Functional Relationship between Leptin and Nitric Oxide in Metabolism

 ,
,  ,
,  ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
1.1. Leptin
1.2. Nitric Oxide
2. Energy Balance
2.1. Leptin and Energy Balance
2.1.1. Food Intake and Body Weight
2.1.2. Thermogenesis
2.2. NO and Energy Balance
2.2.1. Food Intake and Body Weight
2.2.2. Thermogenesis
2.3. Leptin, NO, and Energy Balance
2.3.1. Food Intake and Body Weight
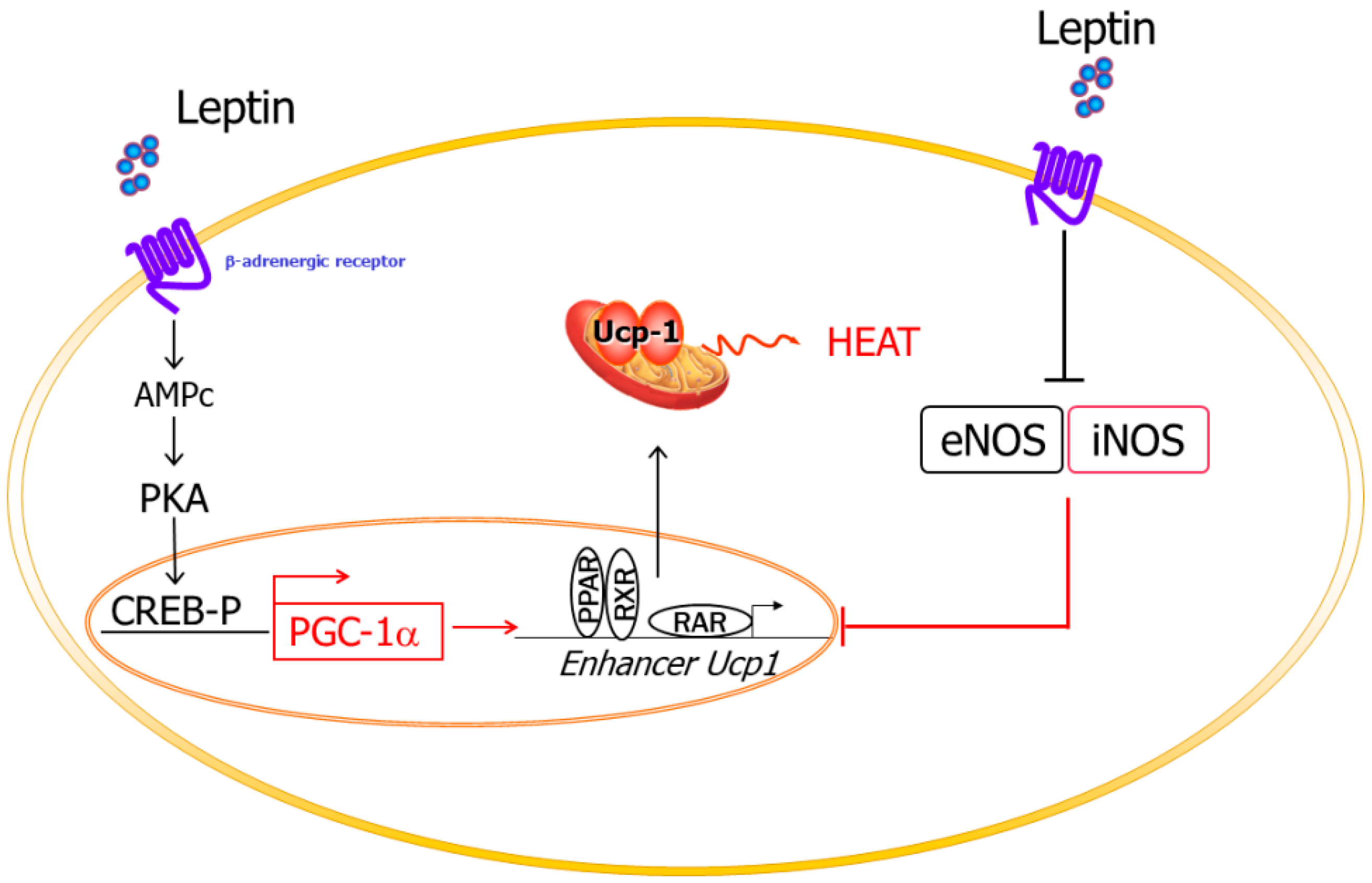
2.3.2. Thermogenesis
3. Glucose Metabolism
3.1. Leptin and Glucose Metabolism
3.2. NO and Glucose Metabolism
3.3. Leptin, NO, and Glucose Metabolism
4. Lipid Metabolism
4.1. Leptin and Lipid Metabolism
4.2. NO and Lipid Metabolism
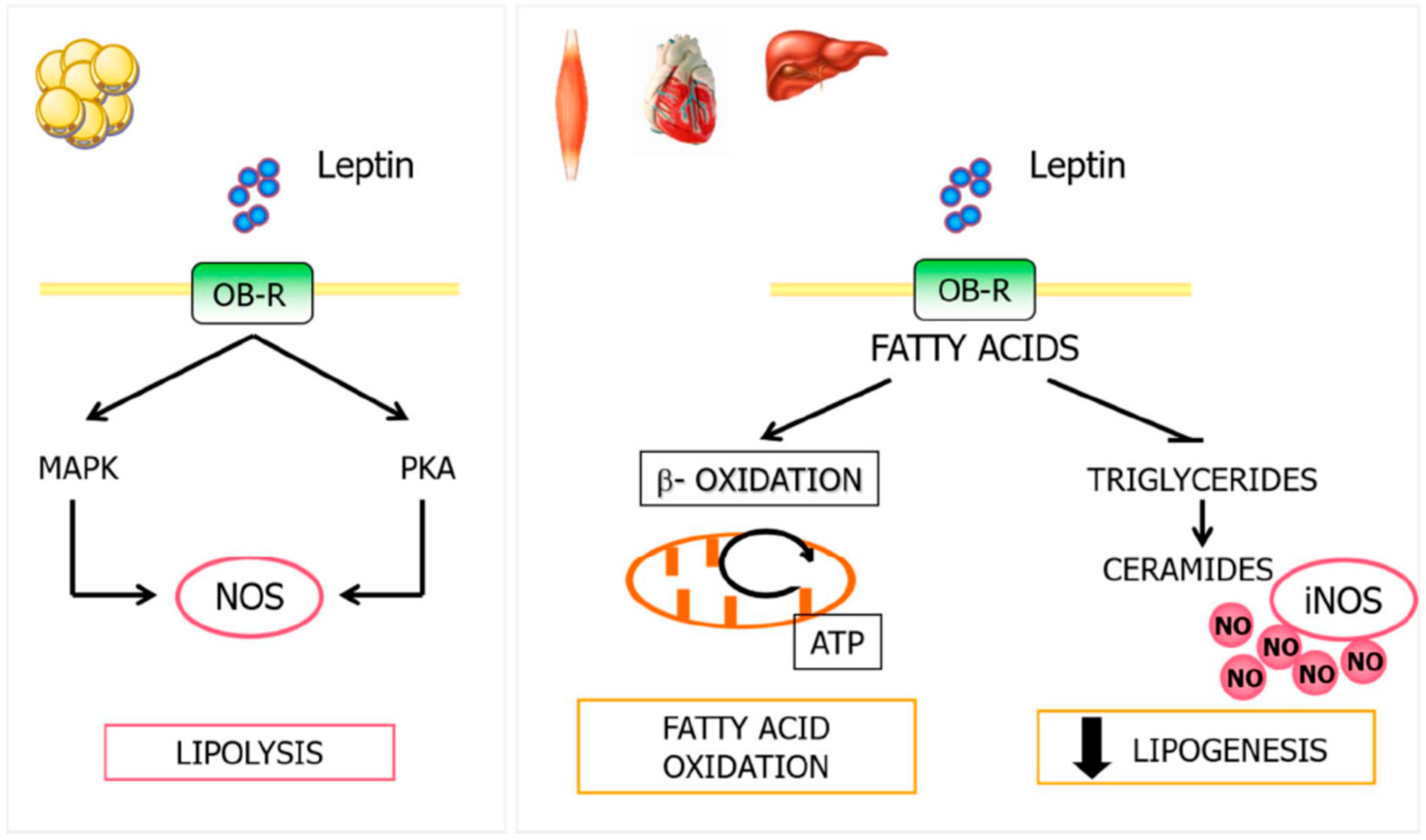
4.3. Leptin, NO, and Lipid Metabolism
5. Cardiovascular System
5.1. Leptin and the Cardiovascular System
5.1.1. Angiogenesis and Wound Healing
5.1.2. Vascular Health
5.1.3. Blood Pressure
5.2. NO and the Cardiovascular System
5.2.1. Angiogenesis and Wound Healing
5.2.2. Vascular Health
5.2.3. Blood Pressure
5.3. Leptin, NO, and Cardiovascular System
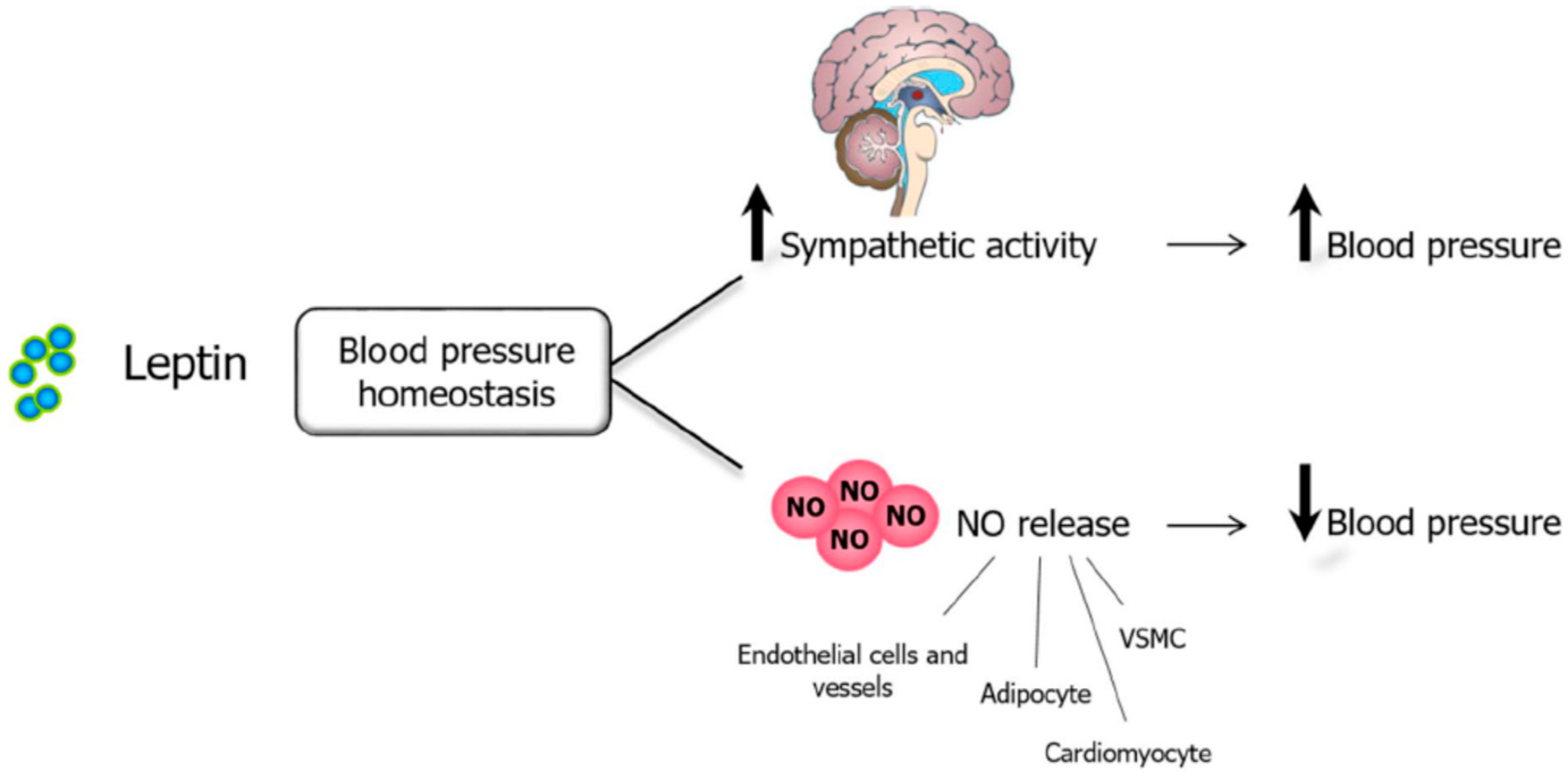
Leptin, NO, and Blood Pressure
5.4. Leptin, NO, and Angiogenesis
6. Bone Metabolism
6.1. Leptin and Bone Metabolism
6.2. NO and Bone Metabolism
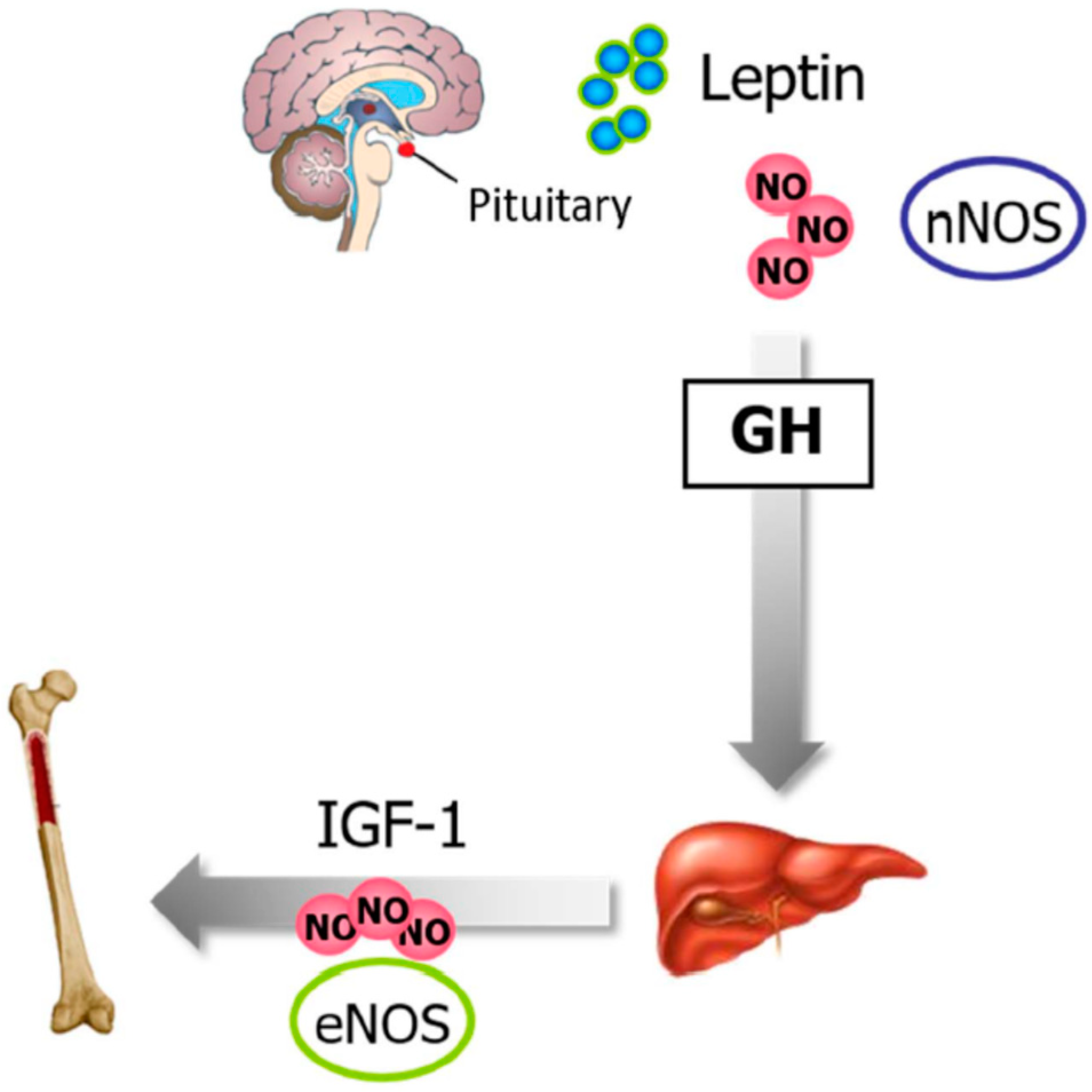
6.3. Leptin, NO, and Bone Metabolism
7. Reproduction
7.1. Leptin and Reproduction
7.2. NO and Reproduction
7.3. Leptin, NO, and Reproduction
8. Immune Response
8.1. Leptin and the Immune Response
8.1.1. Leptin and Hematopoiesis
8.1.2. Leptin and the Innate Immune Response
8.1.3. Leptin and the Adaptive Immune Response
8.1.4. Leptin and Autoimmunity
8.2. NO and the Immune Response
8.2.1. NO and Hematopoiesis
8.2.2. NO and Innate Immune Response
8.2.3. NO and the Adaptive Immune Response
8.2.4. NO and Autoimmunity
8.3. Leptin, NO, and Immune Response
9. Obesity
Leptin, NO, and Obesity
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Abelenda, M.; Nava, M.P.; Fernández, A.; Puerta, M.L. Brown adipose tissue thermogenesis in testosterone-treated rats. Eur. J. Endocrinol. 1992, 126, 434–437. [Google Scholar] [CrossRef]
- Rodríguez, A.; Ezquerro, S.; Méndez-Gimenez, L.; Becerril, S.; Frühbeck, G. Revisiting the adipocyte: A model for integration of cytokine signaling in the regulation of energy metabolism. Am. J. Physiol. Endocrinol. Metab. 2015, 309, E691–E714. [Google Scholar] [CrossRef]
- Zhang, F.; Basinski, M.B.; Beals, J.M.; Briggs, S.L.; Churgay, L.M.; Clawson, D.K.; DiMarchi, R.D.; Furman, T.C.; Hale, J.E.; Hsiung, H.M.; et al. Crystal structure of the obese protein leptin-E100. Nature 1997, 387, 206–209. [Google Scholar] [CrossRef]
- Tartaglia, L.A. The leptin receptor. J. Biol. Chem. 1997, 272, 6093–6096. [Google Scholar] [CrossRef]
- Frühbeck, G.; Gómez-Ambrosi, J. Control of body weight: A physiologic and transgenic perspective. Diabetologia 2003, 46, 143–172. [Google Scholar] [CrossRef]
- Muruzabal, F.J.; Frühbeck, G.; Gomez-Ambrosi, J.; Archanco, M.; Burrell, M. Immunocytochemical detection of leptin in non-mammalian vertebrate stomach. Gen. Comp. Endocrinol. 2002, 128, 149–152. [Google Scholar] [CrossRef]
- Philbrick, K.A.; Wong, C.P.; Branscum, A.J.; Turner, R.T.; Iwaniec, U.T. Leptin Stimulates Bone Formation in ob/ob Mice at Doses Having Minimal Impact on Energy Metabolism. J. Endocrinol. 2017, 232, 461–474. [Google Scholar] [CrossRef]
- Moncada, S.; Higgs, E.A. Molecular mechanisms and therapeutic strategies related to nitric oxide. FASEB J. 1995, 9, 1319–1330. [Google Scholar] [CrossRef]
- Palmer, R.M.J.; Ferrige, A.G.; Moncada, S. Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature 1987, 327, 524–526. [Google Scholar] [CrossRef]
- Higgs, A.; Epstein, F.H.; Moncada, S. The L-Arginine-Nitric Oxide Pathway. N. Engl. J. Med. 1993, 329, 2002–2012. [Google Scholar]
- Tsutsui, M.; Tanimoto, A.; Tamura, M.; Mukae, H.; Yanagihara, N.; Shimokawa, H.; Otsuji, Y. Significance of nitric oxide synthases: Lessons from triple nitric oxide synthases null mice. J. Pharmacol. Sci. 2015, 127, 42–52. [Google Scholar] [CrossRef]
- Zweier, J.L.; Wang, P.; Samouilov, A.; Kuppusamy, P. Enzyme-independent formation of nitric oxide in biological tissues. Nat. Med. 1995, 1, 804–809. [Google Scholar] [CrossRef]
- Larsen, F.J.; Lundberg, J.O.; Ekblom, B.; Sahlin, K.; Weitzberg, E. Effects of Dietary Nitrate on Blood Pressure in Healthy Volunteers. N. Engl. J. Med. 2006, 355, 2792–2793. [Google Scholar] [CrossRef]
- Larsen, F.J.; Schiffer, T.A.; Borniquel, S.; Sahlin, K.; Ekblom, B.; Lundberg, J.O.; Weitzberg, E. Dietary Inorganic Nitrate Improves Mitochondrial Efficiency in Humans. Cell. Metab. 2011, 13, 149–159. [Google Scholar] [CrossRef]
- Richardson, G.; Hicks, S.; O’Byrne, S.; Frost, M.; Moore, K.; Benjamin, N.; McKnight, G. The ingestion of inorganic nitrate increases gastric S-nitrosothiol levels and inhibits platelet function in humans. Nitric Oxide 2002, 7, 24–29. [Google Scholar] [CrossRef]
- Rodríguez-Mateos, A.; Hezel, M.; Aydin, H.; Kelm, M.; Lundberg, J.O.; Weitzberg, E.; Spencer, J.P.; Heiss, C.; Aydın, H. Interactions between cocoa flavanols and inorganic nitrate: Additive effects on endothelial function at achievable dietary amounts. Free. Radic. Boil. Med. 2015, 80, 121–128. [Google Scholar] [CrossRef]
- Ashmore, T.; Roberts, L.D.; Morash, A.J.; Kotwica, A.O.; Finnerty, J.; West, J.A.; Murfitt, S.A.; Fernandez, B.O.; Branco, C.; Cowburn, A.S.; et al. Nitrate enhances skeletal muscle fatty acid oxidation via a nitric oxide-cGMP-PPAR-mediated mechanism. BMC Boil. 2015, 13, 110. [Google Scholar] [CrossRef]
- Ohtake, K.; Nakano, G.; Ehara, N.; Sonoda, K.; Ito, J.; Uchida, H.; Kobayashi, J. Dietary nitrite supplementation improves insulin resistance in type 2 diabetic KKA(y) mice. Nitric Oxide 2015, 44, 31–38. [Google Scholar] [CrossRef]
