Sodium Intake and Hypertension




{kind=link}
Abstract
1. Sodium Intake and Blood Pressure Values
2. Low Sodium Intake and Cardiovascular Risk
3. Hypertension and Salt-Sensitivity
4. Sodium Intake and Sympathetic Activity
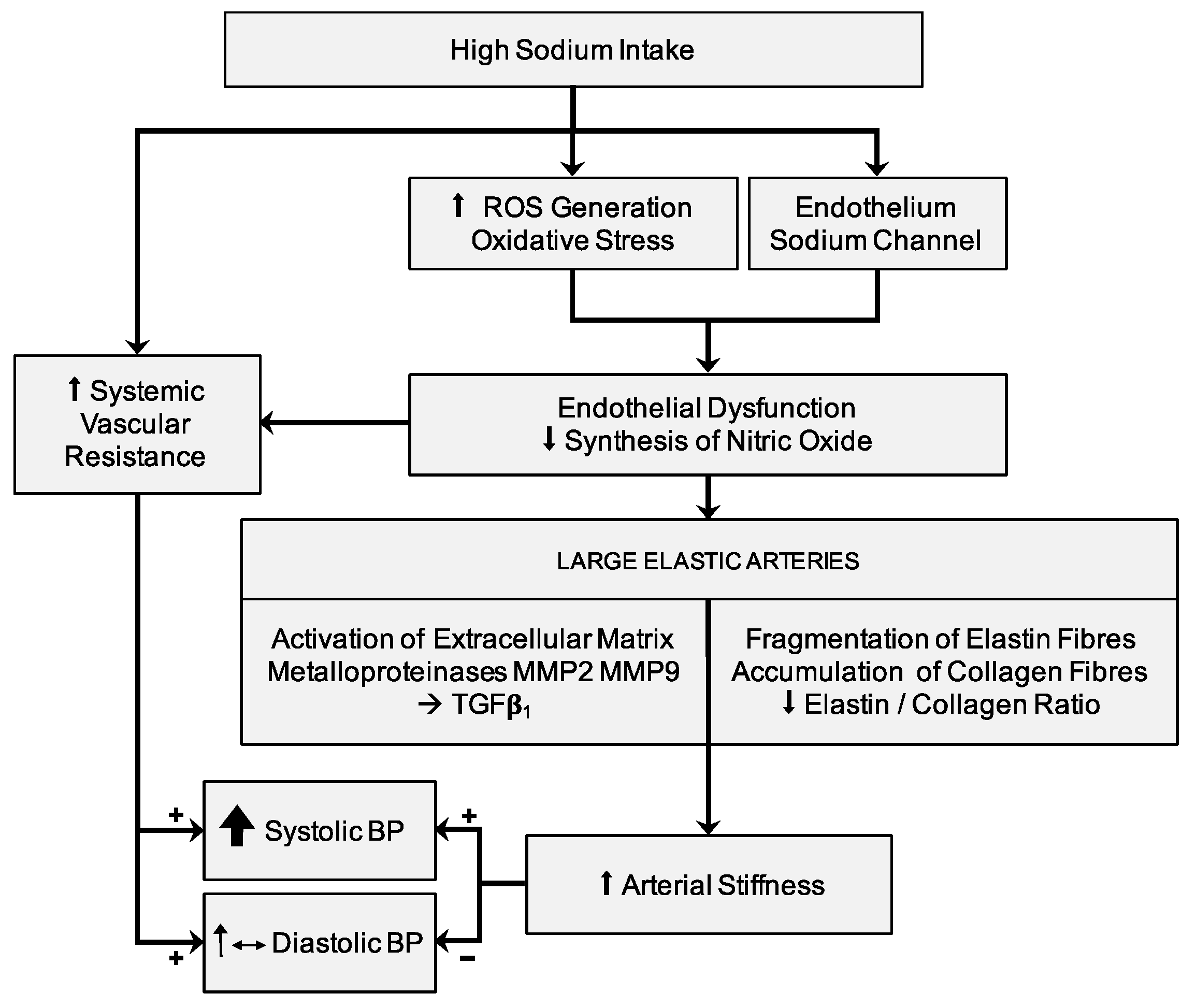
5. Salt-Induced Vasodysfunction
6. Sodium Intake and Arterial Stiffness
7. Conclusions
Funding
Conflicts of Interest
References
- Intersalt Cooperative Research Group. An international study of electrolyte excretion and blood pressure. Results for 24 hour urinary sodium and potassium excretion. BMJ 1988, 297, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Mente, A.; O’Donnell, M.J.; Rangarajan, S.; McQueen, M.J.; Poirier, P.; Wielgosz, A.; Morrison, H.; Li, W.; Wang, X.; Di, C.; et al. Association of urinary sodium and potassium excretion with blood pressure. N. Engl. J. Med. 2014, 371, 601–611. [Google Scholar] [CrossRef] [PubMed]
- He, F.J.; MacGregor, G.A. Effect of modest salt reduction on blood pressure: A meta-analysis of randomized trials. Implications for public health. J. Hum. Hypertens. 2002, 16, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Denton, D.; Weisinger, R.; Mundy, N.I.; Wickings, E.J.; Dixson, A.; Moisson, P.; Pingard, A.M.; Shade, R.; Carey, D.; Ardaillou, R.; et al. The effect of increased salt intake on blood pressure of chimpanzees. Nat. Med. 1995, 1, 1009–1016. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Guideline: Sodium Intake for Adults and Children; World Health Organization: Geneva, Switzerland, 2012. [Google Scholar]
- Weinberger, M.H. Salt sensitivity of blood pressure in humans. Hypertension 1996, 27, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Strazzullo, P.; D’Elia, L.; Kandala, N.B.; Cappuccio, F.P. Salt intake, stroke, and cardiovascular disease: Meta-analysis of prospective studies. BMJ 2009, 339, b4567. [Google Scholar] [CrossRef] [PubMed]
- Whelton, P.K.; He, J. Health effects of sodium and potassium in humans. Curr. Opin. Lipidol. 2014, 25, 75–79. [Google Scholar] [CrossRef]
- He, F.J.; Li, J.; Macgregor, G.A. Effect of longer term modest salt reduction on blood pressure: Cochrane systematic review and meta-analysis of randomised trials. BMJ 2013, 346, f1325. [Google Scholar] [CrossRef]
- Girardin, E.; Caverzasio, J.; Iwai, J.; Bonjour, J.P.; Muller, A.F.; Grandchamp, A. Pressure natriuresis in isolated kidneys from hypertension-prone and hypertension-resistant rats (Dahl rats). Kidney Int. 1980, 18, 10–19. [Google Scholar] [CrossRef]
- Dajnowiec, D.; Langille, B.L. Arterial adaptations to chronic changes in haemodynamic function: Coupling vasomotor tone to structural remodelling. Clin. Sci. 2007, 113, 15–23. [Google Scholar] [CrossRef]
- Dumont, O.; Pinaud, F.; Guihot, A.L.; Baufreton, C.; Loufrani, L.; Henrion, D. Alteration in flow (shear stress)-induced remodelling in rat resistance arteries with aging: Improvement by a treatment with hydralazine. Cardiovasc. Res. 2008, 77, 600–608. [Google Scholar] [CrossRef] [PubMed]
- Marketou, M.E.; Maragkoudakis, S.; Anastasiou, I.; Nakou, H.; Plataki, M.; Vardas, P.E.; Parthenakis, F.I. Salt-induced effects on microvascular function: A critical factor in hypertension mediated organ damage. J. Clin. Hypertens. 2019, 21, 749–757. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, T.W.; DiCarlo, S.E.; Pravenec, M.; Morris, R.C., Jr. The American Heart association scientific statement on salt sensitivity of blood pressure: Prompting consideration of alternative conceptual frameworks for the pathogenesis of salt sensitivity? J. Hypertens. 2017, 35, 2214–2225. [Google Scholar] [CrossRef] [PubMed]
- Elijovich, F.; Weinberger, M.H.; Anderson, C.A.; Appel, L.J.; Bursztyn, M.; Cook, N.R.; Dart, R.A.; Newton-Cheh, C.H.; Sacks, F.M.; Laffer, C.L.; et al. Salt sensitivity of blood pressure: A scientific statement from the american heart association. Hypertension 2016, 68, e7–e46. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.C.; Moran, J.; Forsblom, C.; Harjutsalo, V.; Thorn, L.; Ahola, A.; Waden, J.; Tolonen, N.; Saraheimo, M.; Gordin, D.; et al. The association between dietary sodium intake, ESRD, and all-cause mortality in patients with type 1 diabetes. Diabetes Care 2011, 34, 861–866. [Google Scholar] [CrossRef] [PubMed]
