Targeting Glutathione Metabolism: Partner in Crime in Anticancer Therapy


Abstract
1. Introduction
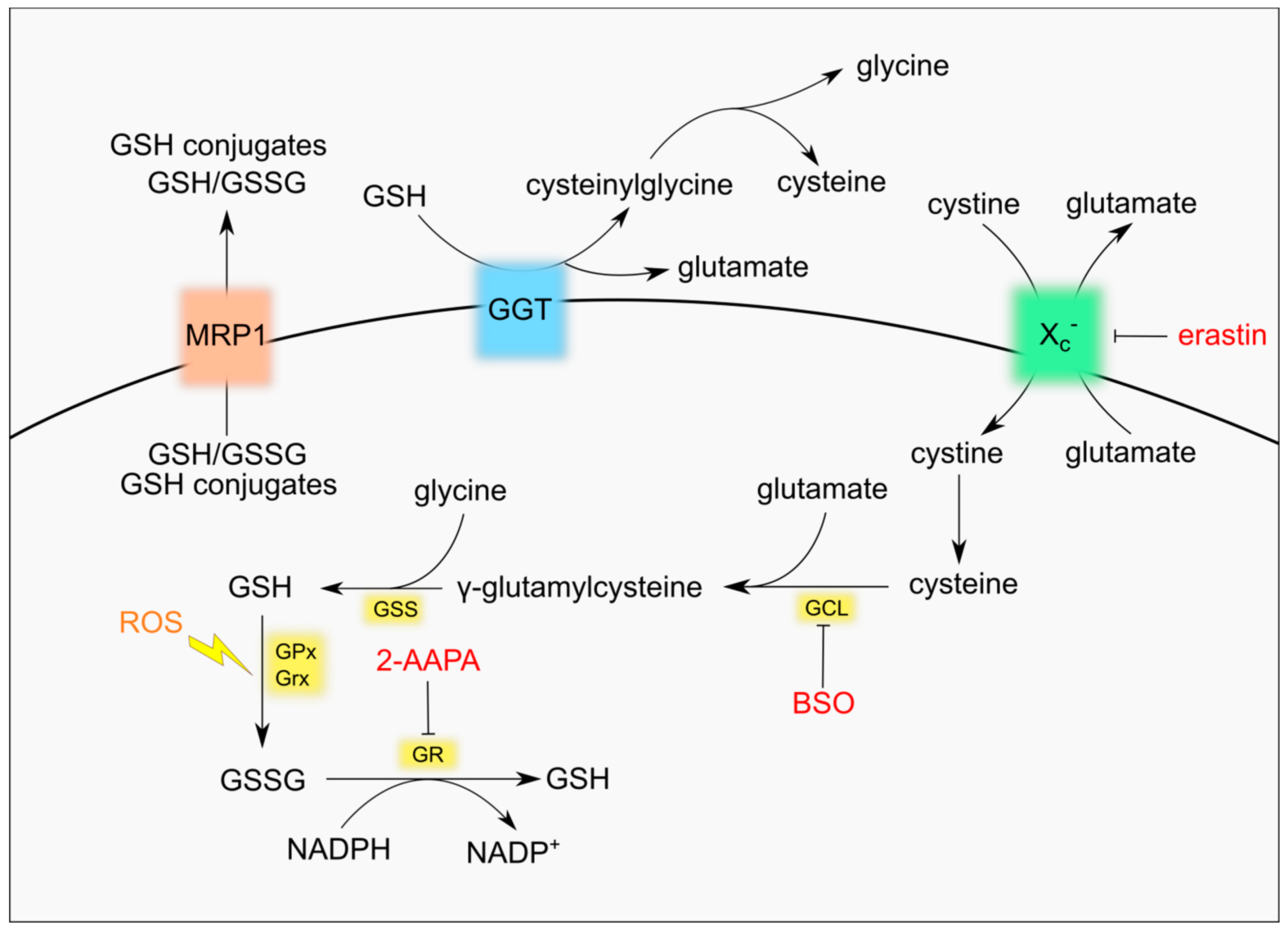
2. GSH Metabolism
3. GSH in Cancer
4. Manipulation of GSH Homeostasis for Cancer Treatment
4.1. Inhibition of GSH Synthesis
4.2. Reduction of GSH Precursors Availability
4.3. Increase of GSSG Levels
4.4. Promotion of GSH Efflux
5. Moving towards Precision Medicine: GSH in Synthetic Lethal Approaches
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Aquilano, K.; Baldelli, S.; Ciriolo, M.R. Glutathione: New roles in redox signaling for an old antioxidant. Front. Pharmacol. 2014, 5, 196. [Google Scholar] [CrossRef] [PubMed]
- Espinosa-Diez, C.; Miguel, V.; Mennerich, D.; Kietzmann, T.; Sánchez-Pérez, P.; Cadenas, S.; Lamas, S. Antioxidant responses and cellular adjustments to oxidative stress. Redox Biol. 2015, 6, 183–197. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Fang, Y.-Z.; Yang, S.; Lupton, J.R.; Turner, N.D. Glutathione metabolism and its implications for health. J. Nutr. 2004, 134, 489–492. [Google Scholar] [CrossRef] [PubMed]
- Scirè, A.; Cianfruglia, L.; Minnelli, C.; Bartolini, D.; Torquato, P.; Principato, G.; Galli, F.; Armeni, T. Glutathione compartmentalization and its role in glutathionylation and other regulatory processes of cellular pathways. BioFactors Oxf. Engl. 2019, 45, 152–168. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, R. Redox outside the box: Linking extracellular redox remodeling with intracellular redox metabolism. J. Biol. Chem. 2012, 287, 4397–4402. [Google Scholar] [CrossRef] [PubMed]
- Hanschmann, E.-M.; Godoy, J.R.; Berndt, C.; Hudemann, C.; Lillig, C.H. Thioredoxins, glutaredoxins, and peroxiredoxins--molecular mechanisms and health significance: From cofactors to antioxidants to redox signaling. Antioxid. Redox Signal. 2013, 19, 1539–1605. [Google Scholar] [CrossRef] [PubMed]
- Popov, D. Protein S-glutathionylation: From current basics to targeted modifications. Arch. Physiol. Biochem. 2014, 120, 123–130. [Google Scholar] [CrossRef]
- Klatt, P.; Molina, E.P.; Lamas, S. Nitric oxide inhibits c-Jun DNA binding by specifically targeted S-glutathionylation. J. Biol. Chem. 1999, 274, 15857–15864. [Google Scholar] [CrossRef]
- Lu, S.C. Glutathione synthesis. Biochim. Biophys. Acta 2013, 1830, 3143–3153. [Google Scholar] [CrossRef]
- Lei, X.G. In vivo antioxidant role of glutathione peroxidase: Evidence from knockout mice. Methods Enzymol. 2002, 347, 213–225. [Google Scholar]
- Winterbourn, C.C.; Metodiewa, D. The reaction of superoxide with reduced glutathione. Arch. Biochem. Biophys. 1994, 314, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Graham, K.S.; Reddy, C.C.; Scholz, R.W. Reduced glutathione effects on alpha-tocopherol concentration of rat liver microsomes undergoing NADPH-dependent lipid peroxidation. Lipids 1989, 24, 909–914. [Google Scholar] [CrossRef] [PubMed]