- Yu, W.H.; Kimura, M.; Walczewska, A.; Karanth, S.; McCann, S.M. Role of leptin in hypothalamic-pituitary function. Proc. Natl. Acad. Sci. USA 1997, 94, 1023–1028. [Google Scholar] [CrossRef]
- Wang, M.-Y.; Koyama, K.; Shimabukuro, M.; Newgard, C.B.; Unger, R.H. OB-Rb gene transfer to leptin-resistant islets reverses diabetogenic phenotype. Proc. Natl. Acad. Sci. USA 1998, 95, 714–718. [Google Scholar] [CrossRef]
- Frühbeck, G. Pivotal role of nitric oxide in the control of blood pressure after leptin administration. Diabetes 1999, 48, 903–908. [Google Scholar] [CrossRef]
- Mastronardi, C.A.; Yu, W.H.; McCann, S.M. Resting and circadian release of nitric oxide is controlled by leptin in male rats. Proc. Natl. Acad. Sci. USA 2002, 99, 5721–5726. [Google Scholar] [CrossRef]
- Di Spiezio, A.; Sandin, E.S.; Dore, R.; Muller-Fielitz, H.; Storck, S.E.; Bernau, M.; Mier, W.; Oster, H.; Johren, O.; Pietrzik, C.U.; et al. The LepR-mediated leptin transport across brain barriers controls food reward. Mol. Metab. 2018, 8, 13–22. [Google Scholar] [CrossRef]
- Cui, H.X.; Lopez, M.; Rahmouni, K. The cellular and molecular bases of leptin and ghrelin resistance in obesity. Nat. Rev. Endocrinol. 2017, 13, 338–351. [Google Scholar] [CrossRef]
- Mele, P.; Zammaretti, F.; Longo, A.; Panzica, G.; Oberto, A.; Eva, C. Sex-dependent regulation of hypothalamic neuropeptide Y-Y1 receptor gene expression in leptin treated obese (ob/ob) or lean mice. Brain Res. 2016, 1649, 102–109. [Google Scholar] [CrossRef]
- Clément, K.; Biebermann, H.; Farooqi, I.S.; Van der, P.L.; Wolters, B.; Poitou, C.; Puder, L.; Fiedorek, F.; Gottesdiener, K.; Kleinau, G.; et al. MC4R agonism promotes durable weight loss in patients with leptin receptor deficiency. Nat. Med 2018, 24, 551–555. [Google Scholar]
- Farooqi, I.S. The severely obese patient—A genetic work-up. Nat. Clin. Pract. Endocrinol. Metab. 2006, 2, 172–177. [Google Scholar] [CrossRef]
- Ayers, K.L.; Glicksberg, B.S.; Garfield, A.S.; Longerich, S.; White, J.A.; Yang, P.; Du, L.; Chittenden, T.W.; Gulcher, J.R.; Roy, S.; et al. Melanocortin 4 Receptor Pathway Dysfunction in Obesity: Patient Stratification Aimed at MC4R Agonist Treatment. J. Clin. Endocrinol. Metab. 2018, 103, 2601–2612. [Google Scholar] [CrossRef]
- Gong, D.W.; He, Y.; Karas, M.; Reitman, M. Uncoupling protein-3 is a mediator of thermogenesis regulated by thyroid hormone, beta3-adrenergic agonists, and leptin. J. Boil. Chem. 1997, 272, 24129–24132. [Google Scholar]
- Gullicksen, P.; Flatt, W.P.; Dean, R.G.; Hartzell, D.L.; Baile, C.A. Energy metabolism and expression of uncoupling proteins 1, 2, and 3 after 21 days of recovery from intracerebroventricular mouse leptin in rats. Physiol. Behav. 2002, 75, 473–482. [Google Scholar] [CrossRef]
- Gómez-Ambrosi, J.; Frühbeck, G.; Martínez, J.A. Rapid in vivo PGC-1 mRNA upregulation in brown adipose tissue of Wistar rats by a beta(3)-adrenergic agonist and lack of effect of leptin. Mol. Cell. Endocrinol. 2001, 176, 85–90. [Google Scholar]
- Becerril, S.; Rodríguez, A.; Catalán, V.; Sáinz, N.; Ramírez, B.; Collantes, M.; Peñuelas, I.; Gómez-Ambrosi, J.; Frühbeck, G. Deletion of Inducible Nitric-Oxide Synthase in Leptin-Deficient Mice Improves Brown Adipose Tissue Function. PLoS ONE 2010, 5, e10962. [Google Scholar] [CrossRef]
- Tajima, D.; Masaki, T.; Hidaka, S.; Kakuma, T.; Sakata, T.; Yoshimatsu, H. Acute central infusion of leptin modulates fatty acid mobilization by affecting lipolysis and mRNA expression for uncoupling proteins. Exp. Boil. Med. 2005, 230, 200–206. [Google Scholar] [CrossRef]
- Lidell, M.E.; Betz, M.J.; Leinhard, O.D.; Heglind, M.; Elander, L.; Slawik, M.; Mussack, T.; Nilsson, D.; Romu, T.; Nuutila, P.; et al. Evidence for two types of brown adipose tissue in humans. Nat. Med. 2013, 19, 631–634. [Google Scholar] [CrossRef]
- Dodd, G.T.; Decherf, S.; Loh, K.; Simonds, S.E.; Wiede, F.; Balland, E.; Merry, T.L.; Münzberg, H.; Zhang, Z.-Y.; Kahn, B.B.; et al. Leptin and Insulin Act on POMC Neurons to Promote the Browning of White Fat. Cell 2015, 160, 88–104. [Google Scholar] [CrossRef]
- Rodríguez, A.; Becerril, S.; Méndez-Giménez, L.; Ramírez, B.; Sainz, N.; Catalán, V.; Gómez-Ambrosi, J.; Frühbeck, G. Leptin administration activates irisin-induced myogenesis via nitric oxide-dependent mechanisms, but reduces its effect on subcutaneous fat browning in mice. Int. J. Obes. 2015, 39, 397–407. [Google Scholar] [CrossRef]
- Frühbeck, G.; Sesma, P.; Burrell, M.A. PRDM16: The interconvertible adipo-myocyte switch. Trends Cell Boil. 2009, 19, 141–146. [Google Scholar] [CrossRef]
- Gan, L.; Liu, Z.; Feng, F.; Wu, T.; Luo, D.; Hu, C.; Sun, C. Foxc2 coordinates inflammation and browning of white adipose by leptin-STAT3-PRDM16 signal in mice. Int. J. Obes. 2018, 42, 252–259. [Google Scholar] [CrossRef]
- Rocha-Rodrigues, S.; Rodríguez, A.; Becerril, S.; Beleza, J.; Ramírez, B.; Gonçalves, I.O.; Frühbeck, G.; Ascensão, A.; Magalhães, J. Physical exercise remodels visceral adipose tissue and mitochondrial lipid metabolism in rats fed a high-fat diet. Clin. Exp. Pharmacol. Physiol. 2017, 44, 386–394. [Google Scholar] [CrossRef]
- Rodríguez, A.; Becerril, S.; Ezquerro, S.; Méndez-Giménez, L.; Frühbeck, G. Crosstalk between adipokines and myokines in fat browning. Acta Physiol. 2017, 219, 362–381. [Google Scholar] [CrossRef]
- Böstrom, P.; Wu, J.; Jedrychowski, M.P.; Korde, A.; Ye, L.; Lo, J.C.; Rasbach, K.A.; Bostrom, E.A.; Choi, J.H.; Long, J.Z.; et al. A PGC1-α-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature 2012, 481, 463–468. [Google Scholar] [CrossRef]
- Pedersen, B.K. Physical activity and muscle–brain crosstalk. Nat. Rev. Endocrinol. 2019, 15, 383–392. [Google Scholar] [CrossRef]
- Sáinz, N.; Rodríguez, A.; Catalán, V.; Becerril, S.; Ramírez, B.; Gómez-Ambrosi, J.; Frühbeck, G. Leptin Administration Favors Muscle Mass Accretion by Decreasing FoxO3a and Increasing PGC-1α in ob/ob Mice. PLoS ONE 2009, 4, e6808. [Google Scholar] [CrossRef]