- Saulnier, P.J.; Gand, E.; Hadjadj, S.; Group, S.S. Sodium and cardiovascular disease. N. Engl. J. Med. 2014, 371, 2135–2136. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, M.; Mente, A.; Rangarajan, S.; McQueen, M.J.; Wang, X.; Liu, L.; Yan, H.; Lee, S.F.; Mony, P.; Devanath, A.; et al. Urinary sodium and potassium excretion, mortality, and cardiovascular events. N. Engl. J. Med. 2014, 371, 612–623. [Google Scholar] [CrossRef]
- Mente, A.; O’Donnell, M.; Rangarajan, S.; Dagenais, G.; Lear, S.; McQueen, M.; Diaz, R.; Avezum, A.; Lopez-Jaramillo, P.; Lanas, F.; et al. Associations of urinary sodium excretion with cardiovascular events in individuals with and without hypertension: A pooled analysis of data from four studies. Lancet 2016, 388, 465–475. [Google Scholar] [CrossRef]
- Graudal, N.; Jurgens, G.; Baslund, B.; Alderman, M.H. Compared with usual sodium intake, low- and excessive-sodium diets are associated with increased mortality: A meta-analysis. Am. J. Hypertens. 2014, 27, 1129–1137. [Google Scholar] [CrossRef]
- Catanozi, S.; Rocha, J.C.; Passarelli, M.; Guzzo, M.L.; Alves, C.; Furukawa, L.N.; Nunes, V.S.; Nakandakare, E.R.; Heimann, J.C.; Quintao, E.C. Dietary sodium chloride restriction enhances aortic wall lipid storage and raises plasma lipid concentration in LDL receptor knockout mice. J. Lipid Res. 2003, 44, 727–732. [Google Scholar] [CrossRef]
- Graudal, N.A.; Galloe, A.M.; Garred, P. Effects of sodium restriction on blood pressure, renin, aldosterone, catecholamines, cholesterols, and triglyceride: A meta-analysis. JAMA 1998, 279, 1383–1391. [Google Scholar] [CrossRef] [PubMed]
- Brunner, H.R.; Laragh, J.H.; Baer, L.; Newton, M.A.; Goodwin, F.T.; Krakoff, L.R.; Bard, R.H.; Buhler, F.R. Essential hypertension: Renin and aldosterone, heart attack and stroke. N. Engl. J. Med. 1972, 286, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Grassi, G.; Dell’Oro, R.; Seravalle, G.; Foglia, G.; Trevano, F.Q.; Mancia, G. Short- and long-term neuroadrenergic effects of moderate dietary sodium restriction in essential hypertension. Circulation 2002, 106, 1957–1961. [Google Scholar] [CrossRef] [PubMed]
- Petrie, J.R.; Morris, A.D.; Minamisawa, K.; Hilditch, T.E.; Elliott, H.L.; Small, M.; McConnell, J. Dietary sodium restriction impairs insulin sensitivity in noninsulin-dependent diabetes mellitus. J. Clin. Endocrinol. Metab. 1998, 83, 1552–1557. [Google Scholar] [CrossRef] [PubMed]
- Garg, R.; Williams, G.H.; Hurwitz, S.; Brown, N.J.; Hopkins, P.N.; Adler, G.K. Low-salt diet increases insulin resistance in healthy subjects. Metabolism 2011, 60, 965–968. [Google Scholar] [CrossRef]
- Nakandakare, E.R.; Charf, A.M.; Santos, F.C.; Nunes, V.S.; Ortega, K.; Lottenberg, A.M.; Mion, D., Jr.; Nakano, T.; Nakajima, K.; D’Amico, E.A.; et al. Dietary salt restriction increases plasma lipoprotein and inflammatory marker concentrations in hypertensive patients. Atherosclerosis 2008, 200, 410–416. [Google Scholar] [CrossRef]
- Grassi, G.; Cattaneo, B.M.; Seravalle, G.; Lanfranchi, A.; Bolla, G.; Mancia, G. Baroreflex impairment by low sodium diet in mild or moderate essential hypertension. Hypertension 1997, 29, 802–807. [Google Scholar] [CrossRef]
- Cook, N.R. Sodium and cardiovascular disease. N. Engl. J. Med. 2014, 371, 2134. [Google Scholar] [CrossRef]
- Batuman, V. Sodium and cardiovascular disease. N. Engl. J. Med. 2014, 371, 2134–2135. [Google Scholar] [CrossRef]
- Hall, J.E.; Guyton, A.C.; Coleman, T.G.; Mizelle, H.L.; Woods, L.L. Regulation of arterial pressure: Role of pressure natriuresis and diuresis. Fed. Proc. 1986, 45, 2897–2903. [Google Scholar]
- Rapp, J.P.; Dene, H. Development and characteristics of inbred strains of Dahl salt-sensitive and salt-resistant rats. Hypertension 1985, 7, 340–349. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, T.; Delea, C.S.; Bartter, F.C.; Smith, H. The effect of high-sodium and low-sodium intakes on blood pressure and other related variables in human subjects with idiopathic hypertension. Am. J. Med. 1978, 64, 193–198. [Google Scholar] [CrossRef]