- Board, P.G.; Menon, D. Glutathione transferases, regulators of cellular metabolism and physiology. Biochim. Biophys. Acta 2013, 1830, 3267–3288. [Google Scholar] [CrossRef] [PubMed]
- Ballatori, N.; Krance, S.M.; Marchan, R.; Hammond, C.L. Plasma membrane glutathione transporters and their roles in cell physiology and pathophysiology. Mol. Aspects Med. 2009, 30, 13–28. [Google Scholar] [CrossRef] [PubMed]
- Cole, S.P.C.; Deeley, R.G. Transport of glutathione and glutathione conjugates by MRP1. Trends Pharmacol. Sci. 2006, 27, 438–446. [Google Scholar] [CrossRef] [PubMed]
- Franco, R.; Cidlowski, J.A. Glutathione efflux and cell death. Antioxid. Redox Signal. 2012, 17, 1694–1713. [Google Scholar] [CrossRef] [PubMed]
- Hanigan, M.H. Gamma-glutamyl transpeptidase: Redox regulation and drug resistance. Adv. Cancer Res. 2014, 122, 103–141. [Google Scholar] [PubMed]
- Bachhawat, A.K.; Yadav, S. The glutathione cycle: Glutathione metabolism beyond the γ-glutamyl cycle. IUBMB Life 2018, 70, 585–592. [Google Scholar] [CrossRef]
- Lewerenz, J.; Hewett, S.J.; Huang, Y.; Lambros, M.; Gout, P.W.; Kalivas, P.W.; Massie, A.; Smolders, I.; Methner, A.; Pergande, M.; et al. The Cystine/Glutamate Antiporter System xc− in Health and Disease: From Molecular Mechanisms to Novel Therapeutic Opportunities. Antioxid. Redox Signal. 2013, 18, 522–555. [Google Scholar] [CrossRef]
- Crawford, R.R.; Prescott, E.T.; Sylvester, C.F.; Higdon, A.N.; Shan, J.; Kilberg, M.S.; Mungrue, I.N. Human CHAC1 Protein Degrades Glutathione, and mRNA Induction Is Regulated by the Transcription Factors ATF4 and ATF3 and a Bipartite ATF/CRE Regulatory Element. J. Biol. Chem. 2015, 290, 15878–15891. [Google Scholar] [CrossRef]
- Kaur, A.; Gautam, R.; Srivastava, R.; Chandel, A.; Kumar, A.; Karthikeyan, S.; Bachhawat, A.K. ChaC2, an Enzyme for Slow Turnover of Cytosolic Glutathione. J. Biol. Chem. 2017, 292, 638–651. [Google Scholar] [CrossRef] [PubMed]
- Galadari, S.; Rahman, A.; Pallichankandy, S.; Thayyullathil, F. Reactive oxygen species and cancer paradox: To promote or to suppress? Free Radic. Biol. Med. 2017, 104, 144–164. [Google Scholar] [CrossRef] [PubMed]
- Ciccarone, F.; Castelli, S.; Ciriolo, M.R. Oxidative Stress-Driven Autophagy acROSs Onset and Therapeutic Outcome in Hepatocellular Carcinoma. Oxid. Med. Cell. Longev. 2019, 2019, 6050123. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.-A.; Lee, J.-W.; Kim, H.; Kim, E.-Y.; Seo, J.-M.; Ko, J.; Kim, J.-H. Pro-survival of estrogen receptor-negative breast cancer cells is regulated by a BLT2-reactive oxygen species-linked signaling pathway. Carcinogenesis 2010, 31, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Cichon, M.A.; Radisky, D.C. ROS-induced epithelial-mesenchymal transition in mammary epithelial cells is mediated by NF-kB-dependent activation of Snail. Oncotarget 2014, 5, 2827–2838. [Google Scholar] [CrossRef] [PubMed]