- Gutierrez-Repiso, C.; Garcia-Serrano, S.; Rodriguez-Pacheco, F.; Garcia-Escobar, E.; Haro-Mora, J.J.; Garcia-Arnes, J.; Valdés, S.; Gonzalo, M.; Soriguer, F.; Moreno-Ruiz, F.J.; et al. FNDC5 could be regulated by leptin in adipose tissue. Eur. J. Clin. Investig. 2014, 44, 918–925. [Google Scholar] [CrossRef]
- Morley, J.E.; Flood, J.F. Evidence that nitric oxide modulates food intake in mice. Life Sci. 1991, 49, 707–711. [Google Scholar] [CrossRef]
- Morley, J.E.; Kumar, V.B.; Mattammal, M.; Villareal, D.T. Measurement of nitric oxide synthase and its mRNA in genetically obese (ob/ob) mice. Life Sci. 1995, 57, 1327–1331. [Google Scholar] [CrossRef]
- Ueta, Y.; Levy, A.; Chowdrey, H.S.; Lightman, S.L. Inhibition of Hypothalamic Nitric Oxide Synthase Gene Expression in the Rat Paraventricular Nucleus by Food Deprivation is Independent of Serotonin Depletion. J. Neuroendocr. 1995, 7, 861–865. [Google Scholar] [CrossRef]
- Nisoli, E. Inducible Nitric Oxide Synthase in Rat Brown Adipocytes: Implications for Blood Flow to Brown Adipose Tissue. Endocrinology 1997, 138, 676–682. [Google Scholar] [CrossRef]
- Nagashima, T.; Ohinata, H.; Kuroshima, A. Involvement of nitric oxide in noradrenaline-induced increase in blood flow through brown adipose tissue. Life Sci. 1994, 54, 17–25. [Google Scholar] [CrossRef]
- Giordano, A.; Tonello, C.; Bulbarelli, A.; Cozzi, V.; Cinti, S.; Carruba, M.O.; Nisoli, E. Evidence for a functional nitric oxide synthase system in brown adipocyte nucleus. FEBS Lett. 2002, 514, 135–140. [Google Scholar] [CrossRef]
- Gómez-Ambrosi, J.; Becerril, S.; Oroz, P.; Zabalza, S.; Rodríguez, A.; Muruzabal, F.J.; Archanco, M.; Gil, M.J.; Burrell, M.A.; Frühbeck, G. Reduced adipose tissue mass and hypoleptinemia in iNOS deficient mice: Effect of LPS on plasma leptin and adiponectin concentrations. FEBS Lett. 2004, 577, 351–356. [Google Scholar]
- Merial, C.; Bouloumie, A.; Trocheris, V.; Lafontan, M.; Galitzky, J. Nitric oxide-dependent downregulation of adipocyte UCP-2 expression by tumor necrosis factor-α. Am. J. Physiol. Physiol. 2000, 279. [Google Scholar] [CrossRef]
- Morley, J.E.; Alshaher, M.M.; Farr, S.A.; Flood, J.F.; Kumar, V.B. Leptin and neuropeptide Y (NPY) modulate nitric oxide synthase: Further evidence for a role of nitric oxide in feeding. Peptides 1999, 20, 595–600. [Google Scholar] [CrossRef]
- Donato, J.; Frazão, R.; Fukuda, M.; Vianna, C.R.; Elias, C.F. Leptin Induces Phosphorylation of Neuronal Nitric Oxide Synthase in Defined Hypothalamic Neurons. Endocrinology 2010, 151, 5415–5427. [Google Scholar] [CrossRef]
- Calapi, G.; Corica, F.; Corsonello, A.; Sautebin, L.; Di Rosa, M.; Campo, G.M.; Buemi, M.; Mauro, V.N.; Caputi, A.P. Leptin increases serotonin turnover by inhibition of brain nitric oxide synthesis. J. Clin. Investig. 1999, 104, 975–982. [Google Scholar] [CrossRef]
- Leshan, R.L.; Greenwald-Yarnell, M.; Patterson, C.M.; Gonzalez, I.E.; Myers, M.G. Leptin action through hypothalamic nitric oxide synthase-1-expressing neurons controls energy balance. Nat. Med. 2012, 18, 820–823. [Google Scholar] [CrossRef]
- Becerril, S.; Rodríguez, A.; Catalán, V.; Sáinz, N.; Ramírez, B.; Gómez-Ambrosi, J.; Frühbeck, G. Transcriptional analysis of brown adipose tissue in leptin-deficient mice lacking inducible nitric oxide synthase: Evidence of the role of Med1 in energy balance. Physiol. Genom. 2012, 44, 678–688. [Google Scholar] [CrossRef]
- Frühbeck, G.; Salvador, J. Relation between leptin and the regulation of glucose metabolism. Diabetologia 2000, 43, 3–12. [Google Scholar] [CrossRef][Green Version]
- Harvey, J.; Ashford, M. Leptin in the CNS: Much more than a satiety signal. Neuropharmacology 2003, 44, 845–854. [Google Scholar] [CrossRef]
- Coppari, R.; Ichinose, M.; Lee, C.E.; Pullen, A.E.; Kenny, C.D.; McGovern, R.A.; Tang, V.; Liu, S.M.; Ludwig, T.; Chua, S.C.; et al. The hypothalamic arcuate nucleus: A key site for mediating leptin’s effects on glucose homeostasis and locomotor activity. Cell Metab. 2005, 1, 63–72. [Google Scholar] [CrossRef]
- Shiuchi, T.; Nakagami, H.; Iwai, M.; Takeda, Y.; Cui, T.-X.; Chen, R.; Minokoshi, Y.; Horiuchi, M. Involvement of Bradykinin and Nitric Oxide in Leptin-Mediated Glucose Uptake in Skeletal Muscle. Endocrinology 2001, 142, 608–612. [Google Scholar] [CrossRef]
- Sáinz, N.; Rodríguez, A.; Catalán, V.; Becerril, S.; Ramírez, B.; Lancha, A.; Burgos-Ramos, E.; Gómez-Ambrosi, J.; Frühbeck, G. Leptin Reduces the Expression and Increases the Phosphorylation of the Negative Regulators of GLUT4 Traffic TBC1D1 and TBC1D4 in Muscle of ob/ob Mice. PLoS ONE 2012, 7, e29389. [Google Scholar] [CrossRef]
- D’Souza, A.M.; Neumann, U.H.; Glavas, M.M.; Kieffer, T.J. The glucoregulatory actions of leptin. Mol. Metab. 2017, 6, 1052–1065. [Google Scholar] [CrossRef]
- Gómez-Ambrosi, J.; Catalán, V.; Frühbeck, G. The adipo-hepato-insular axis in glucose homeostasis. In Peptides in Energy Balance and Obesity; CAB International Oxfordshire: Wallingford, UK, 2009; pp. 163–193. [Google Scholar]
- Murakami, T.; Yamashita, T.; Iida, M.; Kuwajima, M.; Shima, K. A Short Form of Leptin Receptor Performs Signal Transduction. Biochem. Biophys. Res. Commun. 1997, 231, 26–29. [Google Scholar] [CrossRef]
- Tudurí, E.; Marroquí, L.; Soriano, S.; Ropero, A.B.; Batista, T.M.; Piquer, S.; López-Boado, M.A.; Carneiro, E.M.; Gomis, R.; Nadal, A.; et al. Inhibitory Effects of Leptin on Pancreatic α-Cell Function. Diabetes 2009, 58, 1616–1624. [Google Scholar] [CrossRef]
- Frühbeck, G.; López, M.; Diéguez, C. Role of caveolins in body weight and insulin resistance regulation. Trends Endocrinol. Metab. 2007, 18, 177–182. [Google Scholar] [CrossRef]
- Catalán, V.; Gómez-Ambrosi, J.; Rodríguez, A.; Silva, C.; Rotellar, F.; Gil, M.J.; Cienfuegos, J.A.; Salvador, J.; Frühbeck, G. Expression of caveolin-1 in human adipose tissue is upregulated in obesity and obesity-associated type 2 diabetes mellitus and related to inflammation. Clin. Endocrinol. 2008, 68, 213–219. [Google Scholar] [CrossRef]