- Weinberger, M.H.; Miller, J.Z.; Luft, F.C.; Grim, C.E.; Fineberg, N.S. Definitions and characteristics of sodium sensitivity and blood pressure resistance. Hypertension 1986, 8, II127–II134. [Google Scholar] [CrossRef] [PubMed]
- Coruzzi, P.; Parati, G.; Brambilla, L.; Brambilla, V.; Gualerzi, M.; Novarini, A.; Castiglioni, P.; Di Rienzo, M. Effects of salt sensitivity on neural cardiovascular regulation in essential hypertension. Hypertension 2005, 46, 1321–1326. [Google Scholar] [CrossRef] [PubMed]
- Castiglioni, P.; Parati, G.; Lazzeroni, D.; Bini, M.; Faini, A.; Brambilla, L.; Brambilla, V.; Coruzzi, P. Hemodynamic and autonomic response to different salt intakes in normotensive individuals. J. Am. Heart Assoc. 2016, 5, e003736. [Google Scholar] [CrossRef] [PubMed]
- Galletti, F.; Ferrara, I.; Stinga, F.; Iacone, R.; Noviello, F.; Strazzullo, P. Evaluation of a rapid protocol for the assessment of salt sensitivity against the blood pressure response to dietary sodium chloride restriction. Am. J. Hypertens. 1997, 10, 462–466. [Google Scholar] [CrossRef]
- Castiglioni, P.; Parati, G.; Brambilla, L.; Brambilla, V.; Gualerzi, M.; Di Rienzo, M.; Coruzzi, P. Detecting sodium-sensitivity in hypertensive patients: Information from 24-hour ambulatory blood pressure monitoring. Hypertension 2011, 57, 180–185. [Google Scholar] [CrossRef]
- Barba, G.; Galletti, F.; Cappuccio, F.P.; Siani, A.; Venezia, A.; Versiero, M.; Della Valle, E.; Sorrentino, P.; Tarantino, G.; Farinaro, E.; et al. Incidence of hypertension in individuals with different blood pressure salt-sensitivity: Results of a 15-year follow-up study. J. Hypertens. 2007, 25, 1465–1471. [Google Scholar] [CrossRef]
- Bihorac, A.; Tezcan, H.; Ozener, C.; Oktay, A.; Akoglu, E. Association between salt sensitivity and target organ damage in essential hypertension. Am. J. Hypertens. 2000, 13, 864–872. [Google Scholar] [CrossRef]
- Morimoto, A.; Uzu, T.; Fujii, T.; Nishimura, M.; Kuroda, S.; Nakamura, S.; Inenaga, T.; Kimura, G. Sodium sensitivity and cardiovascular events in patients with essential hypertension. Lancet 1997, 350, 1734–1737. [Google Scholar] [CrossRef]
- Elliott, P.; Walker, L.L.; Little, M.P.; Blair-West, J.R.; Shade, R.E.; Lee, D.R.; Rouquet, P.; Leroy, E.; Jeunemaitre, X.; Ardaillou, R.; et al. Change in salt intake affects blood pressure of chimpanzees: Implications for human populations. Circulation 2007, 116, 1563–1568. [Google Scholar] [CrossRef]
- Galletti, F.; Strazzullo, P. The blood pressure-salt sensitivity paradigm: Pathophysiologically sound yet of no practical value. Nephrol. Dial. Transplant. 2016, 31, 1386–1391. [Google Scholar] [CrossRef]
- Aburto, N.J.; Ziolkovska, A.; Hooper, L.; Elliott, P.; Cappuccio, F.P.; Meerpohl, J.J. Effect of lower sodium intake on health: Systematic review and meta-analyses. BMJ 2013, 346, f1326. [Google Scholar] [CrossRef]
- Frohlich, E.D. Hemodynamic differences between black patients and white patients with essential hypertension. State of the art lecture. Hypertension 1990, 15, 675–680. [Google Scholar] [CrossRef]
- Wedler, B.; Brier, M.E.; Wiersbitzky, M.; Gruska, S.; Wolf, E.; Kallwellis, R.; Aronoff, G.R.; Luft, F.C. Sodium kinetics in salt-sensitive and salt-resistant normotensive and hypertensive subjects. J. Hypertens. 1992, 10, 663–669. [Google Scholar] [CrossRef]
- Rocchini, A.P.; Key, J.; Bondie, D.; Chico, R.; Moorehead, C.; Katch, V.; Martin, M. The effect of weight loss on the sensitivity of blood pressure to sodium in obese adolescents. N. Engl. J. Med. 1989, 321, 580–585. [Google Scholar] [CrossRef]
- Strazzullo, P.; Barbato, A.; Galletti, F.; Barba, G.; Siani, A.; Iacone, R.; D’Elia, L.; Russo, O.; Versiero, M.; Farinaro, E.; et al. Abnormalities of renal sodium handling in the metabolic syndrome. Results of the Olivetti heart study. J. Hypertens. 2006, 24, 1633–1639. [Google Scholar] [CrossRef]
- Barba, G.; Russo, O.; Siani, A.; Iacone, R.; Farinaro, E.; Gerardi, M.C.; Russo, P.; Della Valle, E.; Strazzullo, P. Plasma leptin and blood pressure in men: Graded association independent of body mass and fat pattern. Obes. Res. 2003, 11, 160–166. [Google Scholar] [CrossRef]
