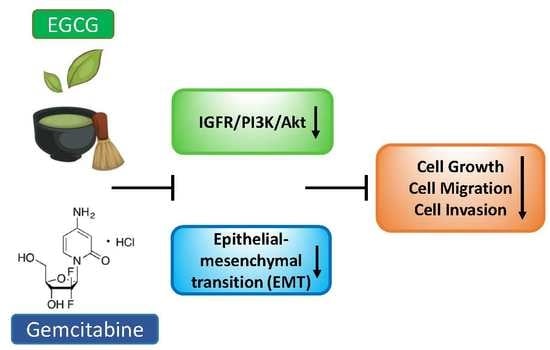
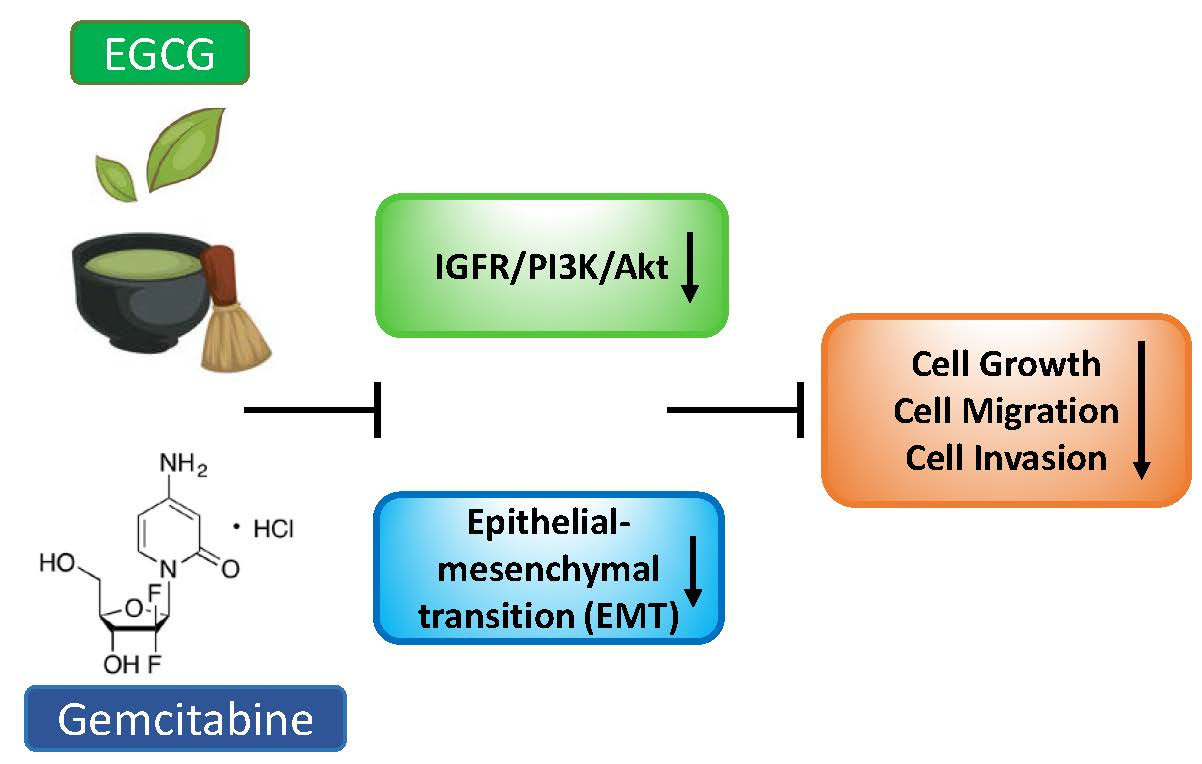
Epigallocatechin-3-Gallate (EGCG) Suppresses Pancreatic Cancer Cell Growth, Invasion, and Migration partly through the Inhibition of Akt Pathway and Epithelial–Mesenchymal Transition: Enhanced Efficacy When Combined with Gemcitabine

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Cell Culture
2.3. Cell Viability
2.4. Wound Healing Assay
2.5. Cell Migration and Invasion
2.6. Western Blot
2.7. Cycloheximide Chasing Assay
2.8. Immunoprecipitation and Ubiquitination Analysis
2.9. Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
2.10. Gene Overexpression
2.11. Clonogenic Assay
2.12. Animal Study
2.13. Immunohistochemistry
2.14. Statistical Analysis
3. Results
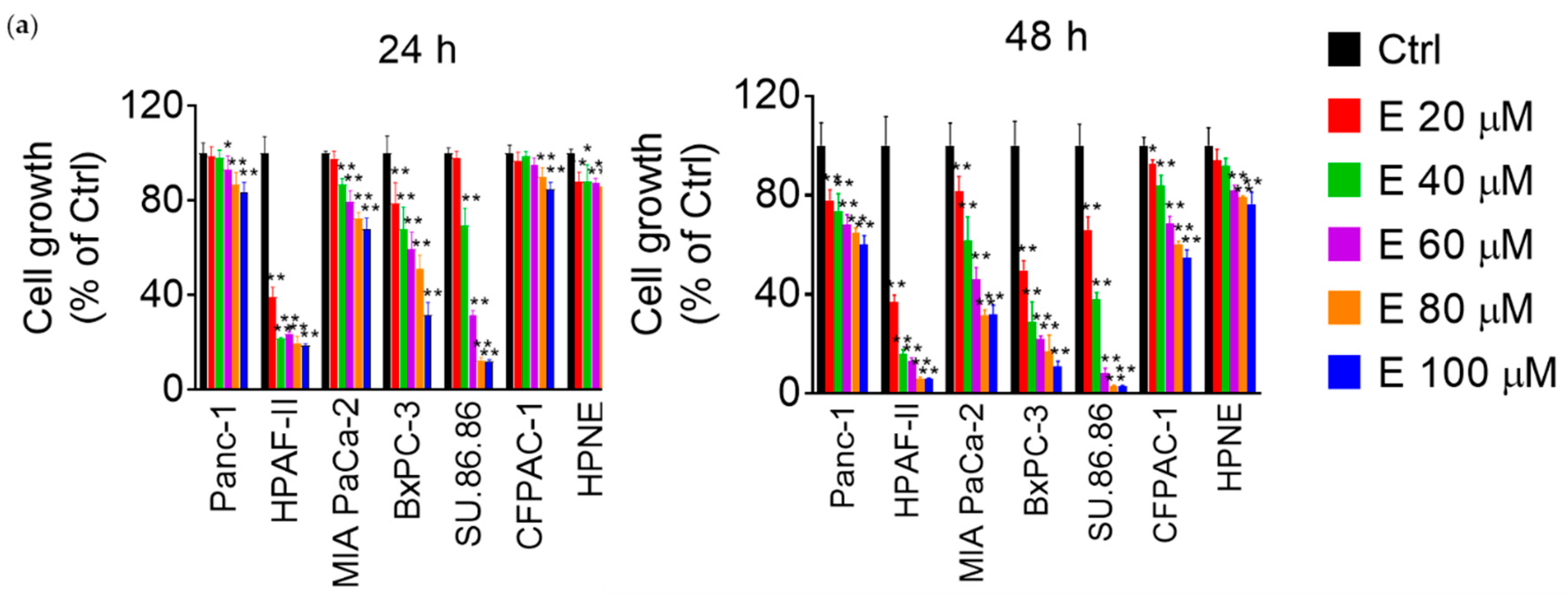
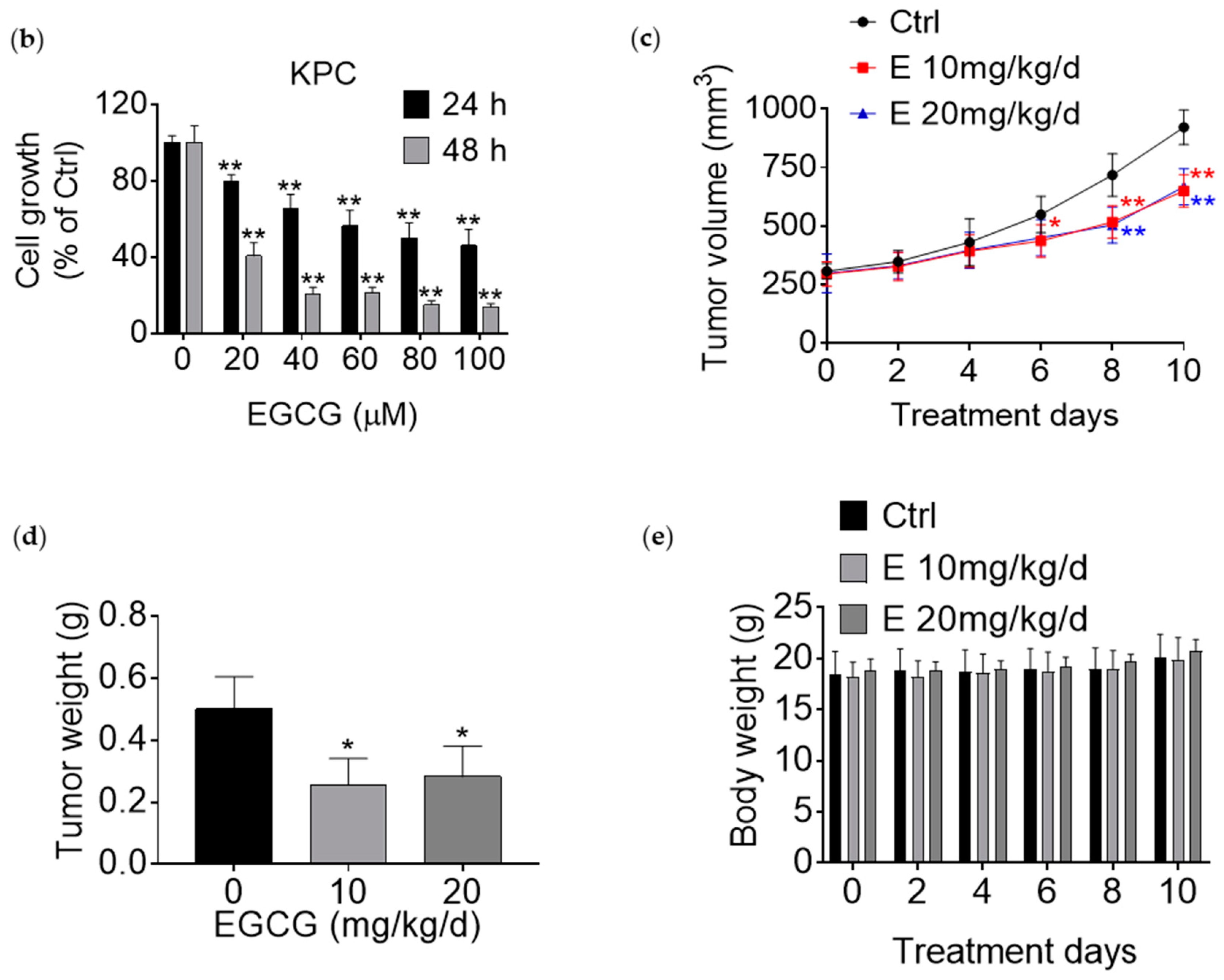
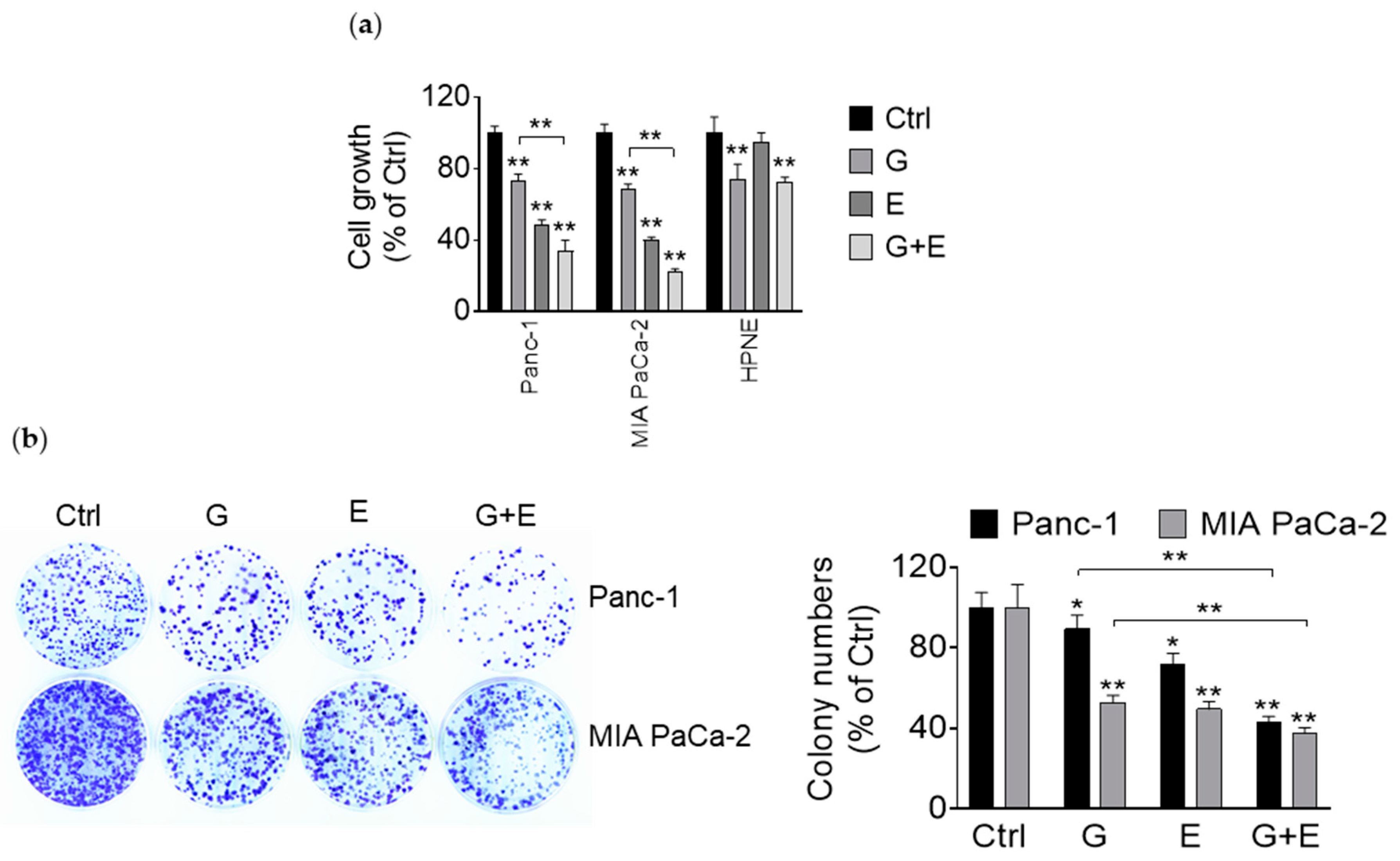
3.1. EGCG Reduces Pancreatic Cancer Cell Growth In Vitro and In Vivo
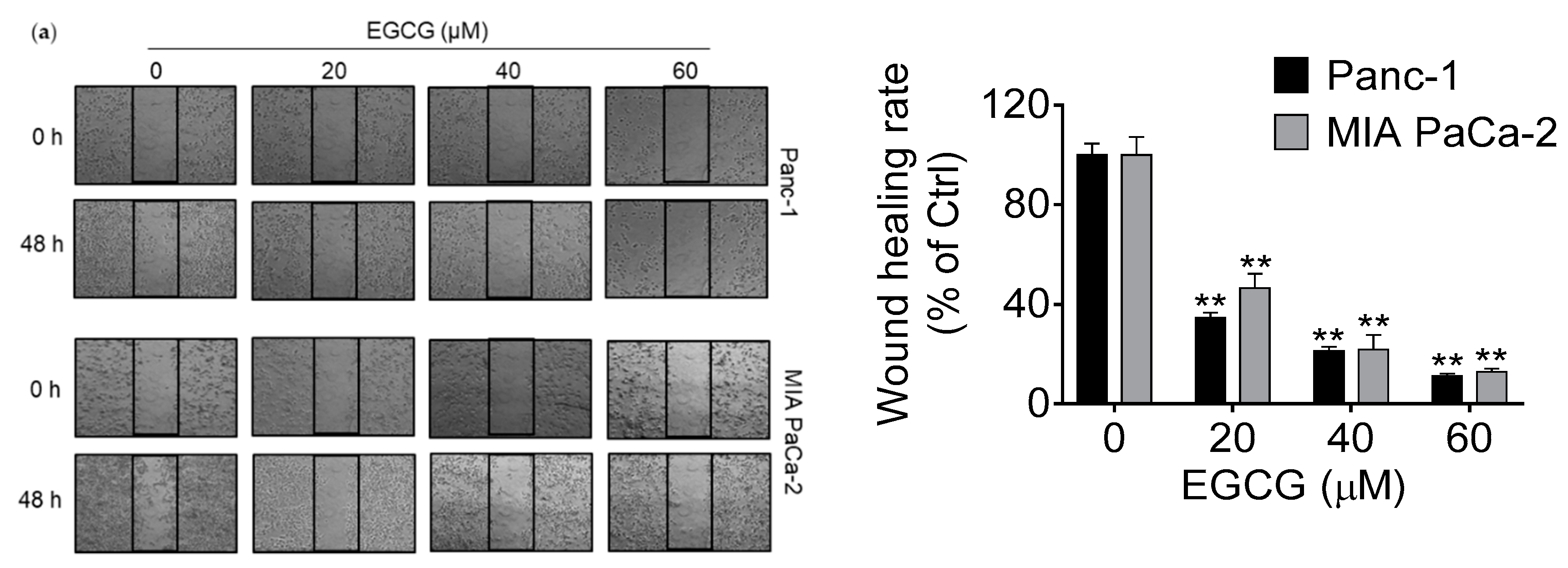
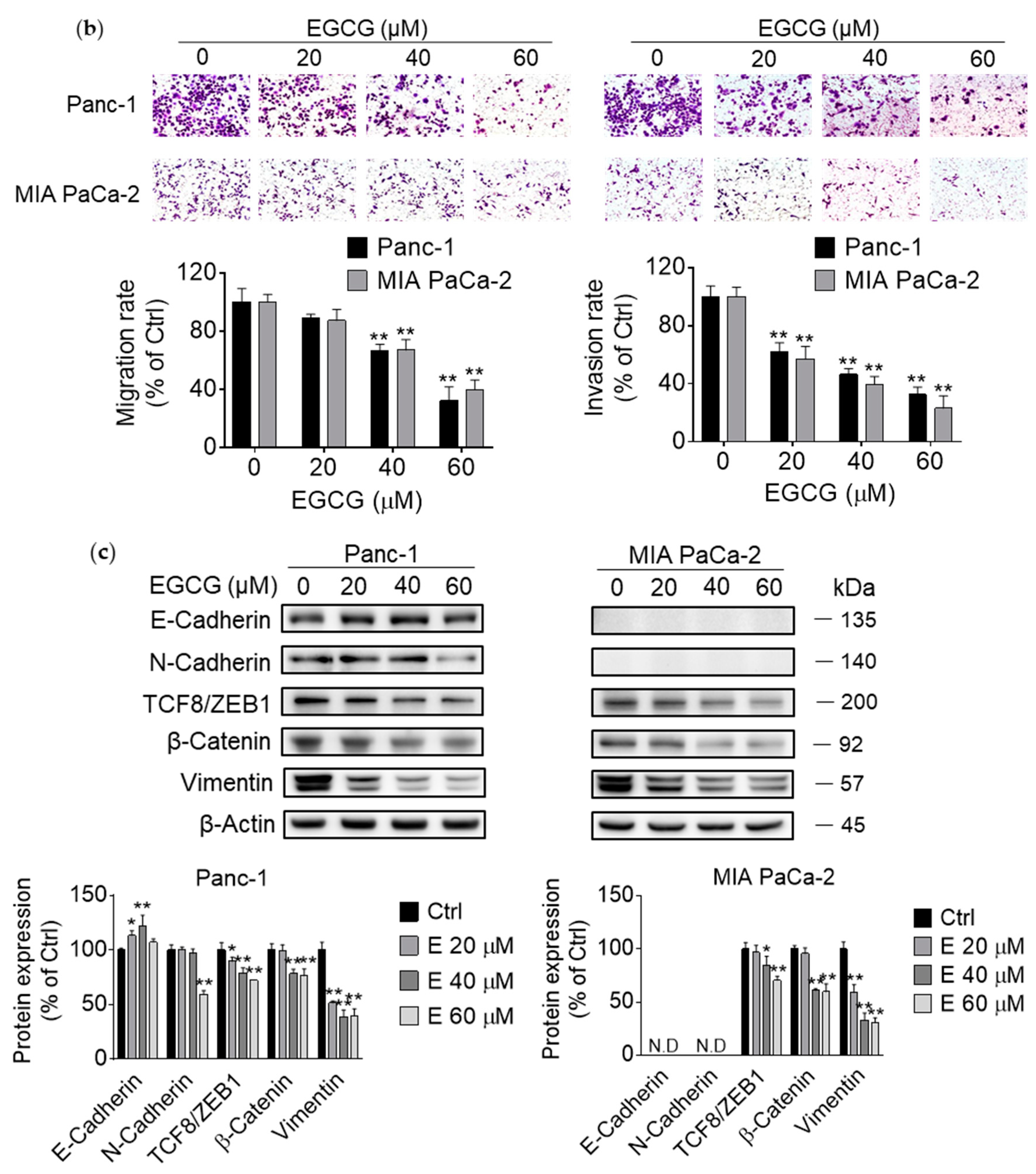
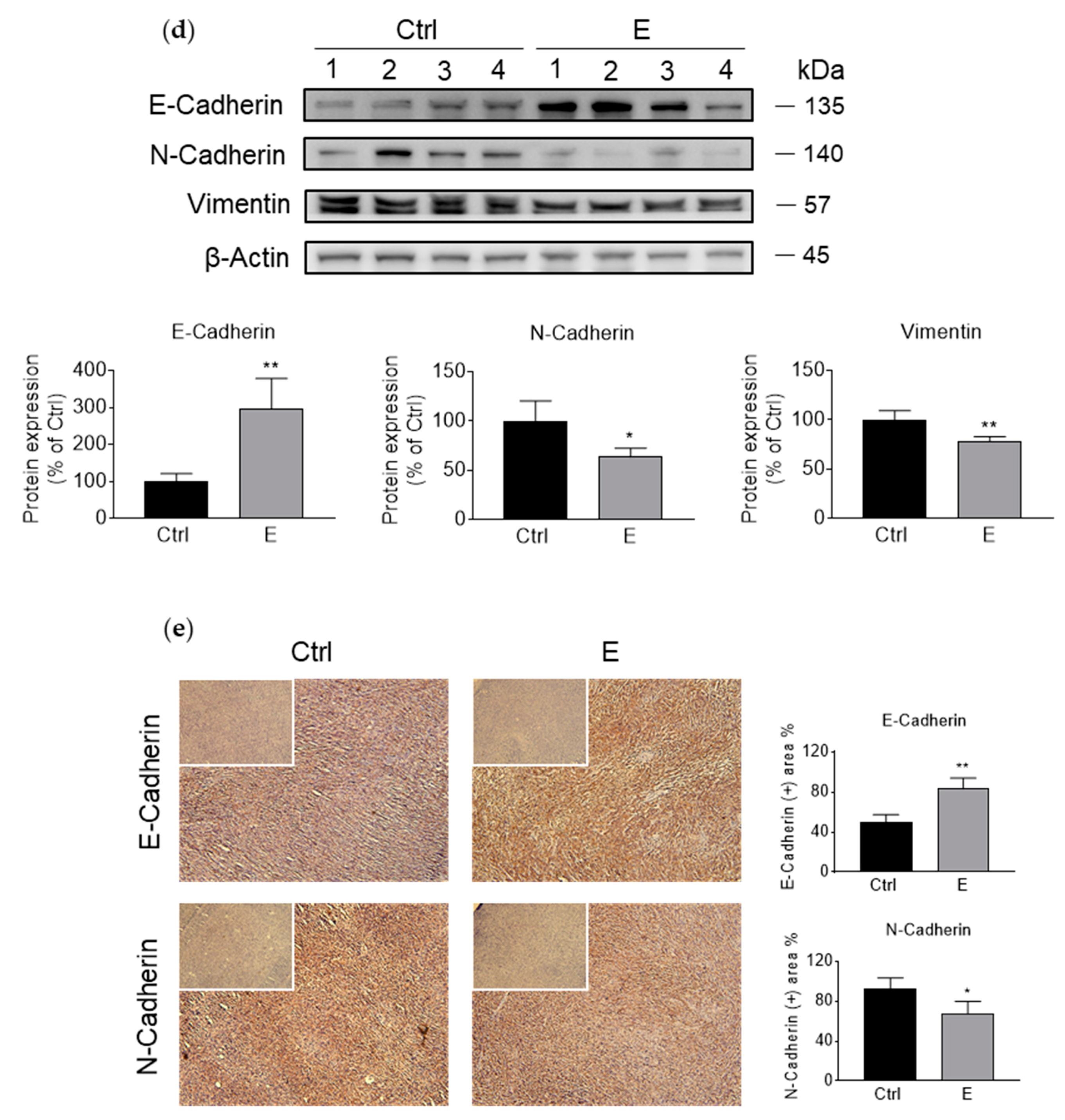
3.2. EGCG Inhibits Pancreatic Cancer Cell Migration and Invasion by Modulating the Epithelial–Mesenchymal Transition (EMT)
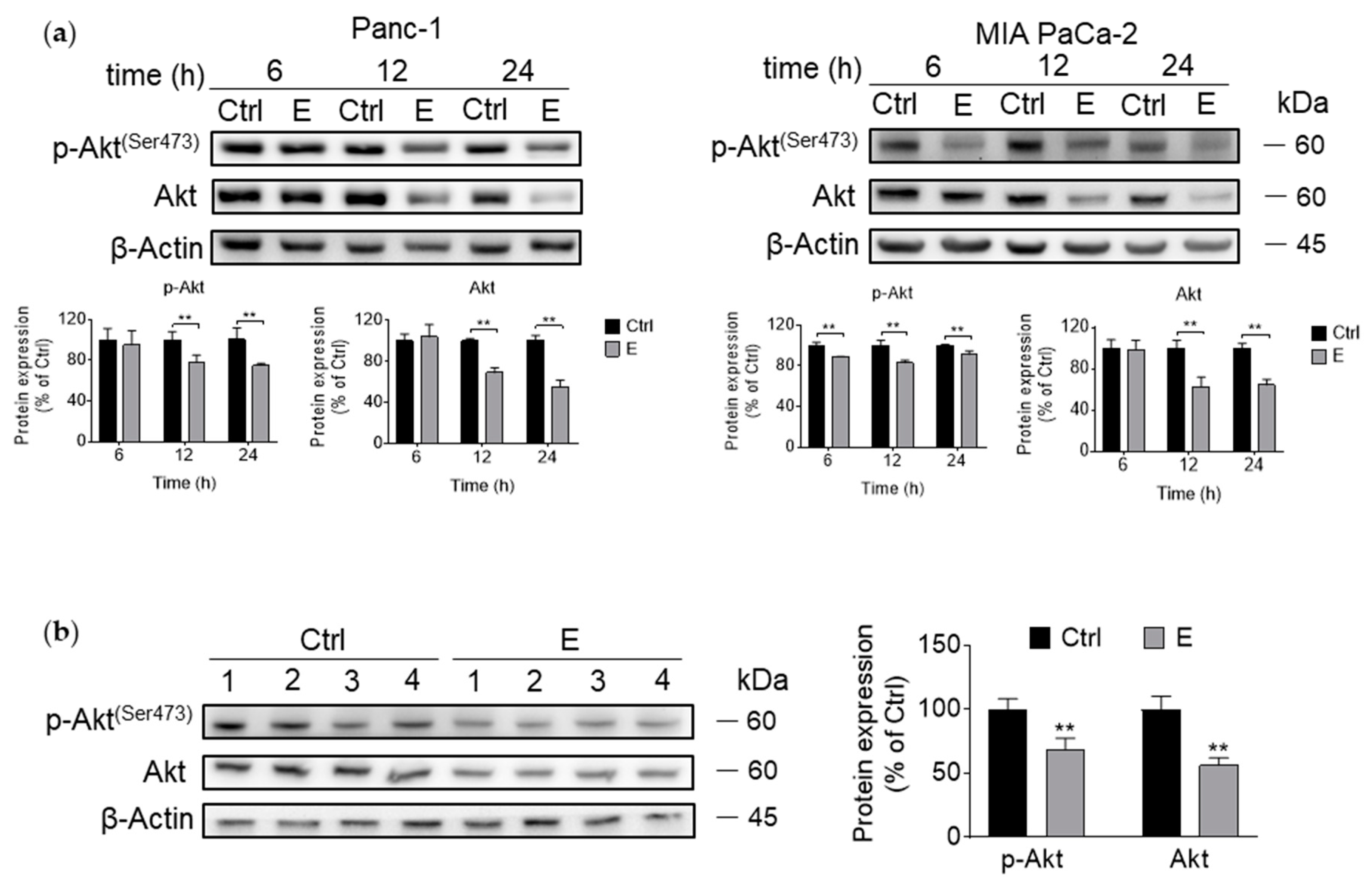
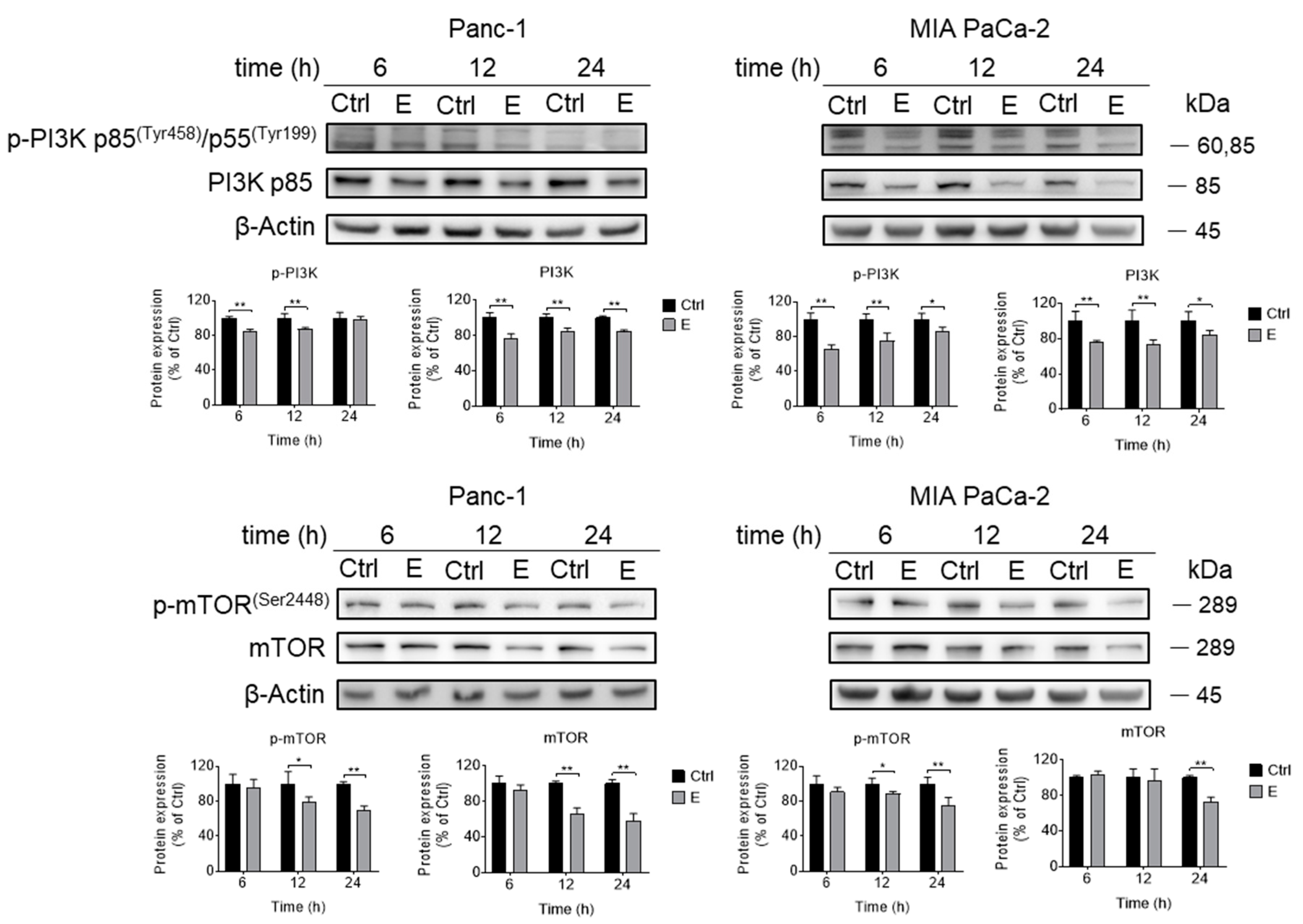
3.3. EGCG Inhibits Akt Signaling Pathway In Vitro and In Vivo
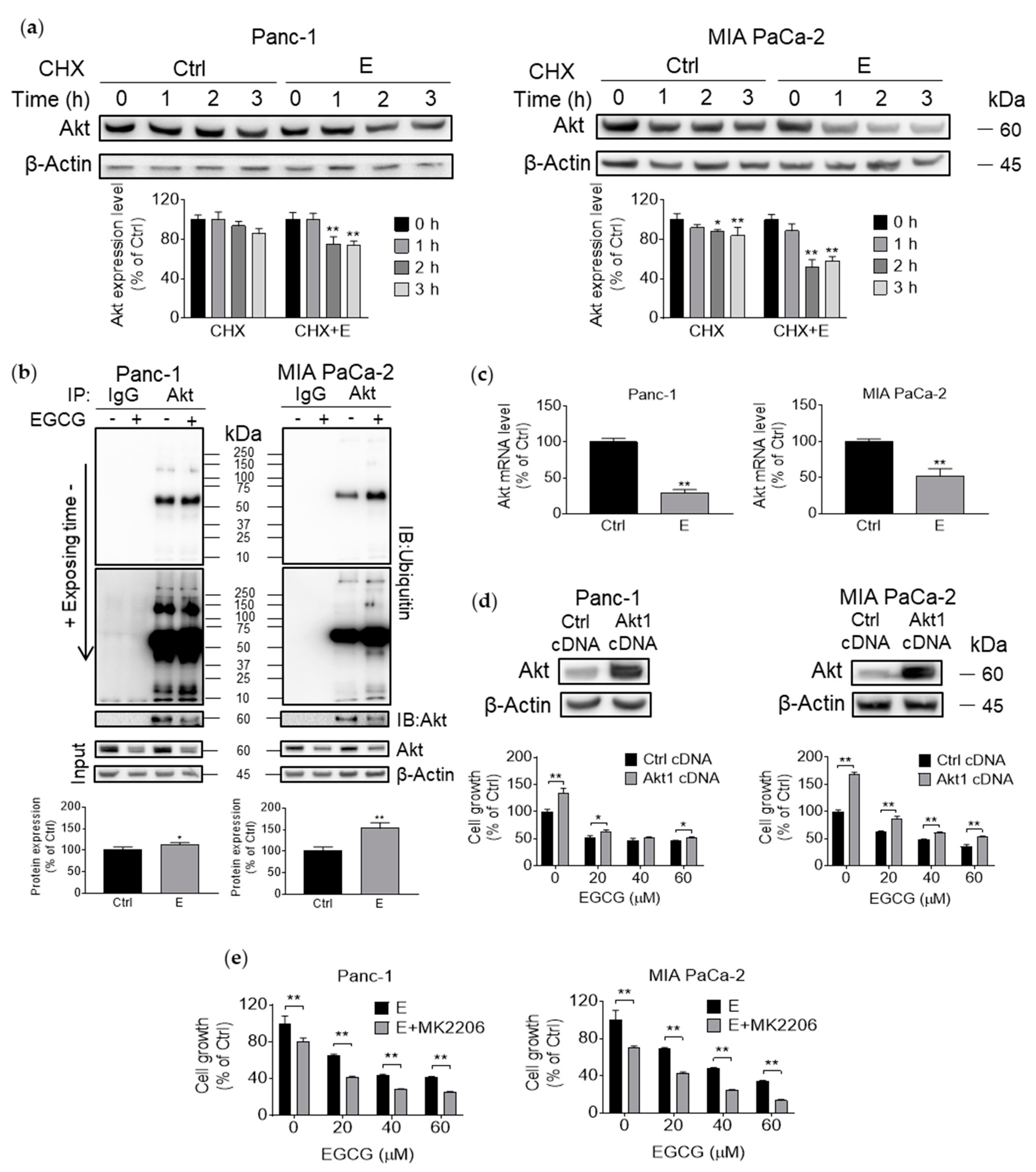
3.4. EGCG Suppresses Akt Protein Expression by Affecting Protein Transcription, Translation, and Degradation
3.5. EGCG Enhances the Growth Inhibitory Effect of Gemcitabine
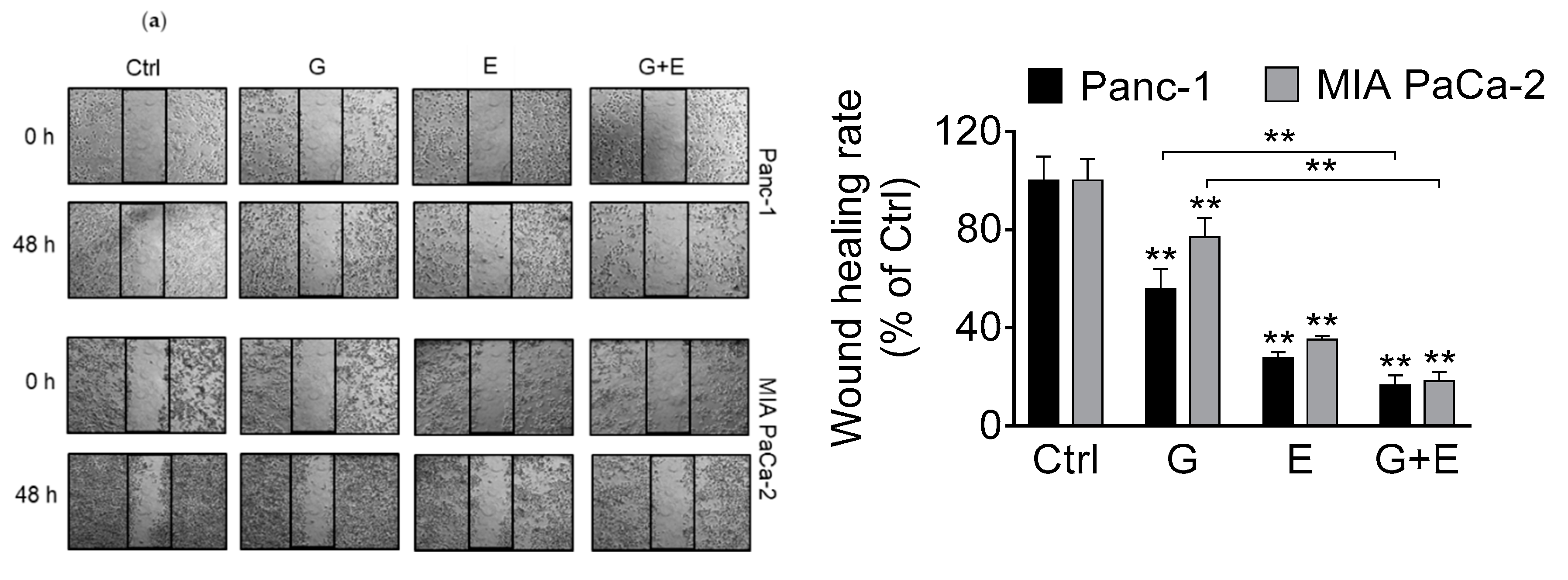
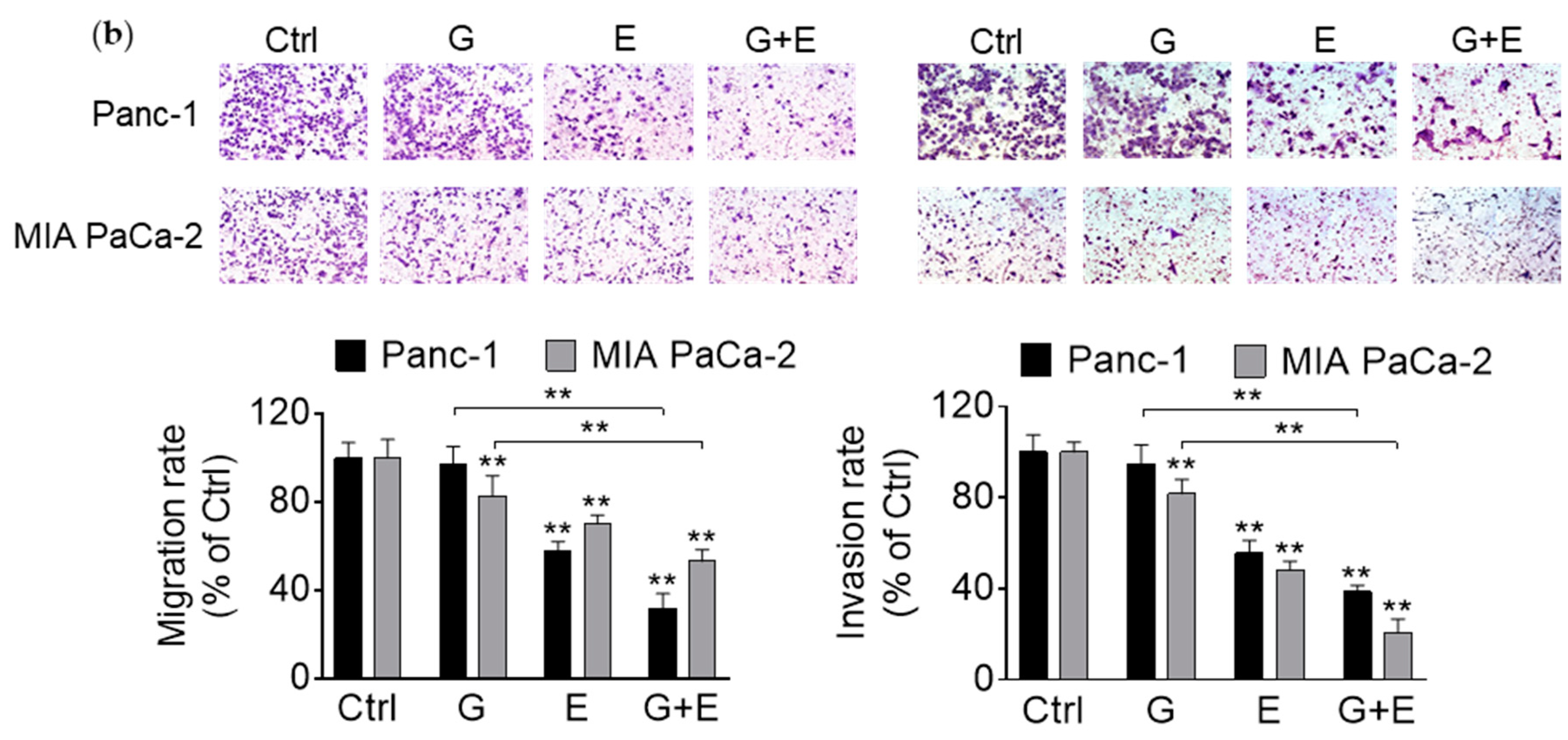
3.6. EGCG Enhances Gemcitabine’s Inhibition of Pancreatic Cancer Cell Migration and Invasion