- Morishita, T.; Tsutsui, M.; Shimokawa, H.; Sabanai, K.; Tasaki, H.; Suda, O.; Nakata, S.; Tanimoto, A.; Wang, K.-Y.; Ueta, Y.; et al. Nephrogenic diabetes insipidus in mice lacking all nitric oxide synthase isoforms. Proc. Natl. Acad. Sci. USA 2005, 102, 10616–10621. [Google Scholar] [CrossRef]
- Perreault, M.; Marette, A. Targeted disruption of inducible nitric oxide synthase protects against obesity-linked insulin resistance in muscle. Nat. Med. 2001, 7, 1138–1143. [Google Scholar] [CrossRef]
- Fujimoto, M.; Shimizu, N.; Kunii, K.; Martyn, J.J.; Ueki, K.; Kaneki, M.; Martyn, J.J. A Role for iNOS in Fasting Hyperglycemia and Impaired Insulin Signaling in the Liver of Obese Diabetic Mice. Diabetes 2005, 54, 1340–1348. [Google Scholar] [CrossRef]
- Shimabukuro, M.; Ohneda, M.; Lee, Y.; Unger, R.H. Role of nitric oxide in obesity-induced beta cell disease. J. Clin. Investig. 1997, 100, 290–295. [Google Scholar] [CrossRef]
- Frühbeck, G.; Aguado, M.; Martınez, J.; Martínez, J. In VitroLipolytic Effect of Leptin on Mouse Adipocytes: Evidence for a Possible Autocrine/Paracrine Role of Leptin. Biochem. Biophys. Res. Commun. 1997, 240, 590–594. [Google Scholar] [CrossRef]
- Frühbeck, G.; Aguado, M.; Gómez-Ambrosi, J.; Martínez, J.A. Lipolytic effect of in vivo leptin administration on adipocytes of lean and ob/ob mice, but not db/db mice. Biochem. Biophys. Res. Commun. 1998, 250, 99–102. [Google Scholar] [CrossRef]
- Frühbeck, G. Leptin-induced lipolysis opposes the tonic inhibition of endogenous adenosine in white adipocytes. FASEB J. 2001, 15, 333–340. [Google Scholar] [CrossRef]
- Frühbeck, G.; Gómez-Ambrosi, J. Modulation of the leptin-induced white adipose tissue lipolysis by nitric oxide. Cell. Signal. 2001, 13, 827–833. [Google Scholar] [CrossRef]
- Harris, R.B. Direct and indirect effects of leptin on adipocyte metabolism. Biochim. Biophys. Acta 2014, 1842, 414–423. [Google Scholar] [CrossRef]
- Ishikawa, Y.; Otsu, K.; Oshikawa, J. Caveolin; different roles for insulin signal? Cell. Signal. 2005, 17, 1175–1182. [Google Scholar] [CrossRef]
- Rodríguez, A.; Moreno, N.R.; Balaguer, I.; Méndez-Giménez, L.; Becerril, S.; Catalán, V.; Gómez-Ambrosi, J.; Portincasa, P.; Calamita, G.; Soveral, G.; et al. Leptin administration restores the altered adipose and hepatic expression of aquaglyceroporins improving the non-alcoholic fatty liver of ob/ob mice. Sci. Rep. 2015, 5, 12067. [Google Scholar] [CrossRef]
- Mallafré, V.C.; Miranda, M.; Chacon, M.R.; Vilarrasa, N.; Megia, A.; Gutiérrez, C.; Fernández-Real, J.M.; Gómez, J.M.; Caubet, E.; Frühbeck, G.; et al. Adipose Tissue Expression of the Glycerol Channel Aquaporin-7 Gene Is Altered in Severe Obesity But Not in Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2007, 92, 3640–3645. [Google Scholar]
- Frühbeck, G. Obesity: Aquaporin enters the picture. Nature 2005, 438, 436–437. [Google Scholar] [CrossRef]
- Unger, R.H.; Orci, L. Lipotoxic diseases of nonadipose tissues in obesity. Int. J. Obes. Relat. Metab. Disord. 2000, 24 (Suppl. 4), S28–S32. [Google Scholar] [CrossRef]
- Ribière, C.; Jaubert, A.; Gaudiot, N.; Sabourault, D.; Marcus, M.; Boucher, J.; Denis-Henriot, D.; Giudicelli, Y. White Adipose Tissue Nitric Oxide Synthase: A Potential Source for NO Production. Biochem. Biophys. Res. Commun. 1996, 222, 706–712. [Google Scholar] [CrossRef]
- Gaudiot, N.; Jaubert, A.-M.; Charbonnier, E.; Sabourault, D.; Lacasa, D.; Giudicelli, Y.; Ribière, C. Modulation of White Adipose Tissue Lipolysis by Nitric Oxide. J. Boil. Chem. 1998, 273, 13475–13481. [Google Scholar] [CrossRef]
- Adam, L.; Bouvier, M.; Jones, T.L.Z. Nitric Oxide Modulates 2-Adrenergic Receptor Palmitoylation and Signaling. J. Boil. Chem. 1999, 274, 26337–26343. [Google Scholar] [CrossRef]
- Klatt, P.; Cacho, J.; Crespo, M.D.; Herrera, E.; Ramos, P. Nitric oxide inhibits isoproterenol-stimulated adipocyte lipolysis through oxidative inactivation of the β-agonist. Biochem. J. 2000, 351, 485. [Google Scholar] [CrossRef]
- Nisoli, E.; Clementi, E.; Paolucci, C.; Cozzi, V.; Tonello, C.; Sciorati, C.; Bracale, R.; Valerio, A.; Francolini, M.; Moncada, S.; et al. Mitochondrial Biogenesis in Mammals: The Role of Endogenous Nitric Oxide. Science 2003, 299, 896–899. [Google Scholar] [CrossRef]
- Penfornis, P.; Marette, A. Inducible nitric oxide synthase modulates lipolysis in adipocytes. J. Lipid Res. 2005, 46, 135–142. [Google Scholar] [CrossRef]
- Greif, D.M.; Kou, R.; Michel, T. Site-Specific Dephosphorylation of Endothelial Nitric Oxide Synthase by Protein Phosphatase 2A: Evidence for Crosstalk between Phosphorylation Sites†. Biochemistry 2002, 41, 15845–15853. [Google Scholar] [CrossRef]
- Winder, W.W.; Hardie, D.G. Inactivation of acetyl-CoA carboxylase and activation of AMP-activated protein kinase in muscle during exercise. Am. J. Physiol. Metab. 1996, 270, E299–E304. [Google Scholar] [CrossRef]
- Schild, L.; Jaroscakova, I.; Lendeckel, U.; Wolf, G.; Keilhoff, G. Neuronal nitric oxide synthase controls enzyme activity pattern of mitochondria and lipid metabolism. FASEB J. 2006, 20, 145–147. [Google Scholar] [CrossRef]
- White, V.; González, E.; Capobianco, E.; Pustovrh, C.; Martínez, N.; Higa, R.; Baier, M.; Jawerbaum, A. Leptin modulates nitric oxide production and lipid metabolism in human placenta. Reprod. Fertil. Dev. 2006, 18, 425–432. [Google Scholar] [CrossRef]
- Vona-Davis, L.; Rose, D.P. Angiogenesis, adipokines and breast cancer. Cytokine Growth Factor Rev. 2009, 20, 193–201. [Google Scholar] [CrossRef]
- Newman, G.; Gonzalez-Perez, R.R. Leptin-cytokine crosstalk in breast cancer. Mol. Cell. Endocrinol. 2014, 382, 570–582. [Google Scholar] [CrossRef]
- Gómez-Ambrosi, J.; Catalán, V.; Rodríguez, A.; Ramírez, B.; Silva, C.; Gil, M.J.; Salvador, J.; Frühbeck, G. Involvement of serum vascular endothelial growth factor family members in the development of obesity in mice and humans☆. J. Nutr. Biochem. 2010, 21, 774–780. [Google Scholar] [CrossRef]