- Guyton, A.C. Blood pressure control—Special role of the kidneys and body fluids. Science 1991, 252, 1813–1816. [Google Scholar] [CrossRef]
- Heer, M.; Baisch, F.; Kropp, J.; Gerzer, R.; Drummer, C. High dietary sodium chloride consumption may not induce body fluid retention in humans. Am. J. Physiol. Physiol. 2000, 278, F585–F595. [Google Scholar] [CrossRef]
- Titze, J.; Bauer, K.; Schafflhuber, M.; Dietsch, P.; Lang, R.; Schwind, K.H.; Luft, F.C.; Eckardt, K.U.; Hilgers, K.F. Internal sodium balance in DOCA-salt rats: A body composition study. Am. J. Physiol. Physiol. 2005, 289, F793–F802. [Google Scholar] [CrossRef]
- Laffer, C.L.; Scott, R.C., 3rd; Titze, J.M.; Luft, F.C.; Elijovich, F. Hemodynamics and salt-and-water balance link sodium storage and vascular dysfunction in salt-sensitive subjects. Hypertension 2016, 68, 195–203. [Google Scholar] [CrossRef]
- Schmidlin, O.; Forman, A.; Leone, A.; Sebastian, A.; Morris, R.C., Jr. Salt sensitivity in blacks: Evidence that the initial pressor effect of NaCl involves inhibition of vasodilatation by asymmetrical dimethylarginine. Hypertension 2011, 58, 380–385. [Google Scholar] [CrossRef]
- Morris, R.C., Jr.; Schmidlin, O.; Sebastian, A.; Tanaka, M.; Kurtz, T.W. Vasodysfunction that involves renal vasodysfunction, not abnormally increased renal retention of sodium, accounts for the initiation of salt-induced hypertension. Circulation 2016, 133, 881–893. [Google Scholar] [CrossRef]
- Kurtz, T.W.; DiCarlo, S.E.; Pravenec, M.; Schmidlin, O.; Tanaka, M.; Morris, R.C., Jr. An alternative hypothesis to the widely held view that renal excretion of sodium accounts for resistance to salt-induced hypertension. Kidney Int. 2016, 90, 965–973. [Google Scholar] [CrossRef]
- Bech, J.N.; Nielsen, C.B.; Ivarsen, P.; Jensen, K.T.; Pedersen, E.B. Dietary sodium affects systemic and renal hemodynamic response to NO inhibition in healthy humans. Am. J. Physiol. 1998, 274, F914–F923. [Google Scholar] [CrossRef]
- Van Paassen, P.; de Zeeuw, D.; Navis, G.; de Jong, P.E. Does the renin-angiotensin system determine the renal and systemic hemodynamic response to sodium in patients with essential hypertension? Hypertension 1996, 27, 202–208. [Google Scholar] [CrossRef]
- Parati, G.; Di Rienzo, M.; Bertinieri, G.; Pomidossi, G.; Casadei, R.; Groppelli, A.; Pedotti, A.; Zanchetti, A.; Mancia, G. Evaluation of the baroreceptor-heart rate reflex by 24-hour intra-arterial blood pressure monitoring in humans. Hypertension 1988, 12, 214–222. [Google Scholar] [CrossRef]
- Di Rienzo, M.; Parati, G.; Castiglioni, P.; Tordi, R.; Mancia, G.; Pedotti, A. Baroreflex effectiveness index: An additional measure of baroreflex control of heart rate in daily life. Am. J. Physiol. Integr. Comp. Physiol. 2001, 280, R744–R751. [Google Scholar] [CrossRef]
- Parati, G.; Saul, J.P.; Di Rienzo, M.; Mancia, G. Spectral analysis of blood pressure and heart rate variability in evaluating cardiovascular regulation. A critical appraisal. Hypertension 1995, 25, 1276–1286. [Google Scholar] [CrossRef]
- Campese, V.M.; Romoff, M.S.; Levitan, D.; Saglikes, Y.; Friedler, R.M.; Massry, S.G. Abnormal relationship between sodium intake and sympathetic nervous system activity in salt-sensitive patients with essential hypertension. Kidney Int. 1982, 21, 371–378. [Google Scholar] [CrossRef]
- Mark, A.; Mancia, G. Cardiopulmonary baroreflexes in humans. In Handbook of Physiology. The Cardiovascular System; Shepherd, J.T., Abboud, F.M., Eds.; American Physiological Society: Bethesda, MD, USA, 1983; pp. 795–813. [Google Scholar]
- Mancia, G.; Parati, G.; Pomidossi, G.; Casadei, R.; Di Rienzo, M.; Zanchetti, A. Arterial baroreflexes and blood pressure and heart rate variabilities in humans. Hypertension 1986, 8, 147–153. [Google Scholar] [CrossRef]
- Parlow, J.; Viale, J.P.; Annat, G.; Hughson, R.; Quintin, L. Spontaneous cardiac baroreflex in humans. Comparison with drug-induced responses. Hypertension 1995, 25, 1058–1068. [Google Scholar] [CrossRef]