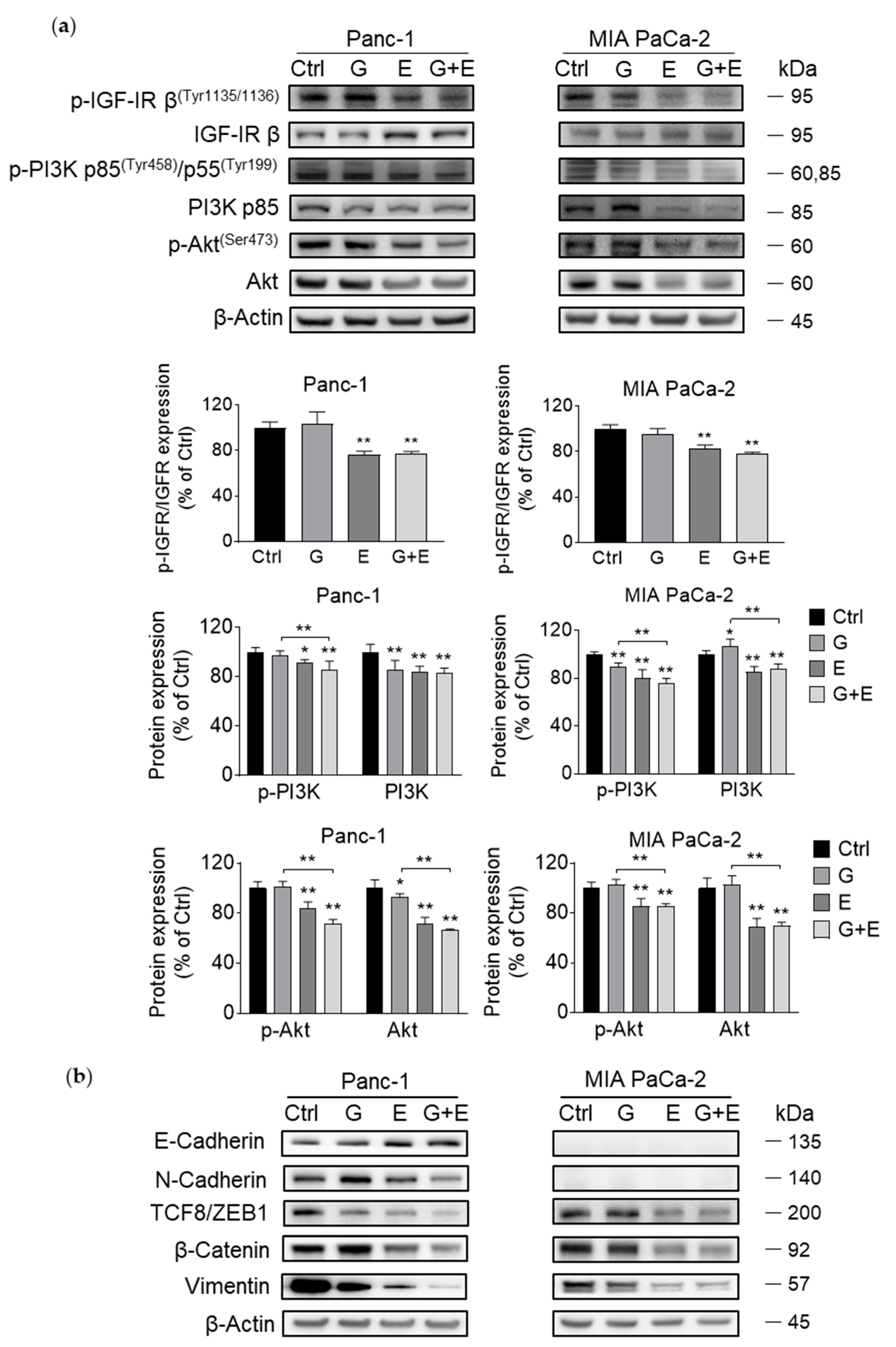
3.7. EGCG Sensitizes Gemcitabine on the Inhibition of Akt Pathway and Epithelial–Mesenchymal Transition Markers
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Aier, I.; Semwal, R.; Sharma, A.; Varadwaj, P.K. A systematic assessment of statistics, risk factors and underlying features involved in pancreatic cancer. Cancer Epidemiol. 2019, 58, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Chaffer, C.L.; Weinberg, R.A. A perspective on cancer cell metastasis. Science 2011, 331, 1559–1564. [Google Scholar] [CrossRef] [PubMed]
- Burris, H.A., III; Moore, M.J.; Andersen, J.; Green, M.R.; Rothenberg, M.L.; Modiano, M.R.; Cripps, M.C.; Portenoy, R.K.; Storniolo, A.M.; Tarassoff, P.; et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: A randomized trial. J. Clin. Oncol. 1997, 15, 2403–2413. [Google Scholar] [CrossRef] [PubMed]
- Asano, T.; Yao, Y.X.; Shin, S.; McCubrey, S.; Abbruzzese, J.L.; Reddy, S.A.G. Insulin receptor substrate is a mediator of phosphoinositide 3-kinase activation in quiescent pancreatic cancer cells. Cancer Res. 2005, 65, 9164–9168. [Google Scholar] [CrossRef]
- Asano, T.; Yao, Y.; Zhu, J.; Li, D.; Abbruzzese, J.L.; Reddy, S.A. The rapamycin analog CCI-779 is a potent inhibitor of pancreatic cancer cell proliferation. Biochem. Biophys. Res. Commun. 2005, 331, 295–302. [Google Scholar] [CrossRef]
- Morran, D.C.; Wu, J.M.; Jamieson, N.B.; Mrowinska, A.; Kalna, G.; Karim, S.A.; Au, A.Y.M.; Scarlett, C.J.; Chang, D.K.; Pajak, M.Z.; et al. Targeting mTOR dependency in pancreatic cancer. Gut 2014, 63, 1481–1489. [Google Scholar] [CrossRef]