- Nwadozi, E.; Ng, A.; Stromberg, A.; Liu, H.Y.; Olsson, K.; Gustafsson, T.; Haas, T.L. Leptin is a physiological regulator of skeletal muscle angiogenesis and is locally produced by PDGFR and PDGFR expressing perivascular cells. Angiogenesis 2019, 22, 103–115. [Google Scholar] [CrossRef]
- Cao, R.; Brakenhielm, E.; Wahlestedt, C.; Thyberg, J.; Cao, Y. Leptin induces vascular permeability and synergistically stimulates angiogenesis with FGF-2 and VEGF. Proc. Natl. Acad. Sci. USA 2001, 98, 6390–6395. [Google Scholar] [CrossRef]
- Park, H.-Y.; Kwon, H.M.; Lim, H.J.; Hong, B.K.; Lee, J.Y.; Park, B.E.; Jang, Y.S.; Cho, S.Y.; Kim, H.-S. Potential role of leptin in angiogenesis: Leptin induces endothelial cell proliferation and expression of matrix metalloproteinases in vivo and in vitro. Exp. Mol. Med. 2001, 33, 95–102. [Google Scholar] [CrossRef]
- Ribatti, D.; Nico, B.; Belloni, A.S.; Vacca, A.; Roncali, L.; Nussdorfer, G.G. Angiogenic activity of leptin in the chick embryo chorioallantoic membrane is in part mediated by endogenous fibroblast growth factor-2. Int. J. Mol. Med. 2001, 8, 265–268. [Google Scholar] [CrossRef]
- Konturek, P.C.; Brzozowski, T.; Sulekova, Z.; Brzozowska, I.; Duda, A.; Meixner, H.; Hahn, E.G.; Konturek, S.J. Role of leptin in ulcer healing. Eur. J. Pharmacol. 2001, 414, 87–97. [Google Scholar] [CrossRef]
- Murad, A.; Nath, A.K.; Cha, S.T.; Demir, E.; Flores-Riveros, J.; Sierra-Honigmann, M.R. Leptin is an autocrine/paracrine regulator of wound healing. FASEB J. 2003, 17, 1895–1897. [Google Scholar] [CrossRef]
- Tadokoro, S.; Ide, S.; Tokuyama, R.; Umeki, H.; Tatehara, S.; Kataoka, S.; Satomura, K. Leptin Promotes Wound Healing in the Skin. PLoS ONE 2015, 10, e0121242. [Google Scholar] [CrossRef]
- Garonna, E.; Botham, K.M.; Birdsey, G.M.; Randi, A.M.; Gonzalez-Perez, R.R.; Wheeler-Jones, C.P.D. Vascular Endothelial Growth Factor Receptor-2 Couples Cyclo-Oxygenase-2 with Pro-Angiogenic Actions of Leptin on Human Endothelial Cells. PLoS ONE 2011, 6, e18823. [Google Scholar] [CrossRef]
- Bouloumie, A.; Marumo, T.; Lafontan, M.; Busse, R. Leptin induces oxidative stress in human endothelial cells. FASEB J. 1999, 13, 1231–1238. [Google Scholar] [CrossRef]
- Singhal, A.; Farooqi, I.S.; Cole, T.J.; O’Rahilly, S.; Fewtrell, M.; Kattenhorn, M.; Lucas, A.; Deanfield, J. Influence of leptin on arterial distensibility–A novel link between obesity and cardiovascular disease? Circulation 2002, 106, 1919–1924. [Google Scholar] [CrossRef]
- Hoffmann, A.; Ebert, T.; Klöting, N.; Dokas, J.; Jeromin, F.; Jessnitzer, B.; Burkhardt, R.; Fasshauer, M.; Kralisch, S. Leptin dose-dependently decreases atherosclerosis by attenuation of hypercholesterolemia and induction of adiponectin. BBA Mol. Basis Dis. 2016, 1862, 113–120. [Google Scholar] [CrossRef]
- Schinzari, F.; Tesauro, M.; Rovella, V.; Di Daniele, N.; Mores, N.; Veneziani, A.; Cardillo, C. Leptin Stimulates Both Endothelin-1 and Nitric Oxide Activity in Lean Subjects But Not in Patients With Obesity-Related Metabolic Syndrome. J. Clin. Endocrinol. Metab. 2013, 98, 1235–1241. [Google Scholar] [CrossRef]
- Hasty, A.H.; Shimano, H.; Osuga, J.-I.; Namatame, I.; Takahashi, A.; Yahagi, N.; Perrey, S.; Iizuka, Y.; Tamura, Y.; Amemiya-Kudo, M.; et al. Severe Hypercholesterolemia, Hypertriglyceridemia, and Atherosclerosis in Mice Lacking Both Leptin and the Low Density Lipoprotein Receptor. J. Boil. Chem. 2001, 276, 37402–37408. [Google Scholar] [CrossRef]
- Eckel, R.H.; Krauss, R.M. American Heart Association Call to Action: Obesity as a Major Risk Factor for Coronary Heart Disease. Circulation 1998, 97, 2099–2100. [Google Scholar] [CrossRef]
- Hildebrandt, D.A.; Kuo, J.; Hall, J.E. Obesity hypertension: Role of leptin and sympathetic nervous system. Am. J. Hypertens. 2001, 14, 103s–115s. [Google Scholar]
- Haynes, W.G.; Sivitz, W.I.; Morgan, D.A.; Walsh, S.A.; Mark, A.L. Sympathetic and Cardiorenal Actions of Leptin. Hypertension 1997, 30, 619–623. [Google Scholar] [CrossRef]
- Villarreal, D.; Reams, G.; Freeman, R.H. Effects of renal denervation on the sodium excretory actions of leptin in hypertensive rats. Kidney Int. 2000, 58, 989–994. [Google Scholar] [CrossRef][Green Version]
- Vecchione, C.; Maffei, A.; Colella, S.; Aretini, A.; Poulet, R.; Frati, G.; Gentile, M.T.; Fratta, L.; Trimarco, V.; Trimarco, B.; et al. Leptin Effect on Endothelial Nitric Oxide Is Mediated Through Akt-Endothelial Nitric Oxide Synthase Phosphorylation Pathway. Diabetes 2002, 51, 168–173. [Google Scholar] [CrossRef]
- Rodríguez, A.; Fortuño, A.; Gómez-Ambrosi, J.; Zalba, G.; Díez, J.; Frühbeck, G. The Inhibitory Effect of Leptin on Angiotensin II-Induced Vasoconstriction in Vascular Smooth Muscle Cells Is Mediated via a Nitric Oxide-Dependent Mechanism. Endocrinology 2007, 148, 324–331. [Google Scholar] [CrossRef]
- Papapetropoulos, A.; García-Cardeña, G.; Madri, J.A.; Sessa, W.C. Nitric oxide production contributes to the angiogenic properties of vascular endothelial growth factor in human endothelial cells. J. Clin. Investig. 1997, 100, 3131–3139. [Google Scholar] [CrossRef]
- Ridnour, L.A.; Isenberg, J.S.; Espey, M.G.; Thomas, D.D.; Roberts, D.D.; Wink, D.A. Nitric oxide regulates angiogenesis through a functional switch involving thrombospondin-1. Proc. Natl. Acad. Sci. USA 2005, 102, 13147–13152. [Google Scholar] [CrossRef]
- Efron, D.T.; Most, D.; Barbul, A. Role of nitric oxide in wound healing. Curr. Opin. Clin. Nutr. Metab. Care 2000, 3, 197–204. [Google Scholar] [CrossRef]
- Debats, I.; Wolfs, T.; Gotoh, T.; Cleutjens, J.; Peutz-Kootstra, C.; Van Der Hulst, R. Role of arginine in superficial wound healing in man. Nitric Oxide 2009, 21, 175–183. [Google Scholar] [CrossRef]