- Luanpitpong, S.; Talbott, S.J.; Rojanasakul, Y.; Nimmannit, U.; Pongrakhananon, V.; Wang, L.; Chanvorachote, P. Regulation of lung cancer cell migration and invasion by reactive oxygen species and caveolin-1. J. Biol. Chem. 2010, 285, 38832–38840. [Google Scholar] [CrossRef] [PubMed]
- Lien, E.C.; Lyssiotis, C.A.; Juvekar, A.; Hu, H.; Asara, J.M.; Cantley, L.C.; Toker, A. Glutathione biosynthesis is a metabolic vulnerability in PI(3)K/Akt-driven breast cancer. Nat. Cell Biol. 2016, 18, 572–578. [Google Scholar] [CrossRef]
- Romero, R.; Sayin, V.I.; Davidson, S.M.; Bauer, M.R.; Singh, S.X.; LeBoeuf, S.E.; Karakousi, T.R.; Ellis, D.C.; Bhutkar, A.; Sánchez-Rivera, F.J.; et al. Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nat. Med. 2017, 23, 1362–1368. [Google Scholar] [CrossRef]
- Lu, H.; Samanta, D.; Xiang, L.; Zhang, H.; Hu, H.; Chen, I.; Bullen, J.W.; Semenza, G.L. Chemotherapy triggers HIF-1–dependent glutathione synthesis and copper chelation that induces the breast cancer stem cell phenotype. Proc. Natl. Acad. Sci. USA 2015, 112, E4600–E4609. [Google Scholar] [CrossRef]
- Yu, D.-D.; Guo, S.-W.; Jing, Y.-Y.; Dong, Y.-L.; Wei, L.-X. A review on hepatocyte nuclear factor-1beta and tumor. Cell Biosci. 2015, 5, 58. [Google Scholar] [CrossRef]
- Lopes-Coelho, F.; Gouveia-Fernandes, S.; Gonçalves, L.G.; Nunes, C.; Faustino, I.; Silva, F.; Félix, A.; Pereira, S.A.; Serpa, J. HNF1β drives glutathione (GSH) synthesis underlying intrinsic carboplatin resistance of ovarian clear cell carcinoma (OCCC). Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2016, 37, 4813–4829. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Qiu, B.; Lee, D.S.M.; Walton, Z.E.; Ochocki, J.D.; Mathew, L.K.; Mancuso, A.; Gade, T.P.F.; Keith, B.; Nissim, I.; et al. Fructose-1, 6-bisphosphatase opposes renal carcinoma progression. Nature 2014, 513, 251–255. [Google Scholar] [CrossRef]
- Bansal, A.; Simon, M.C. Glutathione metabolism in cancer progression and treatment resistance. J. Cell Biol. 2018, 217, 2291–2298. [Google Scholar] [CrossRef] [PubMed]
- Harris, I.S.; Treloar, A.E.; Inoue, S.; Sasaki, M.; Gorrini, C.; Lee, K.C.; Yung, K.Y.; Brenner, D.; Knobbe-Thomsen, C.B.; Cox, M.A.; et al. Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell 2015, 27, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Nichenametla, S.N.; Muscat, J.E.; Liao, J.G.; Lazarus, P.; Richie, J.P. A functional trinucleotide repeat polymorphism in the 5’-untranslated region of the glutathione biosynthetic gene GCLC is associated with increased risk for lung and aerodigestive tract cancers. Mol. Carcinog. 2013, 52, 791–799. [Google Scholar] [CrossRef] [PubMed]