- Tanno, S.; Testa, J.R.; Kohgo, Y. AKT activation up-regulates insulin-like growth factor-1 receptor expression and promotes invasiveness of human pancreatic cancer cells. Gastroenterology 2001, 120, A39–A40. [Google Scholar] [CrossRef]
- Qiao, M.; Sheng, S.J.; Pardee, A.B. Metastasis and AKT activation. Cell Cycle 2008, 7, 2991–2996. [Google Scholar] [CrossRef]
- Larue, L.; Bellacosa, A. Epithelial-mesenchymal transition in development and cancer: role of phosphatidylinositol 3’ kinase/AKT pathways. Oncogene 2005, 24, 7443–7454. [Google Scholar] [CrossRef]
- Sarkar, F.H.; Li, Y.; Wang, Z.; Kong, D. Pancreatic cancer stem cells and EMT in drug resistance and metastasis. Minerva. Chir. 2009, 64, 489–500. [Google Scholar]
- Gan, R.Y.; Li, H.B.; Sui, Z.Q.; Corke, H. Absorption, metabolism, anti-cancer effect and molecular targets of epigallocatechin gallate (EGCG): An updated review. Crit. Rev. Food Sci. Nutr. 2018, 58, 924–941. [Google Scholar] [CrossRef] [PubMed]
- Negri, A.; Naponelli, V.; Rizzi, F.; Bettuzzi, S. Molecular Targets of Epigallocatechin-Gallate (EGCG): A Special Focus on Signal Transduction and Cancer. Nutrients 2018, 10, 1936. [Google Scholar] [CrossRef] [PubMed]
- Granja, A.; Pinheiro, M.; Reis, S. Epigallocatechin Gallate Nanodelivery Systems for Cancer Therapy. Nutrients 2016, 8, 307. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, X.Q.; Zhang, Q.; Zhu, J.Y.; Li, Y.; Xie, C.F.; Li, X.T.; Wu, J.S.; Geng, S.S.; Zhong, C.Y.; et al. Epigallocatechin-3-Gallate Inhibits Colorectal Cancer Stem Cells by Suppressing Wnt/beta-Catenin Pathway. Nutrients 2017, 9, 572. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.Y.; Wu, C.C.; Hsu, H.Y.; Chuang, H.Y.; Huang, S.Y.; Tsai, C.H.; Chang, Y.; Tsao, G.S.; Chen, C.L.; Chen, J.Y. EGCG inhibits proliferation, invasiveness and tumor growth by up-regulation of adhesion molecules, suppression of gelatinases activity and induction of apoptosis in nasopharyngeal carcinoma cells. Int. J. Mol. Sci. 2015, 16, 2530–2558. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.N.; Fu, J.S.; Shankar, S.; Srivastava, R.K. EGCG Enhances the Therapeutic Potential of Gemcitabine and CP690550 by Inhibiting STAT3 Signaling Pathway in Human Pancreatic Cancer. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, G.G.; Queisser, N.; Wolfson, M.L.; Fraga, C.G.; Adamo, A.M.; Oteiza, P.I. Curcumin induces cell-arrest and apoptosis in association with the inhibition of constitutively active NF-kappaB and STAT3 pathways in Hodgkin’s lymphoma cells. Int. J. Cancer 2008, 123, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, G.G.; Huang, L.; Alston, N.; Ouyang, N.; Vrankova, K.; Mattheolabakis, G.; Constantinides, P.P.; Rigas, B. Targeting mitochondrial STAT3 with the novel phospho-valproic acid (MDC-1112) inhibits pancreatic cancer growth in mice. PLoS ONE 2013, 8, e61532. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, G.G.; Keen, C.L.; Oteiza, P.I. Microtubules are required for NF-kappa B nuclear translocation in neuroblastoma IMR-32 cells: modulation by zinc. J. Neurochem. 2006, 99, 402–415. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, T.C.; Lin, C.Y.; Bennett, D.J.; Wu, E.; Wu, J.M. Biochemical and cellular evidence demonstrating AKT-1 as a binding partner for resveratrol targeting protein NQO2. PloS ONE 2014, 9, e101070. [Google Scholar] [CrossRef] [PubMed]