- Tsuda, K.; Kimura, K.; Nishio, I. Leptin improves membrane fluidity of erythrocytes in humans via a nitric oxide-dependent mechanism—an electron paramagnetic resonance investigation. Biochem. Biophys. Res. Commun. 2002, 297, 672–681. [Google Scholar] [CrossRef]
- Yan, Z.-Q.; Yokota, T.; Zhang, W.; Hansson, G.K. Expression of Inducible Nitric Oxide Synthase Inhibits Platelet Adhesion and Restores Blood Flow in the Injured Artery. Circ. Res. 1996, 79, 38–44. [Google Scholar] [CrossRef]
- Rikitake, Y.; Hirata, K.-I.; Kawashima, S.; Akita, H.; Yokoyama, M. Inhibitory effect of inducible type nitric oxide synthase on oxidative modification of low density lipoprotein by vascular smooth muscle cells. Atherosclerosis 1998, 136, 51–57. [Google Scholar] [CrossRef]
- Wilcox, C.S. Oxidative stress and nitric oxide deficiency in the kidney: A critical link to hypertension? Am. J. Physiol. Integr. Comp. Physiol. 2005, 289, R913–R935. [Google Scholar] [CrossRef]
- Sartori, C.; Lepori, M.; Scherrer, U. Interaction between nitric oxide and the cholinergic and sympathetic nervous system in cardiovascular control in humans. Pharmacol. Ther. 2005, 106, 209–220. [Google Scholar] [CrossRef]
- Shabeeh, H.; Khan, S.; Jiang, B.; Brett, S.; Melikian, N.; Casadei, B.; Chowienczyk, P.J.; Shah, A.M. Blood Pressure in Healthy Humans Is Regulated by Neuronal NO Synthase. Hypertension 2017, 69, 970–976. [Google Scholar] [CrossRef]
- Rodríguez, A.; Gómez-Ambrosi, J.; Catalán, V.; Fortuño, A.; Frühbeck, G. Leptin Inhibits the Proliferation of Vascular Smooth Muscle Cells Induced by Angiotensin II through Nitric Oxide-Dependent Mechanisms. Mediat. Inflamm. 2010, 2010, 1–10. [Google Scholar] [CrossRef]
- Benkhoff, S.; Loot, A.E.; Pierson, I.; Sturza, A.; Kohlstedt, K.; Fleming, I.; Shimokawa, H.; Grisk, O.; Brandes, R.P.; Schröder, K. Leptin Potentiates Endothelium-Dependent Relaxation by Inducing Endothelial Expression of Neuronal NO Synthase. Arter. Thromb. Vasc. Boil. 2012, 32, 1605–1612. [Google Scholar] [CrossRef]
- Nickola, M.W.; Wold, L.E.; Colligan, P.B.; Wang, G.-J.; Samson, W.K.; Ren, J. Leptin Attenuates Cardiac Contraction in Rat Ventricular Myocytes. Hypertension 2000, 36, 501–505. [Google Scholar] [CrossRef][Green Version]
- Villarreal, D.; Reams, G.; Samar, H.; Spear, R.; Freeman, R.H. Effects of Chronic Nitric Oxide Inhibition on the Renal Excretory Response to Leptin. Obes. Res. 2004, 12, 1006–1010. [Google Scholar] [CrossRef]
- Knudson, J.D.; Dincer, Ü.D.; Zhang, C.; Swafford, A.N.; Koshida, R.; Picchi, A.; Focardi, M.; Dick, G.M.; Tune, J.D. Leptin receptors are expressed in coronary arteries, and hyperleptinemia causes significant coronary endothelial dysfunction. Am. J. Physiol. Circ. Physiol. 2005, 289, H48–H56. [Google Scholar] [CrossRef]
- Sierra-Honigmann, M.R.; Nath, A.K.; Murakami, C.; García-Cardeña, G.; Papapetropoulos, A.; Sessa, W.C.; Madge, L.A.; Schechner, J.S.; Schwabb, M.B.; Polverini, P.J.; et al. Biological Action of Leptin as an Angiogenic Factor. Science 1998, 281, 1683–1686. [Google Scholar] [CrossRef]
- Rudic, R.D.; Papapetropoulos, A.; Sessa, W.C. Molecular control of nitric oxide synthases in the cardiovascular system. Cardiovasc. Res. 1999, 43, 509–520. [Google Scholar]
- Cao, Y. Angiogenesis modulates adipogenesis and obesity. J. Clin. Investig. 2007, 117, 2362–2368. [Google Scholar] [CrossRef]
- Becerril, S.; Rodríguez, A.; Catalán, V.; Méndez-Giménez, L.; Ramírez, B.; Sainz, N.; Llorente, M.; Unamuno, X.; Gómez-Ambrosi, J.; Frühbeck, G. Targeted disruption of the iNOS gene improves adipose tissue inflammation and fibrosis in leptin-deficient ob/ob mice: Role of tenascin C. Int. J. Obes. 2018, 42, 1458–1470. [Google Scholar] [CrossRef]
- Gordeladze, J.O.; Drevon, C.A.; Syversen, U.; Reseland, J.E. Leptin stimulates human osteoblastic cell proliferation, de novo collagen synthesis, and mineralization: Impact on differentiation markers, apoptosis, and osteoclastic signaling. J. Cell. Biochem. 2002, 85, 825–836. [Google Scholar] [CrossRef]
- Cornish, J.; Callon, K.; Bava, U.; Lin, C.; Naot, D.; Hill, B.; Grey, A.; Broom, N.; Myers, D.; Nicholson, G.; et al. Leptin directly regulates bone cell function in vitro and reduces bone fragility in vivo. J. Endocrinol. 2002, 175, 405–415. [Google Scholar] [CrossRef]
- Takeda, S.; Elefteriou, F.; Levasseur, R.; Liu, X.; Zhao, L.; Parker, K.L.; Armstrong, D.; Ducy, P.; Karsenty, G. Leptin Regulates Bone Formation via the Sympathetic Nervous System. Cell 2002, 111, 305–317. [Google Scholar] [CrossRef]
- Karsenty, G. Convergence between bone and energy homeostases: Leptin regulation of bone mass. Cell Metab. 2006, 4, 341–348. [Google Scholar] [CrossRef]
- Ducy, P.; Amling, M.; Takeda, S.; Priemel, M.; Schilling, A.F.; Beil, F.T.; Shen, J.; Vinson, C.; Rueger, J.M.; Karsenty, G. Leptin inhibits bone formation through a hypothalamic relay: A central control of bone mass. Cell 2000, 100, 197–207. [Google Scholar] [CrossRef]
- Dimitri, P. The Impact of Childhood Obesity on Skeletal Health and Development. J. Obes. Metab. Syndr. 2019, 28, 4–17. [Google Scholar] [CrossRef]
- Chae, H.-J.; Park, R.-K.; Chung, H.-T.; Kang, J.-S.; Kim, M.-S.; Choi, D.-Y.; Bang, B.-G.; Kim, H.-R. Nitric Oxide is a Regulator of Bone Remodelling. J. Pharm. Pharmacol. 1997, 49, 897–902. [Google Scholar] [CrossRef]
- Armour, K.E.; Van’T Hof, R.J.; Grabowski, P.S.; Reid, D.M.; Ralston, S.H. Evidence for a Pathogenic Role of Nitric Oxide in Inflammation-Induced Osteoporosis. J. Bone Miner. Res. 1999, 14, 2137–2142. [Google Scholar] [CrossRef]
- van’t Hof, R.J.; Armour, K.J.; Smith, L.M.; Armour, K.E.; Wei, X.Q.; Liew, F.Y.; Ralston, S.H. Requirement of the inducible nitric oxide synthase pathway for IL-1-induced osteoclastic bone resorption. Proc. Natl. Acad. Sci. USA 2000, 97, 7993–7998. [Google Scholar] [CrossRef]
- van’t Hof, R.J.; Ralston, S.H. Cytokine-Induced Nitric Oxide Inhibits Bone Resorption by Inducing Apoptosis of Osteoclast Progenitors and Suppressing Osteoclast Activity. J. Bone Miner. Res. 1997, 12, 1797–1804. [Google Scholar] [CrossRef]