- Ke, H.-L.; Lin, J.; Ye, Y.; Wu, W.-J.; Lin, H.-H.; Wei, H.; Huang, M.; Chang, D.W.; Dinney, C.P.; Wu, X. Genetic Variations in Glutathione Pathway Genes Predict Cancer Recurrence in Patients Treated with Transurethral Resection and Bacillus Calmette-Guerin Instillation for Non-muscle Invasive Bladder Cancer. Ann. Surg. Oncol. 2015, 22, 4104–4110. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sun, Z.; Chen, J.; Aakre, J.; Marks, R.S.; Garces, Y.Y.; Jiang, R.; Idowu, O.; Cunningham, J.M.; Liu, Y.; Pankratz, V.S.; et al. Genetic variation in glutathione metabolism and DNA repair genes predicts survival of small-cell lung cancer patients. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2010, 21, 2011–2016. [Google Scholar] [CrossRef] [PubMed]
- Corti, A.; Franzini, M.; Paolicchi, A.; Pompella, A. Gamma-glutamyltransferase of Cancer Cells at the Crossroads of Tumor Progression, Drug Resistance and Drug Targeting. Anticancer Res. 2010, 30, 1169–1181. [Google Scholar]
- Cheteh, E.H.; Augsten, M.; Rundqvist, H.; Bianchi, J.; Sarne, V.; Egevad, L.; Bykov, V.J.; Östman, A.; Wiman, K.G. Human cancer-associated fibroblasts enhance glutathione levels and antagonize drug-induced prostate cancer cell death. Cell Death Dis. 2017, 8, e2848. [Google Scholar] [CrossRef]
- Wang, W.; Kryczek, I.; Dostál, L.; Lin, H.; Tan, L.; Zhao, L.; Lu, F.; Wei, S.; Maj, T.; Peng, D.; et al. Effector T Cells Abrogate Stroma-Mediated Chemoresistance in Ovarian Cancer. Cell 2016, 165, 1092–1105. [Google Scholar] [CrossRef]
- Gamcsik, M.P.; Kasibhatla, M.S.; Teeter, S.D.; Colvin, O.M. Glutathione levels in human tumors. Biomark. Biochem. Indic. Expo. Response Susceptibility Chem. 2012, 17, 671–691. [Google Scholar] [CrossRef] [PubMed]
- Harris, I.S.; Endress, J.E.; Coloff, J.L.; Selfors, L.M.; McBrayer, S.K.; Rosenbluth, J.M.; Takahashi, N.; Dhakal, S.; Koduri, V.; Oser, M.G.; et al. Deubiquitinases Maintain Protein Homeostasis and Survival of Cancer Cells upon Glutathione Depletion. Cell Metab. 2019, 29, 1166–1181. [Google Scholar] [CrossRef] [PubMed]
- Rocha, C.R.R.; Garcia, C.C.M.; Vieira, D.B.; Quinet, A.; de Andrade-Lima, L.C.; Munford, V.; Belizário, J.E.; Menck, C.F.M. Glutathione depletion sensitizes cisplatin- and temozolomide-resistant glioma cells in vitro and in vivo. Cell Death Dis. 2014, 5, e1505. [Google Scholar] [CrossRef] [PubMed]
- Fath, M.A.; Ahmad, I.M.; Smith, C.J.; Spence, J.; Spitz, D.R. Enhancement of carboplatin-mediated lung cancer cell killing by simultaneous disruption of glutathione and thioredoxin metabolism. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2011, 17, 6206–6217. [Google Scholar] [CrossRef] [PubMed]
- Miran, T.; Vogg, A.T.J.; Drude, N.; Mottaghy, F.M.; Morgenroth, A. Modulation of glutathione promotes apoptosis in triple-negative breast cancer cells. FASEB J. 2018, 32, 2803–2813. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Mohammad, R.M.; Muqbil, I.; Lowe, L.; Yedjou, C.; Hsu, H.-Y.; Lin, L.-T.; Siegelin, M.D.; Fimognari, C.; Kumar, N.B.; Dou, Q.P.; et al. Broad targeting of resistance to apoptosis in cancer. Semin. Cancer Biol. 2015, 35, S78–S103. [Google Scholar] [CrossRef]
