Dietary and Sentinel Factors Leading to Hemochromatosis


Abstract
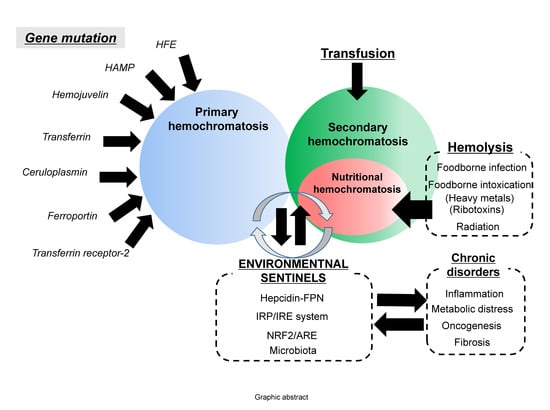
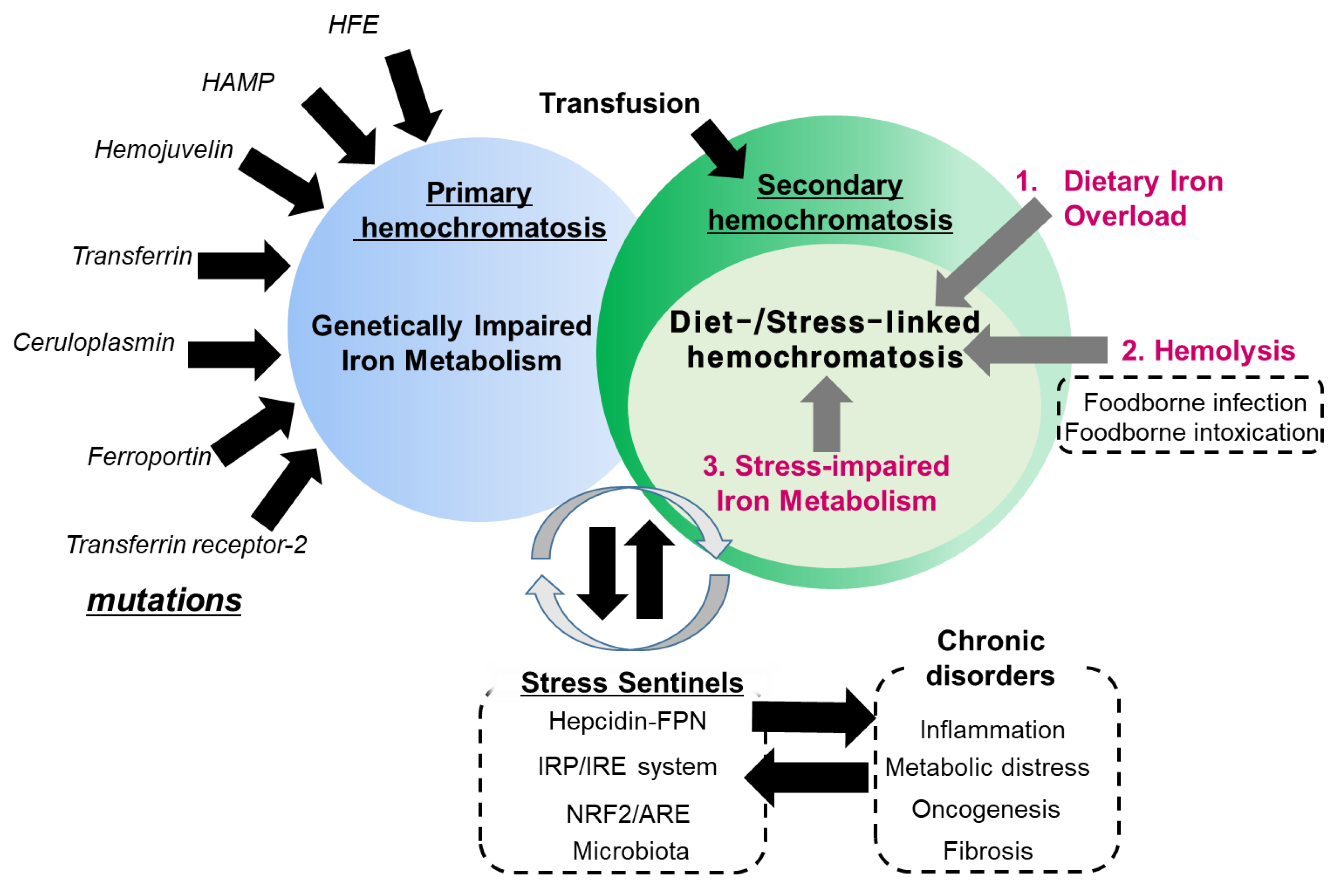
{kind=link}
{kind=link}
{kind=link}
{kind=link}
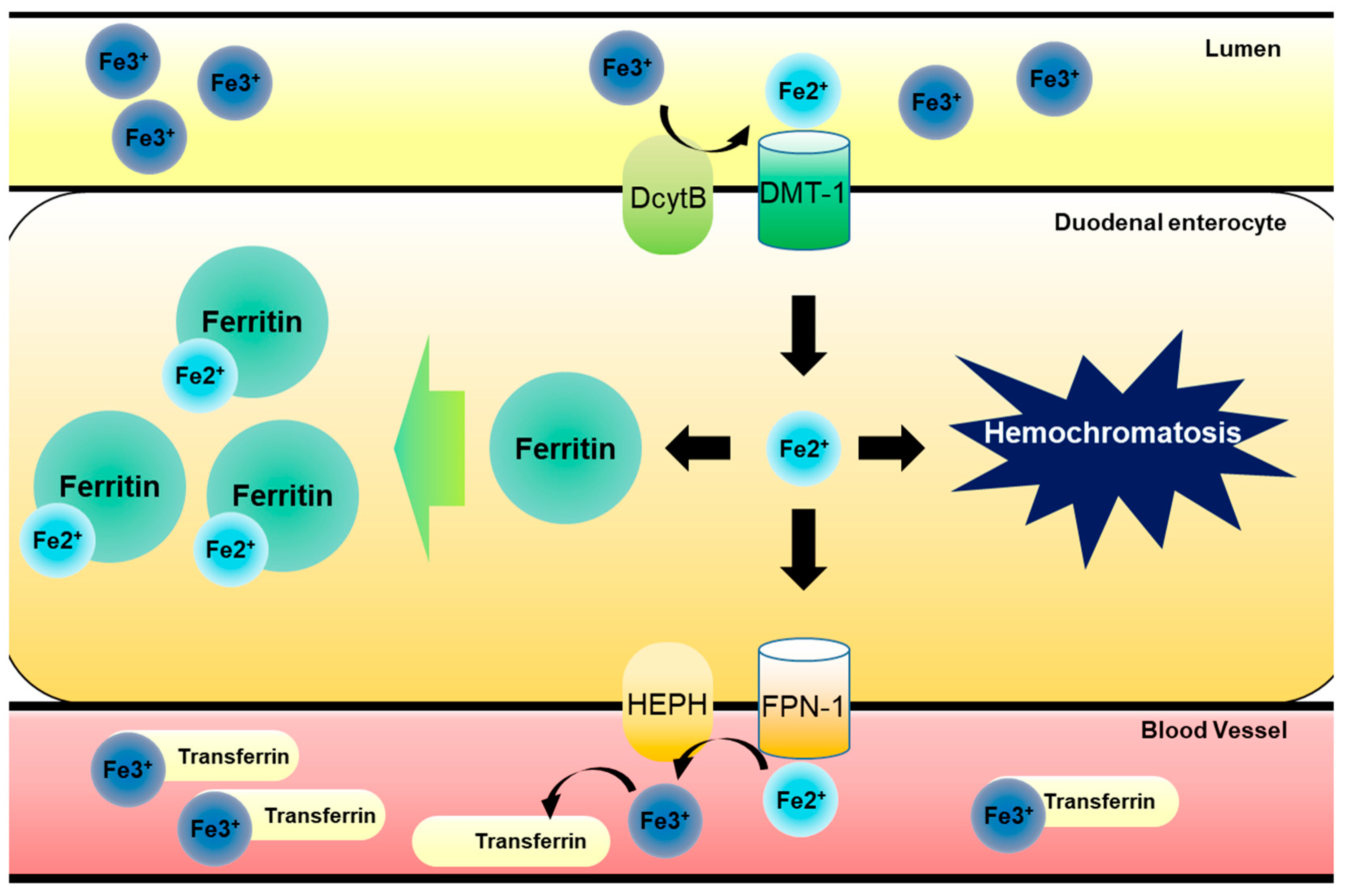
1. Introduction: Regulation of Dietary Iron Metabolism
2. Dietary Iron Overload
2.1. Iron Overload Via Consumption of High Iron-Containing Food
2.2. Gene–Nutrient–Environment Interactions in Hemochromatosis
3. Hemolysis-Associated Hemochromatosis during Foodborne Microbial Infection and Intoxication
4. Stress-Impaired Iron Metabolism
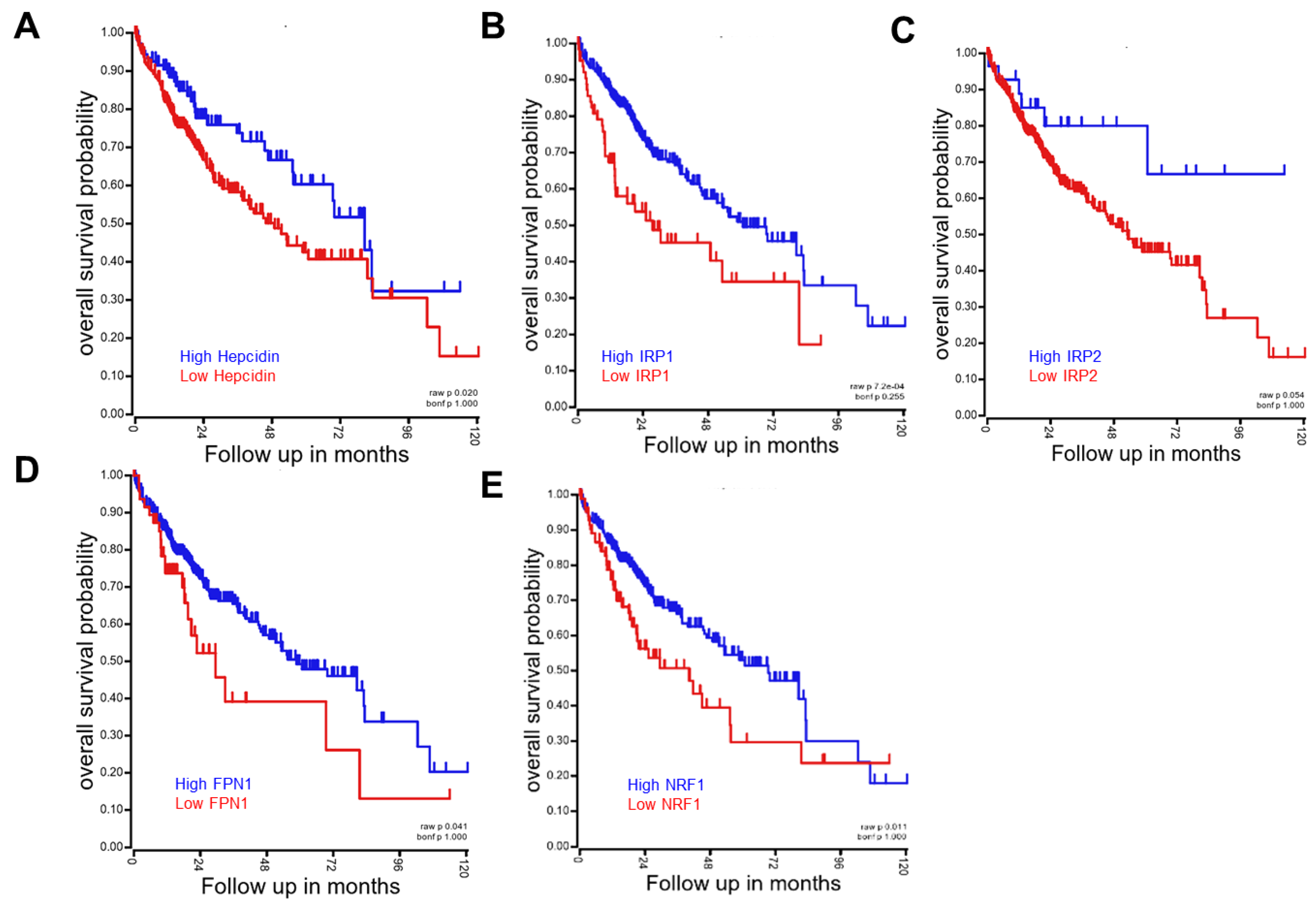
4.1. Hepcidin-FPN Axis as the Environmental Sentinel
4.2. IRP/IRE System as the Environmental Sentinel
4.3. NRF/ARE System as the Environmental Sentinel
4.4. Gut Microbiota, a Crucial Mucosal Sentinel of Hemochromatosis
5. Chronic Predisposing Factors
6. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Siah, C.W.; Ombiga, J.; Adams, L.A.; Trinder, D.; Olynyk, J.K. Normal iron metabolism and the pathophysiology of iron overload disorders. Clin. Biochem. Rev. 2006, 27, 5–16. [Google Scholar] [PubMed]
- Wang, J.; Pantopoulos, K. Regulation of cellular iron metabolism. Biochem. J. 2011, 434, 365–381. [Google Scholar] [CrossRef] [PubMed]
- Orino, K.; Lehman, L.; Tsuji, Y.; Ayaki, H.; Torti, S.V.; Torti, F.M. Ferritin and the response to oxidative stress. Biochem. J. 2001, 357, 241–247. [Google Scholar] [CrossRef]
- von Drygalski, A.; Adamson, J.W. Iron metabolism in man. JPEN J. Parenter Enteral Nutr. 2013, 37, 599–606. [Google Scholar] [CrossRef]