- Che, W.; Bao, Y.; Tang, F. Down-regulation of C35 decreased the cell viability and migration of breast ductal carcinoma cells. PloS ONE 2017, 12, e0183941. [Google Scholar] [CrossRef] [PubMed]
- Wei, R.; Mao, L.; Xu, P.; Zheng, X.; Hackman, R.M.; Mackenzie, G.G.; Wang, Y. Suppressing glucose metabolism with epigallocatechin-3-gallate (EGCG) reduces breast cancer cell growth in preclinical models. Food Funct. 2018, 9, 5682–5696. [Google Scholar] [CrossRef] [PubMed]
- Bartels, L.E.; Mattheolabakis, G.; Vaeth, B.M.; LaComb, J.F.; Wang, R.; Zhi, J.; Komninou, D.; Rigas, B.; Mackenzie, G.G. The novel agent phospho-glycerol-ibuprofen-amide (MDC-330) inhibits glioblastoma growth in mice: an effect mediated by cyclin D1. Carcinogenesis 2016, 37, 420–429. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, G.G.; Ouyang, N.; Xie, G.; Vrankova, K.; Huang, L.; Sun, Y.; Komninou, D.; Kopelovich, L.; Rigas, B. Phospho-sulindac (OXT-328) combined with difluoromethylornithine prevents colon cancer in mice. Cancer Prev. Res. (Phila) 2011, 4, 1052–1060. [Google Scholar] [CrossRef] [PubMed]
- Piacentini, P.; Donadelli, M.; Costanzo, C.; Moore, P.S.; Palmieri, M.; Scarpa, A. Trichostatin A enhances the response of chemotherapeutic agents in inhibiting pancreatic cancer cell proliferation. Virchows Arch. 2006, 448, 797–804. [Google Scholar] [CrossRef] [PubMed]
- Long, L.H.; Clement, M.V.; Halliwell, B. Artifacts in cell culture: Rapid generation of hydrogen peroxide on addition of (-)-epigallocatechin, (-)-epigallocatechin gallate, (+)-catechin, and quercetin to commonly used cell culture media. Biochem. Biophys. Res. Commun. 2000, 273, 50–53. [Google Scholar] [CrossRef] [PubMed]