- Eckberg, D.L.; Drabinsky, M.; Braunwald, E. Defective cardiac parasympathetic control in patients with heart disease. N. Engl. J. Med. 1971, 285, 877–883. [Google Scholar] [CrossRef]
- Pagani, M.; Somers, V.; Furlan, R.; Dell’Orto, S.; Conway, J.; Baselli, G.; Cerutti, S.; Sleight, P.; Malliani, A. Changes in autonomic regulation induced by physical training in mild hypertension. Hypertension 1988, 12, 600–610. [Google Scholar] [CrossRef]
- Berntson, G.G.; Bigger, J.T., Jr.; Eckberg, D.L.; Grossman, P.; Kaufmann, P.G.; Malik, M.; Nagaraja, H.N.; Porges, S.W.; Saul, J.P.; Stone, P.H.; et al. Heart rate variability: Origins, methods, and interpretive caveats. Psychophysiology 1997, 34, 623–648. [Google Scholar] [CrossRef]
- Hansen-Smith, F.M.; Morris, L.W.; Greene, A.S.; Lombard, J.H. Rapid microvessel rarefaction with elevated salt intake and reduced renal mass hypertension in rats. Circ. Res. 1996, 79, 324–330. [Google Scholar] [CrossRef]
- Frisbee, J.C.; Lombard, J.H. Development and reversibility of altered skeletal muscle arteriolar structure and reactivity with high salt diet and reduced renal mass hypertension. Microcirculation 1999, 6, 215–225. [Google Scholar] [CrossRef]
- Zhu, J.; Drenjancevic-Peric, I.; McEwen, S.; Friesema, J.; Schulta, D.; Yu, M.; Roman, R.J.; Lombard, J.H. Role of superoxide and angiotensin II suppression in salt-induced changes in endothelial Ca2+ signaling and NO production in rat aorta. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H929–H938. [Google Scholar] [CrossRef]
- Wang, J.; Roman, R.J.; Falck, J.R.; de la Cruz, L.; Lombard, J.H. Effects of high-salt diet on CYP450-4A omega-hydroxylase expression and active tone in mesenteric resistance arteries. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H1557–H1565. [Google Scholar] [CrossRef]
- Lukaszewicz, K.M.; Falck, J.R.; Manthati, V.L.; Lombard, J.H. Introgression of Brown Norway CYP4A genes on to the Dahl salt-sensitive background restores vascular function in SS-5(BN) consomic rats. Clin. Sci. 2013, 124, 333–342. [Google Scholar] [CrossRef]
- Abularrage, C.J.; Sidawy, A.N.; Aidinian, G.; Singh, N.; Weiswasser, J.M.; Arora, S. Evaluation of the microcirculation in vascular disease. J. Vasc. Surg. 2005, 42, 574–581. [Google Scholar] [CrossRef]
- Tzemos, N.; Lim, P.O.; Wong, S.; Struthers, A.D.; MacDonald, T.M. Adverse cardiovascular effects of acute salt loading in young normotensive individuals. Hypertension 2008, 51, 1525–1530. [Google Scholar] [CrossRef]
- Greaney, J.L.; DuPont, J.J.; Lennon-Edwards, S.L.; Sanders, P.W.; Edwards, D.G.; Farquhar, W.B. Dietary sodium loading impairs microvascular function independent of blood pressure in humans: Role of oxidative stress. J. Physiol. 2012, 590, 5519–5528. [Google Scholar] [CrossRef]
- Cavka, A.; Jukic, I.; Ali, M.; Goslawski, M.; Bian, J.T.; Wang, E.; Drenjancevic, I.; Phillips, S.A. Short-term high salt intake reduces brachial artery and microvascular function in the absence of changes in blood pressure. J. Hypertens. 2016, 34, 676–684. [Google Scholar] [CrossRef]
- Rorije, N.M.G.; Olde Engberink, R.H.G.; Chahid, Y.; van Vlies, N.; van Straalen, J.P.; van den Born, B.H.; Verberne, H.J.; Vogt, L. Microvascular permeability after an acute and chronic salt load in healthy subjects: A randomized open-label crossover intervention study. Anesthesiology 2018, 128, 352–360. [Google Scholar] [CrossRef]
- Schmidlin, O.; Sebastian, A.F.; Morris, R.C., Jr. What initiates the pressor effect of salt in salt-sensitive humans? Observations in normotensive blacks. Hypertension 2007, 49, 1032–1039. [Google Scholar] [CrossRef]
- Jablonski, K.L.; Racine, M.L.; Geolfos, C.J.; Gates, P.E.; Chonchol, M.; McQueen, M.B.; Seals, D.R. Dietary sodium restriction reverses vascular endothelial dysfunction in middle-aged/older adults with moderately elevated systolic blood pressure. J. Am. Coll. Cardiol. 2013, 61, 335–343. [Google Scholar] [CrossRef]
- Kopp, C.; Linz, P.; Dahlmann, A.; Hammon, M.; Jantsch, J.; Muller, D.N.; Schmieder, R.E.; Cavallaro, A.; Eckardt, K.U.; Uder, M.; et al. 23Na magnetic resonance imaging-determined tissue sodium in healthy subjects and hypertensive patients. Hypertension 2013, 61, 635–640. [Google Scholar] [CrossRef]
