epstein, C.J.; Fleming, M.D.; Eisenstein, R.S. Iron-responsive degradation of iron-regulatory protein 1 does not require the Fe-S cluster. EMBO J. 2006, 25, 544–553. [Google Scholar] [CrossRef] [PubMed]
- Luo, Q.Q.; Qian, Z.M.; Zhou, Y.F.; Zhang, M.W.; Wang, D.; Zhu, L.; Ke, Y. Expression of Iron Regulatory Protein 1 Is Regulated not only by HIF-1 but also pCREB under Hypoxia. Int. J. Biol. Sci. 2016, 12, 1191–1202. [Google Scholar] [CrossRef] [PubMed]
- Godoy, P.R.; Mello, S.S.; Magalhaes, D.A.; Donaires, F.S.; Nicolucci, P.; Donadi, E.A.; Passos, G.A.; Sakamoto-Hojo, E.T. Ionizing radiation-induced gene expression changes in TP53 proficient and deficient glioblastoma cell lines. Mutat. Res. 2013, 756, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Dallman, P.R. Biochemical basis for the manifestations of iron deficiency. Annu. Rev. Nutr. 1986, 6, 13–40. [Google Scholar] [CrossRef]
- Rensvold, J.W.; Krautkramer, K.A.; Dowell, J.A.; Denu, J.M.; Pagliarini, D.J. Iron Deprivation Induces Transcriptional Regulation of Mitochondrial Biogenesis. J. Biol. Chem. 2016, 291, 20827–20837. [Google Scholar] [CrossRef]
- Lill, R.; Dutkiewicz, R.; Freibert, S.A.; Heidenreich, T.; Mascarenhas, J.; Netz, D.J.; Paul, V.D.; Pierik, A.J.; Richter, N.; Stumpfig, M.; et al. The role of mitochondria and the CIA machinery in the maturation of cytosolic and nuclear iron-sulfur proteins. Eur. J. Cell. Biol. 2015, 94, 280–291. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, L.; Teng, Z.P.; Zeserson, E.; Patel, M.; Regan, R.F. Hemin induces an iron-dependent, oxidative injury to human neuron-like cells. J. Neurosci. Res. 2003, 73, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Jian, Z.; Li, K.; Liu, L.; Zhang, Y.; Zhou, Z.; Li, C.; Gao, T. Heme oxygenase-1 protects human melanocytes from H2O2-induced oxidative stress via the Nrf2-ARE pathway. J. Investig. Dermatol. 2011, 131, 1420–1427. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, D.; Portales-Casamar, E.; Singh, A.; Srivastava, S.; Arenillas, D.; Happel, C.; Shyr, C.; Wakabayashi, N.; Kensler, T.W.; Wasserman, W.W.; et al. Global mapping of binding sites for Nrf2 identifies novel targets in cell survival response through ChIP-Seq profiling and network analysis. Nucleic Acids Res. 2010, 38, 5718–5734. [Google Scholar] [CrossRef] [PubMed]
- Lok, C.N.; Ponka, P. Identification of a hypoxia response element in the transferrin receptor gene. J. Biol. Chem. 1999, 274, 24147–24152. [Google Scholar] [CrossRef] [PubMed]
- Tacchini, L.; Bianchi, L.; Bernelli-Zazzera, A.; Cairo, G. Transferrin receptor induction by hypoxia. HIF-1-mediated transcriptional activation and cell-specific post-transcriptional regulation. J. Biol. Chem. 1999, 274, 24142–24146. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, L.; Tacchini, L.; Cairo, G. HIF-1-mediated activation of transferrin receptor gene transcription by iron chelation. Nucleic Acids Res. 1999, 27, 4223–4227. [Google Scholar] [CrossRef]
- Simpson, R.J.; McKie, A.T. Iron and oxygen sensing: A tale of 2 interacting elements? Metallomics 2015, 7, 223–231. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Clarke, S.L.; Thompson, L.R.; Dandekar, E.; Srinivasan, A.; Montgomery, M.R. Distinct TP53 Mutation Subtypes Differentially Influence Cellular Iron Metabolism. Nutrients 2019, 11, 2144. https://doi.org/10.3390/nu11092144
Clarke SL, Thompson LR, Dandekar E, Srinivasan A, Montgomery MR. Distinct TP53 Mutation Subtypes Differentially Influence Cellular Iron Metabolism. Nutrients. 2019; 11(9):2144. https://doi.org/10.3390/nu11092144
Chicago/Turabian StyleClarke, Stephen L., Laurie R. Thompson, Eshan Dandekar, Aishwarya Srinivasan, and McKale R. Montgomery. 2019. "Distinct TP53 Mutation Subtypes Differentially Influence Cellular Iron Metabolism" Nutrients 11, no. 9: 2144. https://doi.org/10.3390/nu11092144
APA StyleClarke, S. L., Thompson, L. R., Dandekar, E., Srinivasan, A., & Montgomery, M. R. (2019). Distinct TP53 Mutation Subtypes Differentially Influence Cellular Iron Metabolism. Nutrients, 11(9), 2144. https://doi.org/10.3390/nu11092144