- Lu, J.P.; Hayashi, K. Selective iron deposition in pancreatic islet B cells of transfusional iron-overloaded autopsy cases. Pathol. Int. 1994, 44, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Pietrangelo, A. Hereditary hemochromatosis--a new look at an old disease. N. Engl. J. Med. 2004, 350, 2383–2397. [Google Scholar] [CrossRef]
- Mendes, A.I.; Ferro, A.; Martins, R.; Picanco, I.; Gomes, S.; Cerqueira, R.; Correia, M.; Nunes, A.R.; Esteves, J.; Fleming, R.; et al. Non-classical hereditary hemochromatosis in Portugal: Novel mutations identified in iron metabolism-related genes. Ann. Hematol. 2009, 88, 229–234. [Google Scholar] [CrossRef]
- Finch, S.C.; Finch, C.A. Idiopathic hemochromatosis, an iron storage disease. A. Iron metabolism in hemochromatosis. Medicine 1955, 34, 381–430. [Google Scholar] [CrossRef]
- European Association For The Study Of The Liver. EASL clinical practice guidelines for HFE hemochromatosis. J. Hepatol. 2010, 53, 3–22. [Google Scholar] [CrossRef]
- Nemeth, E.; Ganz, T. Regulation of iron metabolism by hepcidin. Annu. Rev. Nutr. 2006, 26, 323–342. [Google Scholar] [CrossRef] [PubMed]
- Njajou, O.T.; Vaessen, N.; Joosse, M.; Berghuis, B.; van Dongen, J.W.; Breuning, M.H.; Snijders, P.J.; Rutten, W.P.; Sandkuijl, L.A.; Oostra, B.A.; et al. A mutation in SLC11A3 is associated with autosomal dominant hemochromatosis. Nat. Genet. 2001, 28, 213–214. [Google Scholar] [CrossRef]