- Macarulla, T.; Pazo-Cid, R.; Guillen-Ponce, C.; Lopez, R.; Vera, R.; Reboredo, M.; Munoz Martin, A.; Rivera, F.; Diaz Beveridge, R.; La Casta, A.; et al. Phase I/II Trial to Evaluate the Efficacy and Safety of Nanoparticle Albumin-Bound Paclitaxel in Combination With Gemcitabine in Patients with Pancreatic Cancer and an ECOG Performance Status of 2. J Clin. Oncol. 2019, 37, 230–238. [Google Scholar] [CrossRef]
- Tong, M.T.; Wang, J.; Zhang, H.L.; Xing, H.B.; Wang, Y.L.; Fang, Y.; Pan, H.M.; Li, D. Efficacy and safety of gemcitabine plus anti-angiogenesis therapy for advanced pancreatic cancer: a systematic review and meta-analysis of clinical randomized phase III trials. J. Cancer 2019, 10, 968–978. [Google Scholar] [CrossRef]
- Xu, W.T.; Yang, Z.; Lu, N.H. A new role for the PI3K/Akt signaling pathway in the epithelial-mesenchymal transition. Cell Adhes. Migr. 2015, 9, 317–324. [Google Scholar] [CrossRef]
- Gloushankova, N.A.; Zhitnyak, I.Y.; Rubtsova, S.N. Role of Epithelial-Mesenchymal Transition in Tumor Progression. Biochemistry (Mosc.) 2018, 83, 1469–1476. [Google Scholar] [CrossRef]
- Kourtidis, A.; Lu, R.; Pence, L.J.; Anastasiadis, P.Z. A central role for cadherin signaling in cancer. Exp. Cell Res. 2017, 358, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, R.A.; Delic, S.; Herrmann, H.; Snider, N.T. Vimentin on the move: New developments in cell migration. F1000 Res. 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, M.; Neilson, E.G. Biomarkers for epithelial-mesenchymal transitions. J. Clin. Invest. 2009, 119, 1429–1437. [Google Scholar] [CrossRef] [PubMed]