- Tagde, A.; Singh, H.; Kang, M.H.; Reynolds, C.P. The glutathione synthesis inhibitor buthionine sulfoximine synergistically enhanced melphalan activity against preclinical models of multiple myeloma. Blood Cancer J. 2014, 4, e229. [Google Scholar] [CrossRef]
- Villablanca, J.G.; Volchenboum, S.L.; Cho, H.; Kang, M.H.; Cohn, S.L.; Anderson, C.P.; Marachelian, A.; Groshen, S.; Tsao-Wei, D.; Matthay, K.K.; et al. A Phase I New Approaches to Neuroblastoma Therapy Study of Buthionine Sulfoximine and Melphalan With Autologous Stem Cells for Recurrent/Refractory High-Risk Neuroblastoma. Pediatr. Blood Cancer 2016, 63, 1349–1356. [Google Scholar] [CrossRef]
- Bailey, H.H.; Mulcahy, R.T.; Tutsch, K.D.; Arzoomanian, R.Z.; Alberti, D.; Tombes, M.B.; Wilding, G.; Pomplun, M.; Spriggs, D.R. Phase I clinical trial of intravenous L-buthionine sulfoximine and melphalan: An attempt at modulation of glutathione. J. Clin. Oncol. 1994, 12, 194–205. [Google Scholar] [CrossRef]
- Hamilton, D.; Wu, J.H.; Batist, G. Structure-based identification of novel human gamma-glutamylcysteine synthetase inhibitors. Mol. Pharmacol. 2007, 71, 1140–1147. [Google Scholar] [CrossRef] [PubMed]
- Robe, P.A.; Martin, D.H.; Nguyen-Khac, M.T.; Artesi, M.; Deprez, M.; Albert, A.; Vanbelle, S.; Califice, S.; Bredel, M.; Bours, V. Early termination of ISRCTN45828668, a phase 1/2 prospective, randomized study of sulfasalazine for the treatment of progressing malignant gliomas in adults. BMC Cancer 2009, 9, 372. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-S.; Lee, D.-H.; Jeong, S.Y.; Park, S.H.; Oh, S.C.; Park, Y.S.; Yu, J.; Choudry, H.A.; Bartlett, D.L.; Lee, Y.J. Ferroptosis-inducing agents enhance TRAIL-induced apoptosis through upregulation of death receptor 5. J. Cell. Biochem. 2019, 120, 928–939. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Lin, Z.; Jiang, D.; Yu, Y.; Yang, D.; Zhou, H.; Zhan, D.; Liu, S.; Peng, G.; Chen, Z.; et al. Erastin decreases radioresistance of NSCLC cells partially by inducing GPX4-mediated ferroptosis. Oncol. Lett. 2019, 17, 3001–3008. [Google Scholar] [CrossRef] [PubMed]
- Joyce-Brady, M.; Hiratake, J. Inhibiting Glutathione Metabolism in Lung Lining Fluid as a Strategy to Augment Antioxidant Defense. Curr. Enzyme Inhib. 2011, 7, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Couto, N.; Wood, J.; Barber, J. The role of glutathione reductase and related enzymes on cellular redox homoeostasis network. Free Radic. Biol. Med. 2016, 95, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Seefeldt, T.; Chen, W.; Carlson, L.; Stoebner, A.; Hanson, S.; Foll, R.; Matthees, D.P.; Palakurthi, S.; Guan, X. Increase in thiol oxidative stress via glutathione reductase inhibition as a novel approach to enhance cancer sensitivity to X-ray irradiation. Free Radic. Biol. Med. 2009, 47, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Catanzaro, D.; Gaude, E.; Orso, G.; Giordano, C.; Guzzo, G.; Rasola, A.; Ragazzi, E.; Caparrotta, L.; Frezza, C.; Montopoli, M. Inhibition of glucose-6-phosphate dehydrogenase sensitizes cisplatin-resistant cells to death. Oncotarget 2015, 6, 30102–30114. [Google Scholar] [CrossRef] [PubMed]