- Pietrangelo, A. Non-HFE hemochromatosis. Hepatology 2004, 39, 21–29. [Google Scholar] [CrossRef]
- Camaschella, C.; Fargion, S.; Sampietro, M.; Roetto, A.; Bosio, S.; Garozzo, G.; Arosio, C.; Piperno, A. Inherited HFE-unrelated hemochromatosis in Italian families. Hepatology 1999, 29, 1563–1564. [Google Scholar] [CrossRef]
- Adams, P.C. Hemochromatosis case definition: Out of focus? Nat. Clin. Pract. Gastroenterol. Hepatol. 2006, 3, 178–179. [Google Scholar] [CrossRef]
- Beaton, M.D.; Adams, P.C. The myths and realities of hemochromatosis. Can. J. Gastroenterol. 2007, 21, 101–104. [Google Scholar] [CrossRef]
- Jaeger, M.; Aul, C.; Sohngen, D.; Germing, U.; Schneider, W. Secondary hemochromatosis in polytransfused patients with myelodysplastic syndromes. Beitr. Infusionsther. 1992, 30, 464–468. [Google Scholar]
- Lichtman, S.M.; Attivissimo, L.; Goldman, I.S.; Schuster, M.W.; Buchbinder, A. Secondary hemochromatosis as a long-term complication of the treatment of hematologic malignancies. Am. J. Hematol 1999, 61, 262–264. [Google Scholar] [CrossRef]
- Wallerstein, R.O.; Robbins, S.L. Hemochromatosis after prolonged oral iron therapy in a patient with chronic hemolytic anemia. Am. J. Med. 1953, 14, 256–260. [Google Scholar] [CrossRef]
- Piperno, A. Classification and diagnosis of iron overload. Haematologica 1998, 83, 447–455. [Google Scholar]
- Banner, W., Jr.; Tong, T.G. Iron poisoning. Pediatr. Clin. N. Am. 1986, 33, 393–409. [Google Scholar] [CrossRef]