- Nijst, P.; Verbrugge, F.H.; Grieten, L.; Dupont, M.; Steels, P.; Tang, W.H.W.; Mullens, W. The pathophysiological role of interstitial sodium in heart failure. J. Am. Coll. Cardiol. 2015, 65, 378–388. [Google Scholar] [CrossRef]
- Selvarajah, V.; Connolly, K.; McEniery, C.; Wilkinson, I. Skin sodium and hypertension: A paradigm shift? Curr. Hypertens. Rep. 2018, 20, 94. [Google Scholar] [CrossRef]
- Titze, J.; Krause, H.; Hecht, H.; Dietsch, P.; Rittweger, J.; Lang, R.; Kirsch, K.A.; Hilgers, K.F. Reduced osmotically inactive Na storage capacity and hypertension in the Dahl model. Am. J. Physiol. Physiol. 2002, 283, F134–F141. [Google Scholar] [CrossRef]
- Titze, J.; Lang, R.; Ilies, C.; Schwind, K.H.; Kirsch, K.A.; Dietsch, P.; Luft, F.C.; Hilgers, K.F. Osmotically inactive skin Na+ storage in rats. Am. J. Physiol. Physiol. 2003, 285, F1108–F1117. [Google Scholar] [CrossRef]
- Titze, J.; Shakibaei, M.; Schafflhuber, M.; Schulze-Tanzil, G.; Porst, M.; Schwind, K.H.; Dietsch, P.; Hilgers, K.F. Glycosaminoglycan polymerization may enable osmotically inactive Na+ storage in the skin. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H203–H208. [Google Scholar] [CrossRef]
- Machnik, A.; Neuhofer, W.; Jantsch, J.; Dahlmann, A.; Tammela, T.; Machura, K.; Park, J.K.; Beck, F.X.; Muller, D.N.; Derer, W.; et al. Macrophages regulate salt-dependent volume and blood pressure by a vascular endothelial growth factor-C-dependent buffering mechanism. Nat. Med. 2009, 15, 545–552. [Google Scholar] [CrossRef]
- Wiig, H.; Schroder, A.; Neuhofer, W.; Jantsch, J.; Kopp, C.; Karlsen, T.V.; Boschmann, M.; Goss, J.; Bry, M.; Rakova, N.; et al. Immune cells control skin lymphatic electrolyte homeostasis and blood pressure. J. Clin. Investig. 2013, 123, 2803–2815. [Google Scholar] [CrossRef]
- Machnik, A.; Dahlmann, A.; Kopp, C.; Goss, J.; Wagner, H.; van Rooijen, N.; Eckardt, K.U.; Muller, D.N.; Park, J.K.; Luft, F.C.; et al. Mononuclear phagocyte system depletion blocks interstitial tonicity-responsive enhancer binding protein/vascular endothelial growth factor C expression and induces salt-sensitive hypertension in rats. Hypertension 2010, 55, 755–761. [Google Scholar] [CrossRef]
- He, F.J.; Marciniak, M.; Markandu, N.D.; Antonios, T.F.; MacGregor, G.A. Effect of modest salt reduction on skin capillary rarefaction in white, black, and Asian individuals with mild hypertension. Hypertension 2010, 56, 253–259. [Google Scholar] [CrossRef]
- Helle, F.; Karlsen, T.V.; Tenstad, O.; Titze, J.; Wiig, H. High-salt diet increases hormonal sensitivity in skin pre-capillary resistance vessels. Acta Physiol. 2013, 207, 577–581. [Google Scholar] [CrossRef]
- Zhu, Q.; Hu, J.; Han, W.Q.; Zhang, F.; Li, P.L.; Wang, Z.; Li, N. Silencing of HIF prolyl-hydroxylase 2 gene in the renal medulla attenuates salt-sensitive hypertension in Dahl S rats. Am. J. Hypertens. 2014, 27, 107–113. [Google Scholar] [CrossRef]
- Safar, M.; Laurent, S.; Safavian, A.; Pannier, B.; Asmar, R. Sodium and large arteries in hypertension. Effects of indapamide. Am. J. Med. 1988, 84, 15–19. [Google Scholar] [CrossRef]
- Blaustein, M.P. Sodium ions, calcium ions, blood pressure regulation, and hypertension: A reassessment and a hypothesis. Am. J. Physiol. 1977, 232, C165–C173. [Google Scholar] [CrossRef]
- Avolio, A.P.; Deng, F.Q.; Li, W.Q.; Luo, Y.F.; Huang, Z.D.; Xing, L.F.; O’Rourke, M.F. Effects of aging on arterial distensibility in populations with high and low prevalence of hypertension: Comparison between urban and rural communities in China. Circulation 1985, 71, 202–210. [Google Scholar] [CrossRef]
- Avolio, A.P.; Clyde, K.M.; Beard, T.C.; Cooke, H.M.; Ho, K.K.; O’Rourke, M.F. Improved arterial distensibility in normotensive subjects on a low salt diet. Arteriosclerosis 1986, 6, 166–169. [Google Scholar] [CrossRef]
- Todd, A.S.; Macginley, R.J.; Schollum, J.B.; Johnson, R.J.; Williams, S.M.; Sutherland, W.H.; Mann, J.I.; Walker, R.J. Dietary salt loading impairs arterial vascular reactivity. Am. J. Clin. Nutr. 2010, 91, 557–564. [Google Scholar] [CrossRef]