- Pazoles, C.J.; Gerstein, H. NOV-002, a chemoprotectant/immunomodulator, added to first-line carboplatin/paclitaxel in advanced non-small cell lung cancer (NSCLC): A randomized Phase 1/2, open-label, controlled study. J. Clin. Oncol. 2006, 24, 17021. [Google Scholar]
- Fidias, P.; Ciuleanu, T.A.; Gladkov, O.; Manikhas, G.M.; Bondarenko, I.N.; Pluzanska, A.; Ramlau, R.; Lynch, T.J. A randomized, open-label, phase III trial of NOV-002 in combination with paclitaxel (P) and carboplatin (C) versus paclitaxel and carboplatin alone for the treatment of advanced non-small cell lung cancer (NSCLC). J. Clin. Oncol. 2010, 28, LBA7007. [Google Scholar] [CrossRef]
- Montero, A.J.; Diaz-Montero, C.M.; Deutsch, Y.E.; Hurley, J.; Koniaris, L.G.; Rumboldt, T.; Yasir, S.; Jorda, M.; Garret-Mayer, E.; Avisar, E.; et al. Phase 2 study of neoadjuvant treatment with NOV-002 in combination with doxorubicin and cyclophosphamide followed by docetaxel in patients with HER-2 negative clinical stage II-IIIc breast cancer. Breast Cancer Res. Treat. 2012, 132, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Munoz, M.; Henderson, M.; Haber, M.; Norris, M. Role of the MRP1/ABCC1 multidrug transporter protein in cancer. IUBMB Life 2007, 59, 752–757. [Google Scholar] [CrossRef] [PubMed]
- Cole, S.P.C. Multidrug resistance protein 1 (MRP1, ABCC1), a “multitasking” ATP-binding cassette (ABC) transporter. J. Biol. Chem. 2014, 289, 30880–30888. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.Y.; Poddar, A.; Magtanong, L.; Lumb, J.H.; Mileur, T.R.; Reid, M.A.; Dovey, C.M.; Wang, J.; Locasale, J.W.; Stone, E.; et al. A Genome-wide Haploid Genetic Screen Identifies Regulators of Glutathione Abundance and Ferroptosis Sensitivity. Cell Rep. 2019, 26, 1544–1556.e8. [Google Scholar] [CrossRef] [PubMed]
- Desideri, E.; Filomeni, G.; Ciriolo, M.R. Glutathione participates in the modulation of starvation-induced autophagy in carcinoma cells. Autophagy 2012, 8, 1769–1781. [Google Scholar] [CrossRef] [PubMed]
- Elgendy, M.; Cirò, M.; Hosseini, A.; Weiszmann, J.; Mazzarella, L.; Ferrari, E.; Cazzoli, R.; Curigliano, G.; DeCensi, A.; Bonanni, B.; et al. Combination of Hypoglycemia and Metformin Impairs Tumor Metabolic Plasticity and Growth by Modulating the PP2A-GSK3β-MCL-1 Axis. Cancer Cell 2019, 35, 798–815.e5. [Google Scholar] [CrossRef] [PubMed]
- Pietrocola, F.; Pol, J.; Vacchelli, E.; Rao, S.; Enot, D.P.; Baracco, E.E.; Levesque, S.; Castoldi, F.; Jacquelot, N.; Yamazaki, T.; et al. Caloric Restriction Mimetics Enhance Anticancer Immunosurveillance. Cancer Cell 2016, 30, 147–160. [Google Scholar] [CrossRef] [PubMed]
- Di Biase, S.; Lee, C.; Brandhorst, S.; Manes, B.; Buono, R.; Cheng, C.-W.; Cacciottolo, M.; Martin-Montalvo, A.; de Cabo, R.; Wei, M.; et al. Fasting-Mimicking Diet Reduces HO-1 to Promote T Cell-Mediated Tumor Cytotoxicity. Cancer Cell 2016, 30, 136–146. [Google Scholar] [CrossRef] [PubMed]