- Ponka, P. Tissue-specific regulation of iron metabolism and heme synthesis: Distinct control mechanisms in erythroid cells. Blood 1997, 89, 1–25. [Google Scholar] [PubMed]
- Bottomley, S.S. Porphyrin and iron metabolism in sideroblastic anemia. Semin. Hematol. 1977, 14, 169–185. [Google Scholar] [PubMed]
- Rotaru, I.; Gaman, A.; Gaman, G. Secondary haemochromatosis in a patient with thalassemia intermedia. Curr. Health Sci. J. 2014, 40, 67–70. [Google Scholar] [CrossRef] [PubMed]
- Gangaidzo, I.T.; Gordeuk, V.R. Hepatocellular carcinoma and African iron overload. Gut 1995, 37, 727–730. [Google Scholar] [CrossRef] [PubMed]
- Gordeuk, V.R. African iron overload. Semin. Hematol. 2002, 39, 263–269. [Google Scholar] [CrossRef] [PubMed]
- MacPhail, A.P.; Mandishona, E.M.; Bloom, P.D.; Paterson, A.C.; Rouault, T.A.; Gordeuk, V.R. Measurements of iron status and survival in African iron overload. S. Afr. Med. J. 1999, 89, 966–972. [Google Scholar]
- Brink, B.; Disler, P.; Lynch, S.; Jacobs, P.; Charlton, R.; Bothwell, T. Patterns of iron storage in dietary iron overload and idiopathic hemochromatosis. J. Lab. Clin. Med. 1976, 88, 725–731. [Google Scholar]
- Kowdley, K.V. Iron, hemochromatosis, and hepatocellular carcinoma. Gastroenterology 2004, 127, S79–S86. [Google Scholar] [CrossRef] [PubMed]
- Park, C.H.; Bacon, B.R.; Brittenham, G.M.; Tavill, A.S. Pathology of dietary carbonyl iron overload in rats. Lab. Investig. 1987, 57, 555–563. [Google Scholar]