- van’t Hof, R.J.; Ralston, S.H. Nitric oxide and bone. Immunology 2001, 103, 255–261. [Google Scholar] [CrossRef]
- Baratta, M.; Saleri, R.; Mainardi, G.L.; Valle, D.; Giustina, A.; Tamanini, C. Leptin Regulates GH Gene Expression and Secretion and Nitric Oxide Production in Pig Pituitary Cells. Endocrinology 2002, 143, 551–557. [Google Scholar] [CrossRef]
- Rodríguez-Pacheco, F.; Luque, R.M.; Tena-Sempere, M.; Malagón, M.M.; Castaño, J.P. Ghrelin induces growth hormone secretion via a nitric oxide/cGMP signalling pathway. J. Neuroendocrinol. 2008, 20, 406–412. [Google Scholar] [CrossRef]
- Lagumdzija, A.; Ou, G.; Petersson, M.; Bucht, E.; Gonon, A.; Pernow, Y. Inhibited anabolic effect of insulin-like growth factor-I on stromal bone marrow cells in endothelial nitric oxide synthase-knockout mice. Acta Physiol. Scand. 2004, 182, 29–35. [Google Scholar] [CrossRef]
- Caprio, M.; Fabbrini, E.; Isidori, A.M.; Aversa, A.; Fabbri, A. Leptin in reproduction. Trends Endocrinol. Metab. 2001, 12, 65–72. [Google Scholar] [CrossRef]
- Brann, D.W.; Wade, M.F.; Dhandapani, K.M.; Mahesh, V.B.; Buchanan, C.D. Leptin and reproduction. Steroids 2002, 67, 95–104. [Google Scholar] [CrossRef]
- Archanco, M.; Muruzábal, F.J.; Llopiz, D.; Garayoa, M.; Gómez-Ambrosi, J.; Frühbeck, G.; Burrell, M.A. Leptin expression in the rat ovary depends on estrous cycle. J. Histochem. Cytochem. 2003, 51, 1269–1277. [Google Scholar] [CrossRef]
- Ashworth, C.J.; Hoggard, N.; Thomas, L.; Mercer, J.G.; Wallace, J.M.; Lea, R.G. Placental leptin. Rev. Reprod. 2000, 5, 18–24. [Google Scholar] [CrossRef]
- McCann, S.M.; Haens, G.; Mastronardi, C.; Walczewska, A.; Karanth, S.; Rettori, V.; Yu, W.H. The role of nitric oxide (NO) in control of LHRH release that mediates gonadotropin release and sexual behavior. Curr Pharm Design 2003, 9, 381–390. [Google Scholar] [CrossRef]
- Klein, S.L.; Carnovale, D.; Burnett, A.L.; Wallach, E.E.; Zacur, H.A.; Crone, J.K.; Dawson, V.L.; Nelson, R.J.; Dawson, T.M. Impaired Ovulation in Mice with Targeted Deletion of the Neuronal Isoform of Nitric Oxide Synthase. Mol. Med. 1998, 4, 658–664. [Google Scholar] [CrossRef]
- Chun, S.Y. Interleukin-1 beta suppresses apoptosis in rat ovarian follicles by increasing nitric oxide production. Endocrinology 1995, 136, 3120–3127. [Google Scholar] [CrossRef]
- Jablonka-Shariff, A. The Role of Nitric Oxide in Oocyte Meiotic Maturation and Ovulation: Meiotic Abnormalities of Endothelial Nitric Oxide Synthase Knock-Out Mouse Oocytes. Endocrinology 1998, 139, 2944–2954. [Google Scholar] [CrossRef]
- Purcell, T.; Given, R.; Garfield, R.; Chwalisz, K. Nitric oxide synthase distribution during implantation in the mouse. Mol. Hum. Reprod. 1999, 5, 467–475. [Google Scholar] [CrossRef]
- Burnett, T.; Tash, J.; Hunt, J. Investigation of the role of nitric oxide synthase 2 in pregnancy using mutant mice. Reproduction 2002, 124, 49–57. [Google Scholar] [CrossRef]
- Lee, N.P.; Cheng, C.Y. Nitric Oxide/Nitric Oxide Synthase, Spermatogenesis, and Tight Junction Dynamics1. Boil. Reprod. 2004, 70, 267–276. [Google Scholar] [CrossRef]
- Quennell, J.H.; Mulligan, A.C.; Tups, A.; Liu, X.; Phipps, S.J.; Kemp, C.J.; Herbison, A.E.; Grattan, D.R.; Anderson, G.M. Leptin Indirectly Regulates Gonadotropin-Releasing Hormone Neuronal Function. Endocrinology 2009, 150, 2805–2812. [Google Scholar] [CrossRef]
- Szwarcfarb, B.; Ponzo, O.; Moguilevsky, J.A.; Scacchi, P.; Reynoso, R.; Cardoso, N.; Carbone, S. Nitric Oxide Synthase Inhibition Prevents Leptin Induced Gn-RH Release in Prepubertal and Peripubertal Female Rats. Exp. Clin. Endocrinol. Diabetes 2007, 115, 423–427. [Google Scholar]
- Lampiao, F.; Du Plessis, S.S. Insulin and leptin enhance human sperm motility, acrosome reaction and nitric oxide production. Asian J. Androl. 2008, 10, 799–807. [Google Scholar] [CrossRef]
- Lord, G.M.; Matarese, G.; Howard, J.K.; Baker, R.J.; Bloom, S.R.; Lechler, R.I. Leptin modulates the T-cell immune response and reverses starvation-induced immunosuppression. Nature 1998, 394, 897–901. [Google Scholar] [CrossRef]
- Faggioni, R.; Fantuzzi, G.; Gabay, C.; Moser, A.; Dinarello, C.A.; Feingold, K.R.; Grunfeld, C. Leptin deficiency enhances sensitivity to endotoxin-induced lethality. Am. J. Physiol. Integr. Comp. Physiol. 1999, 276, R136–R142. [Google Scholar] [CrossRef]
- Cioffi, J.A.; Shafer, A.W.; Zupancic, T.J.; Smith-Gbur, J.; Mikhail, A.; Platika, D.; Snodgrass, H.R. Novel B219/OB receptor isoforms: Possible role of leptin in hematopoiesis and reproduction. Nat. Med. 1996, 2, 585–589. [Google Scholar] [CrossRef]
- Gainsford, T.; Willson, T.A.; Metcalf, D.; Handman, E.; McFarlane, C.; Ng, A.; Nicola, N.A.; Alexander, W.S.; Hilton, D.J. Leptin can induce proliferation, differentiation, and functional activation of hemopoietic cells. Proc. Natl. Acad. Sci. USA 1996, 93, 14564–14568. [Google Scholar] [CrossRef]
- Bennett, B.D.; Solar, G.P.; Yuan, J.Q.; Mathias, J.; Thomas, G.; Matthews, W. A role for leptin and its cognate receptor in hematopoiesis. Curr. Boil. 1996, 6, 1170–1180. [Google Scholar] [CrossRef]
- Mancuso, P.; Gottschalk, A.; Phare, S.M.; Peters-Golden, M.; Lukacs, N.W.; Huffnagle, G.B. Leptin-Deficient Mice Exhibit Impaired Host Defense in Gram-Negative Pneumonia. J. Immunol. 2002, 168, 4018–4024. [Google Scholar] [CrossRef]
- Dixit, V.D.; Mielenz, M.; Taub, D.D.; Parvizi, N. Leptin Induces Growth Hormone Secretion from Peripheral Blood Mononuclear Cells via a Protein Kinase C- and Nitric Oxide-Dependent Mechanism. Endocrinology 2003, 144, 5595–5603. [Google Scholar] [CrossRef]
- Caldefie-Chezet, F.; Poulin, A.; Tridon, A.; Sion, B.; Vasson, M.P. Leptin: A potential regulator of polymorphonuclear neutrophil bactericidal action? J. Leukoc. Boil. 2001, 69, 414–418. [Google Scholar]