- McMahon, E.J.; Bauer, J.D.; Hawley, C.M.; Isbel, N.M.; Stowasser, M.; Johnson, D.W.; Campbell, K.L. A randomized trial of dietary sodium restriction in CKD. J. Am. Soc. Nephrol. 2013, 24, 2096–2103. [Google Scholar] [CrossRef]
- Jablonski, K.L.; Fedorova, O.V.; Racine, M.L.; Geolfos, C.J.; Gates, P.E.; Chonchol, M.; Fleenor, B.S.; Lakatta, E.G.; Bagrov, A.Y.; Seals, D.R. Dietary sodium restriction and association with urinary marinobufagenin, blood pressure, and aortic stiffness. Clin. J. Am. Soc. Nephrol. 2013, 8, 1952–1959. [Google Scholar] [CrossRef]
- He, F.J.; Marciniak, M.; Visagie, E.; Markandu, N.D.; Anand, V.; Dalton, R.N.; MacGregor, G.A. Effect of modest salt reduction on blood pressure, urinary albumin, and pulse wave velocity in white, black, and Asian mild hypertensives. Hypertension 2009, 54, 482–488. [Google Scholar] [CrossRef]
- Todd, A.S.; Macginley, R.J.; Schollum, J.B.; Williams, S.M.; Sutherland, W.H.; Mann, J.I.; Walker, R.J. Dietary sodium loading in normotensive healthy volunteers does not increase arterial vascular reactivity or blood pressure. Nephrology 2012, 17, 249–256. [Google Scholar] [CrossRef]
- Dickinson, K.M.; Keogh, J.B.; Clifton, P.M. Effects of a low-salt diet on flow-mediated dilatation in humans. Am. J. Clin. Nutr. 2009, 89, 485–490. [Google Scholar] [CrossRef]
- Dickinson, K.M.; Clifton, P.M.; Keogh, J.B. A reduction of 3 g/day from a usual 9 g/day salt diet improves endothelial function and decreases endothelin-1 in a randomised cross_over study in normotensive overweight and obese subjects. Atherosclerosis 2014, 233, 32–38. [Google Scholar] [CrossRef]
- Van der Graaf, A.M.; Paauw, N.D.; Toering, T.J.; Feelisch, M.; Faas, M.M.; Sutton, T.R.; Minnion, M.; Lefrandt, J.D.; Scherjon, S.A.; Franx, A.; et al. Impaired sodium-dependent adaptation of arterial stiffness in formerly preeclamptic women: The RETAP-vascular study. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, H1827–H1833. [Google Scholar] [CrossRef]
- Suckling, R.J.; He, F.J.; Markandu, N.D.; MacGregor, G.A. Modest salt reduction lowers blood pressure and albumin excretion in impaired glucose tolerance and type 2 diabetes mellitus: A randomized double-blind trial. Hypertension 2016, 67, 1189–1195. [Google Scholar] [CrossRef]
- Gijsbers, L.; Dower, J.I.; Mensink, M.; Siebelink, E.; Bakker, S.J.; Geleijnse, J.M. Effects of sodium and potassium supplementation on blood pressure and arterial stiffness: A fully controlled dietary intervention study. J. Hum. Hypertens. 2015, 29, 592–598. [Google Scholar] [CrossRef]
- Pimenta, E.; Gaddam, K.K.; Oparil, S.; Aban, I.; Husain, S.; Dell’Italia, L.J.; Calhoun, D.A. Effects of dietary sodium reduction on blood pressure in subjects with resistant hypertension: Results from a randomized trial. Hypertension 2009, 54, 475–481. [Google Scholar] [CrossRef]
- D’Elia, L.; Galletti, F.; La Fata, E.; Sabino, P.; Strazzullo, P. Effect of dietary sodium restriction on arterial stiffness: Systematic review and meta-analysis of the randomized controlled trials. J. Hypertens. 2018, 36, 734–743. [Google Scholar] [CrossRef]
- Salvi, P.; Palombo, C.; Salvi, G.M.; Labat, C.; Parati, G.; Benetos, A. Left ventricular ejection time, not heart rate, is an independent correlate of aortic pulse wave velocity. J. Appl. Physiol. 2013, 115, 1610–1617. [Google Scholar] [CrossRef]
- Salvi, P. Pulse Waves. How Vascular Hemodynamics Affects Blood Pressure, 2nd ed.; Springer Nature: Heidelberg, Germany, 2017. [Google Scholar]
- Matrougui, K.; Schiavi, P.; Guez, D.; Henrion, D. High sodium intake decreases pressure-induced (myogenic) tone and flow-induced dilation in resistance arteries from hypertensive rats. Hypertension 1998, 32, 176–179. [Google Scholar] [CrossRef]
- Ying, W.Z.; Sanders, P.W. Dietary salt increases endothelial nitric oxide synthase and TGF-beta1 in rat aortic endothelium. Am. J. Physiol. 1999, 277, H1293–H1298. [Google Scholar]
- Edwards, D.G.; Farquhar, W.B. Vascular effects of dietary salt. Curr. Opin. Nephrol. Hypertens. 2015, 24, 8–13. [Google Scholar] [CrossRef]
- Harvey, A.; Montezano, A.C.; Lopes, R.A.; Rios, F.; Touyz, R.M. Vascular fibrosis in aging and hypertension: Molecular mechanisms and clinical implications. Can. J. Cardiol. 2016, 32, 659–668. [Google Scholar] [CrossRef]