- Raffaghello, L.; Lee, C.; Safdie, F.M.; Wei, M.; Madia, F.; Bianchi, G.; Longo, V.D. Starvation-dependent differential stress resistance protects normal but not cancer cells against high-dose chemotherapy. Proc. Natl. Acad. Sci. USA 2008, 105, 8215–8220. [Google Scholar] [CrossRef] [PubMed]
- O’Neil, N.J.; Bailey, M.L.; Hieter, P. Synthetic lethality and cancer. Nat. Rev. Genet. 2017, 18, 613–623. [Google Scholar] [CrossRef]
- Bhattacharjee, S.; Nandi, S. Synthetic lethality in DNA repair network: A novel avenue in targeted cancer therapy and combination therapeutics. IUBMB Life 2017, 69, 929–937. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, S.; Nandi, S. Rare Genetic Diseases with Defects in DNA Repair: Opportunities and Challenges in Orphan Drug Development for Targeted Cancer Therapy. Cancers 2018, 10, 298. [Google Scholar] [CrossRef] [PubMed]
- Dolma, S.; Lessnick, S.L.; Hahn, W.C.; Stockwell, B.R. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell 2003, 3, 285–296. [Google Scholar] [CrossRef]
- Kerins, M.J.; Milligan, J.; Wohlschlegel, J.A.; Ooi, A. Fumarate hydratase inactivation in hereditary leiomyomatosis and renal cell cancer is synthetic lethal with ferroptosis induction. Cancer Sci. 2018, 109, 2757–2766. [Google Scholar] [CrossRef] [PubMed]
- Ogiwara, H.; Takahashi, K.; Sasaki, M.; Kuroda, T.; Yoshida, H.; Watanabe, R.; Maruyama, A.; Makinoshima, H.; Chiwaki, F.; Sasaki, H.; et al. Targeting the Vulnerability of Glutathione Metabolism in ARID1A-Deficient Cancers. Cancer Cell 2019, 35, 177–190.e8. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.S.; Duong, C.P.; Haupt, S.; Montgomery, K.G.; House, C.M.; Azar, W.J.; Pearson, H.B.; Fisher, O.M.; Read, M.; Guerra, G.R.; et al. Inhibiting the system xC-/glutathione axis selectively targets cancers with mutant-p53 accumulation. Nat. Commun. 2017, 8, 14844. [Google Scholar] [CrossRef] [PubMed]
- Sayin, V.I.; LeBoeuf, S.E.; Singh, S.X.; Davidson, S.M.; Biancur, D.; Guzelhan, B.S.; Alvarez, S.W.; Wu, W.L.; Karakousi, T.R.; Zavitsanou, A.M.; et al. Activation of the NRF2 antioxidant program generates an imbalance in central carbon metabolism in cancer. eLife 2017, 6, e28083. [Google Scholar] [CrossRef] [PubMed]
- Galan-Cobo, A.; Sitthideatphaiboon, P.; Qu, X.; Poteete, A.; Pisegna, M.A.; Tong, P.; Chen, P.-H.; Boroughs, L.K.; Rodriguez, M.L.M.; Zhang, W.; et al. LKB1 and KEAP1/NRF2 Pathways Cooperatively Promote Metabolic Reprogramming with Enhanced Glutamine Dependence in KRAS-Mutant Lung Adenocarcinoma. Cancer Res. 2019, 79, 3251–3267. [Google Scholar] [CrossRef]
- Mandal, P.K.; Schneider, M.; Kölle, P.; Kuhlencordt, P.; Förster, H.; Beck, H.; Bornkamm, G.W.; Conrad, M. Loss of thioredoxin reductase 1 renders tumors highly susceptible to pharmacologic glutathione deprivation. Cancer Res. 2010, 70, 9505–9514. [Google Scholar] [CrossRef]