- Tian, Z.; Sun, R.; Wei, H.; Gao, B. Impaired natural killer (NK) cell activity in leptin receptor deficient mice: Leptin as a critical regulator in NK cell development and activation. Biochem. Biophys. Res. Commun. 2002, 298, 297–302. [Google Scholar] [CrossRef]
- Lam, Q.L.; Liu, S.; Cao, X.; Lu, L. Involvement of leptin signaling in the survival and maturation of bone marrow-derived dendritic cells. Eur. J. Immunol. 2006, 36, 3118–3130. [Google Scholar] [CrossRef]
- La Cava, A.; Matarese, G. The weight of leptin in immunity. Nat. Rev. Immunol. 2004, 4, 371–379. [Google Scholar] [CrossRef]
- Matarese, G.; Moschos, S.; Mantzoros, C.S. Leptin in immunology. J. Immunol. 2005, 174, 3137–3142. [Google Scholar] [CrossRef]
- Bogdan, C. Nitric oxide and the immune response. Nat. Immunol. 2001, 2, 907–916. [Google Scholar] [CrossRef]
- Michurina, T.; Krasnov, P.; Balazs, A.; Nakaya, N.; Vasilieva, T.; Kuzin, B.; Khrushchov, N.; Mulligan, R.C.; Enikolopov, G. Nitric Oxide Is a Regulator of Hematopoietic Stem Cell Activity. Mol. Ther. 2004, 10, 241–248. [Google Scholar] [CrossRef]
- Krasnov, P.; Michurina, T.; Packer, M.A.; Stasiv, Y.; Nakaya, N.; Moore, K.A.; Drazan, K.E.; Enikolopov, G. Neuronal nitric oxide synthase contributes to the regulation of hematopoiesis. Mol. Med. 2008, 14, 141–149. [Google Scholar] [CrossRef]
- Aicher, A.; Heeschen, C.; Mildner-Rihm, C.; Urbich, C.; Ihling, C.; Technau-Ihling, K.; Zeiher, A.M.; Dimmeler, S. Essential role of endothelial nitric oxide synthase for mobilization of stem and progenitor cells. Nat. Med. 2003, 9, 1370–1376. [Google Scholar] [CrossRef]
- MacMicking, J.; Xie, Q.-W.; Nathan, C. NITRIC OXIDE AND MACROPHAGE FUNCTION. Annu. Rev. Immunol. 1997, 15, 323–350. [Google Scholar] [CrossRef]
- Coleman, J.W. Nitric oxide in immunity and inflammation. Int. Immunopharmacol. 2001, 1, 1397–1406. [Google Scholar] [CrossRef]
- Bredt, D.S.; Snyder, S.H. Nitric Oxide: A Physiologic Messenger Molecule. Annu. Rev. Biochem. 1994, 63, 175–195. [Google Scholar] [CrossRef]
- Brunet, L.R. Nitric oxide in parasitic infections. Int. Immunopharmacol. 2001, 1, 1457–1467. [Google Scholar] [CrossRef]
- Niedbala, W.; Wei, X.-Q.; Campbell, C.; Thomson, D.; Komai-Koma, M.; Liew, F.Y. Nitric oxide preferentially induces type 1 T cell differentiation by selectively up-regulating IL-12 receptor β2 expression via cGMP. Proc. Natl. Acad. Sci. USA 2002, 99, 16186–16191. [Google Scholar] [CrossRef]
- Vladutiu, A.O. Role of Nitric Oxide in Autoimmunity. Clin. Immunol. Immunopathol. 1995, 76, 1–11. [Google Scholar] [CrossRef]
- Kolb, H.; Kolb-Bachofen, V. Nitric oxide in autoimmune disease: Cytotoxic or regulatory mediator? Immunol. Today 1998, 19, 556–561. [Google Scholar] [CrossRef]
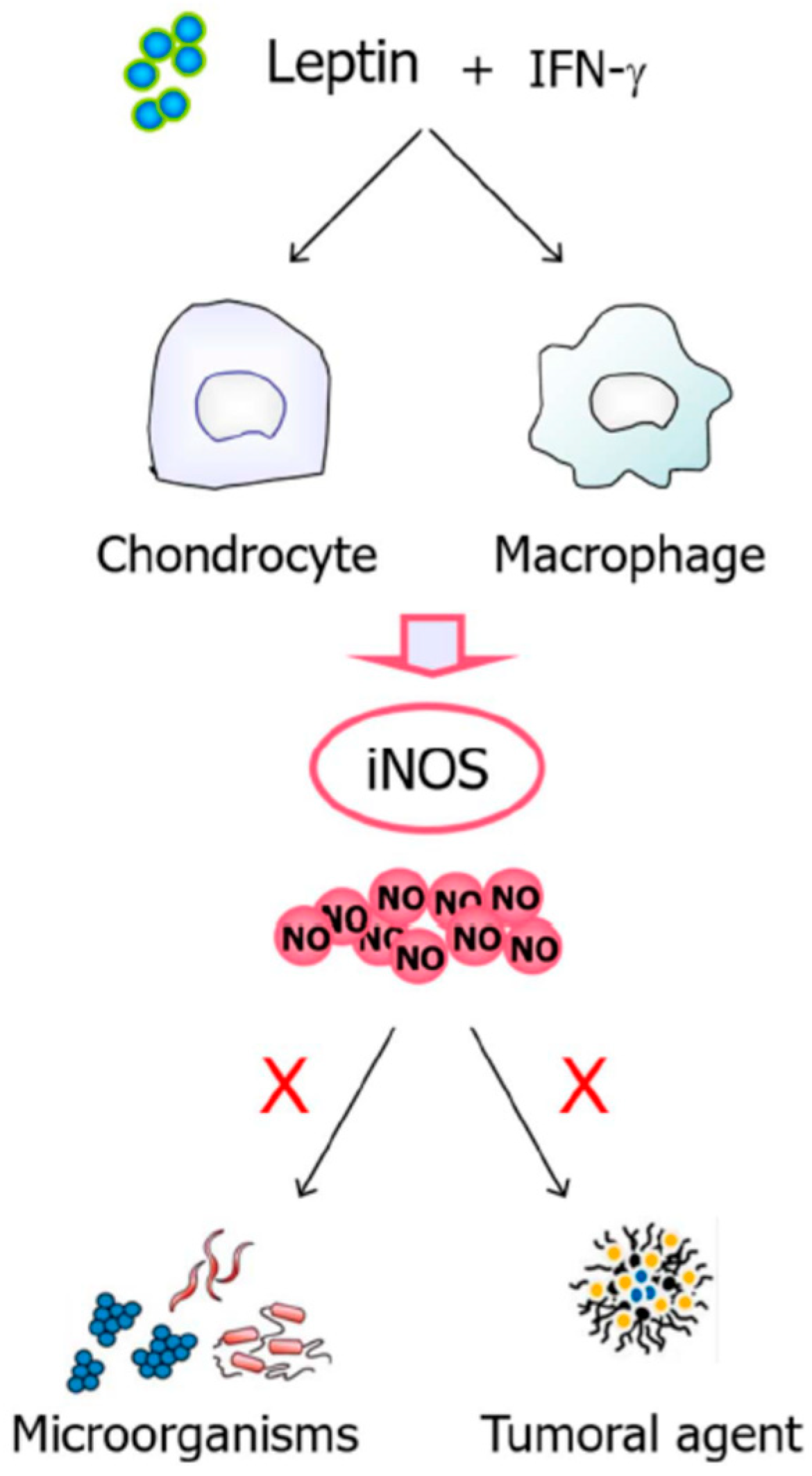
- Raso, G.M.; Pacilio, M.; Esposito, E.; Coppola, A.; Di Carlo, R.; Meli, R. Leptin potentiates IFN-γ-induced expression of nitric oxide synthase and cyclo-oxygenase-2 in murine macrophage J774A.1. Br. J. Pharmacol. 2002, 137, 799–804. [Google Scholar] [CrossRef]
- Otero, M.; Lago, R.; Gómez, R.; Lago, F.; Gómez-Reino, J.J.; Gualillo, O. Phosphatidylinositol 3-kinase, MEK-1 and p38 mediate leptin/interferon-gamma synergistic NOS type II induction in chondrocytes. Life Sci. 2007, 81, 1452–1460. [Google Scholar] [CrossRef]
- Lawler, H.M.; Underkofler, C.M.; Kern, P.A.; Erickson, C.; Bredbeck, B.; Rasouli, N. Adipose Tissue Hypoxia, Inflammation, and Fibrosis in Obese Insulin-Sensitive and Obese Insulin-Resistant Subjects. J. Clin. Endocrinol. Metab. 2016, 101, 1422–1428. [Google Scholar] [CrossRef]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Investig. 2003, 112, 1796–1808. [Google Scholar] [CrossRef]
- Catalán, V.; Gómez-Ambrosi, J.; Rodríguez, A.; Ramírez, B.; Rotellar, F.; Valentí, V.; Silva, C.; Gil, M.J.; Salvador, J.; Frühbeck, G. Increased Tenascin C And Toll-Like Receptor 4 Levels in Visceral Adipose Tissue as a Link between Inflammation and Extracellular Matrix Remodeling in Obesity. J. Clin. Endocrinol. Metab. 2012, 97, E1880–E1889. [Google Scholar] [CrossRef]
- Chatterjee, S.; Ganini, D.; Tokar, E.J.; Kumar, A.; Das, S.; Corbett, J.; Kadiiska, M.B.; Waalkes, M.P.; Diehl, A.M.; Mason, R.P. Leptin is key to peroxynitrite-mediated oxidative stress and Kupffer cell activation in experimental non-alcoholic steatohepatitis. J. Hepatol. 2013, 58, 778–784. [Google Scholar] [CrossRef]
- Becerril, S.; Rodríguez, A.; Catalán, V.; Ramírez, B.; Unamuno, X.; Gómez-Ambrosi, J.; Frühbeck, G. iNOS Gene Ablation Prevents Liver Fibrosis in Leptin-Deficient ob/ob Mice. Genes 2019, 10, 184. [Google Scholar] [CrossRef]
- Vuolteenaho, K.; Koskinen, A.; Kukkonen, M.; Nieminen, R.; Paivarinta, U.; Moilanen, T.; Moilanen, E. Leptin enhances synthesis of proinflammatory mediators in human osteoarthritic cartilage--mediator role of NO in leptin-induced PGE2, IL-6, and IL-8 production. Mediators Inflamm. 2009, 345838. [Google Scholar]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Becerril, S.; Rodríguez, A.; Catalán, V.; Ramírez, B.; Unamuno, X.; Portincasa, P.; Gómez-Ambrosi, J.; Frühbeck, G. Functional Relationship between Leptin and Nitric Oxide in Metabolism. Nutrients 2019, 11, 2129. https://doi.org/10.3390/nu11092129
Becerril S, Rodríguez A, Catalán V, Ramírez B, Unamuno X, Portincasa P, Gómez-Ambrosi J, Frühbeck G. Functional Relationship between Leptin and Nitric Oxide in Metabolism. Nutrients. 2019; 11(9):2129. https://doi.org/10.3390/nu11092129
Chicago/Turabian StyleBecerril, Sara, Amaia Rodríguez, Victoria Catalán, Beatriz Ramírez, Xabier Unamuno, Piero Portincasa, Javier Gómez-Ambrosi, and Gema Frühbeck. 2019. "Functional Relationship between Leptin and Nitric Oxide in Metabolism" Nutrients 11, no. 9: 2129. https://doi.org/10.3390/nu11092129
APA StyleBecerril, S., Rodríguez, A., Catalán, V., Ramírez, B., Unamuno, X., Portincasa, P., Gómez-Ambrosi, J., & Frühbeck, G. (2019). Functional Relationship between Leptin and Nitric Oxide in Metabolism. Nutrients, 11(9), 2129. https://doi.org/10.3390/nu11092129