- Wang, M.; Zhao, D.; Spinetti, G.; Zhang, J.; Jiang, L.Q.; Pintus, G.; Monticone, R.; Lakatta, E.G. Matrix metalloproteinase 2 activation of transforming growth factor-beta1 (TGF-beta1) and TGF-beta1-type II receptor signaling within the aged arterial wall. Arter. Thromb. Vasc. Biol. 2006, 26, 1503–1509. [Google Scholar] [CrossRef]
- Duncan, M.R.; Frazier, K.S.; Abramson, S.; Williams, S.; Klapper, H.; Huang, X.; Grotendorst, G.R. Connective tissue growth factor mediates transforming growth factor beta-induced collagen synthesis: Down-regulation by cAMP. FASEB J. 1999, 13, 1774–1786. [Google Scholar] [CrossRef]
- Safar, M.E.; Thuilliez, C.; Richard, V.; Benetos, A. Pressure-independent contribution of sodium to large artery structure and function in hypertension. Cardiovasc. Res. 2000, 46, 269–276. [Google Scholar] [CrossRef]
- Prakobwong, S.; Yongvanit, P.; Hiraku, Y.; Pairojkul, C.; Sithithaworn, P.; Pinlaor, P.; Pinlaor, S. Involvement of MMP-9 in peribiliary fibrosis and cholangiocarcinogenesis via Rac1-dependent DNA damage in a hamster model. Int. J. Cancer 2010, 127, 2576–2587. [Google Scholar] [CrossRef]
- Newby, A.C. Dual role of matrix metalloproteinases (matrixins) in intimal thickening and atherosclerotic plaque rupture. Physiol. Rev. 2005, 85, 1–31. [Google Scholar] [CrossRef]
- Wang, M.; Kim, S.H.; Monticone, R.E.; Lakatta, E.G. Matrix metalloproteinases promote arterial remodeling in aging, hypertension, and atherosclerosis. Hypertension 2015, 65, 698–703. [Google Scholar] [CrossRef]
- Pons, M.; Cousins, S.W.; Alcazar, O.; Striker, G.E.; Marin-Castano, M.E. Angiotensin II-induced MMP-2 activity and MMP-14 and basigin protein expression are mediated via the angiotensin II receptor type 1-mitogen-activated protein kinase 1 pathway in retinal pigment epithelium: Implications for age-related macular degeneration. Am. J. Pathol. 2011, 178, 2665–2681. [Google Scholar] [CrossRef]
- Savoia, C.; Touyz, R.M.; Amiri, F.; Schiffrin, E.L. Selective mineralocorticoid receptor blocker eplerenone reduces resistance artery stiffness in hypertensive patients. Hypertension 2008, 51, 432–439. [Google Scholar] [CrossRef]
- Wang, D.H.; Du, Y. Regulation of vascular type 1 angiotensin II receptor in hypertension and sodium loading: Role of angiotensin II. J. Hypertens. 1998, 16, 467–475. [Google Scholar] [CrossRef]
- Benetos, A.; Gautier, S.; Ricard, S.; Topouchian, J.; Asmar, R.; Poirier, O.; Larosa, E.; Guize, L.; Safar, M.; Soubrier, F.; et al. Influence of angiotensin-converting enzyme and angiotensin II type 1 receptor gene polymorphisms on aortic stiffness in normotensive and hypertensive patients. Circulation 1996, 94, 698–703. [Google Scholar] [CrossRef]
- Pojoga, L.; Gautier, S.; Blanc, H.; Guyene, T.T.; Poirier, O.; Cambien, F.; Benetos, A. Genetic determination of plasma aldosterone levels in essential hypertension. Am. J. Hypertens. 1998, 11, 856–860. [Google Scholar] [CrossRef]
- Mercier, N.; Labat, C.; Louis, H.; Cattan, V.; Benetos, A.; Safar, M.E.; Lacolley, P. Sodium, arterial stiffness, and cardiovascular mortality in hypertensive rats. Am. J. Hypertens. 2007, 20, 319–325. [Google Scholar] [CrossRef][Green Version]
- Safar, M.E.; Temmar, M.; Kakou, A.; Lacolley, P.; Thornton, S.N. Sodium intake and vascular stiffness in hypertension. Hypertension 2009, 54, 203–209. [Google Scholar] [CrossRef]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grillo, A.; Salvi, L.; Coruzzi, P.; Salvi, P.; Parati, G. Sodium Intake and Hypertension. Nutrients 2019, 11, 1970. https://doi.org/10.3390/nu11091970
Grillo A, Salvi L, Coruzzi P, Salvi P, Parati G. Sodium Intake and Hypertension. Nutrients. 2019; 11(9):1970. https://doi.org/10.3390/nu11091970
Chicago/Turabian StyleGrillo, Andrea, Lucia Salvi, Paolo Coruzzi, Paolo Salvi, and Gianfranco Parati. 2019. "Sodium Intake and Hypertension" Nutrients 11, no. 9: 1970. https://doi.org/10.3390/nu11091970
APA StyleGrillo, A., Salvi, L., Coruzzi, P., Salvi, P., & Parati, G. (2019). Sodium Intake and Hypertension. Nutrients, 11(9), 1970. https://doi.org/10.3390/nu11091970