- Aigner, K.; Dampier, B.; Descovich, L.; Mikula, M.; Sultan, A.; Schreiber, M.; Mikulits, W.; Brabletz, T.; Strand, D.; Obrist, P.; et al. The transcription factor ZEB1 (deltaEF1) promotes tumour cell dedifferentiation by repressing master regulators of epithelial polarity. Oncogene 2007, 26, 6979–6988. [Google Scholar] [CrossRef] [PubMed]
- Brabletz, T.; Hlubek, F.; Spaderna, S.; Schmalhofer, O.; Hiendlmeyer, E.; Jung, A.; Kirchner, T. Invasion and metastasis in colorectal cancer: epithelial-mesenchymal transition, mesenchymal-epithelial transition, stem cells and beta-catenin. Cells Tissues Organs 2005, 179, 56–65. [Google Scholar] [CrossRef]
- Kim, D.; Dan, H.C.; Park, S.; Yang, L.; Liu, Q.Y.; Kaneko, S.; Ning, J.Y.; He, L.L.; Yang, H.; Sun, M.; et al. Akt/PKB signaling mechanisms in cancer and chemoresistance. Front. Biosci. Landmark 2005, 10, 975–987. [Google Scholar] [CrossRef]
- Borse, V.; Al Aameri, R.F.H.; Sheehan, K.; Sheth, S.; Kaur, T.; Mukherjea, D.; Tupal, S.; Lowy, M.; Ghosh, S.; Dhukhwa, A.; et al. Epigallocatechin-3-gallate, a prototypic chemopreventative agent for protection against cisplatin-based ototoxicity. Cell Death Dis. 2017, 8, e2921. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Alanine Transaminase U/L | Albumin g/dL | Alkaline Phosphatase U/L | Aspartate Transaminase U/L | Blood Urea Nitrogen mg/dL | Creatinine mg/dL | Total Bilirubin mg/dL | Total Protein g/dL | |
|---|---|---|---|---|---|---|---|---|
| Reference Range | 0–403 | 2.9–4.0 | 49–172 | 0–552 | 15.2–34.7 | 0.0–0.3 | 0.0–0.2 | 4.7–6.1 |
| Ctrl | 38.6 ± 12.5 | 3.1 ± 0.1 | 72.4 ± 2.0 | 98.8 ± 17.0 | 19.8 ± 5.0 | 0.1 ± 0.1 | 0.1 ± 0.0 | 4.6 ± 0.2 |
| E 10mg/kg/d | 40.3 ± 17.2 | 3.3 ± 0.4 | 118.3 ± 44.5 | 119.6 ± 58.7 | 26.0 ± 5.2 | 0.1 ± 0.0 | 0.1 ± 0.1 | 4.9 ± 0.2 |