- Yan, X.; Zhang, X.; Wang, L.; Zhang, R.; Pu, X.; Wu, S.; Li, L.; Tong, P.; Wang, J.; Meng, Q.H.; et al. Inhibition of Thioredoxin/Thioredoxin Reductase Induces Synthetic Lethality in Lung Cancers with Compromised Glutathione Homeostasis. Cancer Res. 2019, 79, 125–132. [Google Scholar] [CrossRef]
- Roh, J.-L.; Jang, H.; Kim, E.H.; Shin, D. Targeting of the Glutathione, Thioredoxin, and Nrf2 Antioxidant Systems in Head and Neck Cancer. Antioxid. Redox Signal. 2017, 27, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, S.; Shintani, S.; Hirata, Y.; Suina, K.; Semba, T.; Yamasaki, J.; Umene, K.; Ishikawa, M.; Saya, H.; Nagano, O. Synthetic lethality of the ALDH3A1 inhibitor dyclonine and xCT inhibitors in glutathione deficiency-resistant cancer cells. Oncotarget 2018, 9, 33832–33843. [Google Scholar] [CrossRef] [PubMed]
- Ortega, A.L.; Mena, S.; Estrela, J.M. Glutathione in cancer cell death. Cancers 2011, 3, 1285–1310. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Drug | Activity | Synthetic Lethal Partner | Effects | Cancer Model | Ref |
|---|---|---|---|---|---|
| Erastin | Inhibition of xc− activity | HRASV12 | Increased lipid peroxidation/activation of ferroptosis | Engineered tumorigenic fibroblasts | [73] |
| Accumulation of mutant p53 | Oesophageal adenocarcinoma | [76] | |||
| Inactivation of fumarate hydratase (FH) | Hereditary leiomyomatosis and renal cell cancer | [74] | |||
| BSO Sulfasalazine | Inhibition of GCLC Inhibition of xc− activity | Inactivating mutation/ablation of ARID1A | Induction of ROS dependent apoptosis | Ovarian Cancer, endometrial carcinoma | [75] |
| BPTES B-839 | Inhibition of glutaminase | Inactivating mutation of KEAP1 | Suppression of cell growth | KRAS-mutant lung adenocarcinoma | [78] |
| BSO | Inhibition of GCLC | Inhibition of TrxR | Cancer cell death | Breast cancer, lung adenocarcinoma | [34] |
| DUBs inhibition | Proteotoxic and ER stress-driven cell death | Breast cancer | [42] | ||
| Sulfasalazine | Inhibition of xc− activity | Inhibitor of ALDH | Suppression of cell growth | Head and neck squamous cell carcinoma | [82] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Desideri, E.; Ciccarone, F.; Ciriolo, M.R. Targeting Glutathione Metabolism: Partner in Crime in Anticancer Therapy. Nutrients 2019, 11, 1926. https://doi.org/10.3390/nu11081926
Desideri E, Ciccarone F, Ciriolo MR. Targeting Glutathione Metabolism: Partner in Crime in Anticancer Therapy. Nutrients. 2019; 11(8):1926. https://doi.org/10.3390/nu11081926
Chicago/Turabian StyleDesideri, Enrico, Fabio Ciccarone, and Maria Rosa Ciriolo. 2019. "Targeting Glutathione Metabolism: Partner in Crime in Anticancer Therapy" Nutrients 11, no. 8: 1926. https://doi.org/10.3390/nu11081926
APA StyleDesideri, E., Ciccarone, F., & Ciriolo, M. R. (2019). Targeting Glutathione Metabolism: Partner in Crime in Anticancer Therapy. Nutrients, 11(8), 1926. https://doi.org/10.3390/nu11081926