| E 20mg/kg/d | 35.3 ± 18.3 | 3.6 ± 0.1 | 132.0 ± 6.7 | 93.1 ± 11.5 | 27.0 ± 4.2 | 0.1 ± 0.0 | 0.1 ± 0.0 | 5.0 ± 0.3 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, R.; Cortez Penso, N.E.; Hackman, R.M.; Wang, Y.; Mackenzie, G.G. Epigallocatechin-3-Gallate (EGCG) Suppresses Pancreatic Cancer Cell Growth, Invasion, and Migration partly through the Inhibition of Akt Pathway and Epithelial–Mesenchymal Transition: Enhanced Efficacy When Combined with Gemcitabine. Nutrients 2019, 11, 1856. https://doi.org/10.3390/nu11081856
Wei R, Cortez Penso NE, Hackman RM, Wang Y, Mackenzie GG. Epigallocatechin-3-Gallate (EGCG) Suppresses Pancreatic Cancer Cell Growth, Invasion, and Migration partly through the Inhibition of Akt Pathway and Epithelial–Mesenchymal Transition: Enhanced Efficacy When Combined with Gemcitabine. Nutrients. 2019; 11(8):1856. https://doi.org/10.3390/nu11081856
Chicago/Turabian StyleWei, Ran, Natalia E. Cortez Penso, Robert M. Hackman, Yuefei Wang, and Gerardo G. Mackenzie. 2019. "Epigallocatechin-3-Gallate (EGCG) Suppresses Pancreatic Cancer Cell Growth, Invasion, and Migration partly through the Inhibition of Akt Pathway and Epithelial–Mesenchymal Transition: Enhanced Efficacy When Combined with Gemcitabine" Nutrients 11, no. 8: 1856. https://doi.org/10.3390/nu11081856
APA StyleWei, R., Cortez Penso, N. E., Hackman, R. M., Wang, Y., & Mackenzie, G. G. (2019). Epigallocatechin-3-Gallate (EGCG) Suppresses Pancreatic Cancer Cell Growth, Invasion, and Migration partly through the Inhibition of Akt Pathway and Epithelial–Mesenchymal Transition: Enhanced Efficacy When Combined with Gemcitabine. Nutrients, 11(8), 1856. https://doi.org/10.3390/nu11081856