- Pearson, E.G.; Hedstrom, O.R.; Poppenga, R.H. Hepatic cirrhosis and hemochromatosis in three horses. J. Am. Vet. Med. Assoc. 1994, 204, 1053–1056. [Google Scholar]
- Sabelli, M.; Montosi, G.; Garuti, C.; Caleffi, A.; Oliveto, S.; Biffo, S.; Pietrangelo, A. Human macrophage ferroportin biology and the basis for the ferroportin disease. Hepatology 2017, 65, 1512–1525. [Google Scholar] [CrossRef]
- Fu, S.; Li, F.; Zhou, J.; Liu, Z. The Relationship Between Body Iron Status, Iron Intake And Gestational Diabetes: A Systematic Review and Meta-Analysis. Medicine 2016, 95, e2383. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Li, X.; Gao, Z.; Li, H. Iron increases diabetes-induced kidney injury and oxidative stress in rats. Biol. Trace Elem. Res. 2014, 160, 368–375. [Google Scholar] [CrossRef] [PubMed]
- Sukumaran, A.; Chang, J.; Han, M.; Mintri, S.; Khaw, B.A.; Kim, J. Iron overload exacerbates age-associated cardiac hypertrophy in a mouse model of hemochromatosis. Sci. Rep. 2017, 7, 5756. [Google Scholar] [CrossRef]
- Beutler, E.; Barton, J.C.; Felitti, V.J.; Gelbart, T.; West, C.; Lee, P.L.; Waalen, J.; Vulpe, C. Ferroportin 1 (SCL40A1) variant associated with iron overload in African-Americans. Blood Cells Mol. Dis. 2003, 31, 305–309. [Google Scholar] [CrossRef]
- Smith, B.C.; Gorve, J.; Guzail, M.A.; Day, C.P.; Daly, A.K.; Burt, A.D.; Bassendine, M.F. Heterozygosity for hereditary hemochromatosis is associated with more fibrosis in chronic hepatitis C. Hepatology 1998, 27, 1695–1699. [Google Scholar] [CrossRef]
- Kazemi-Shirazi, L.; Datz, C.; Maier-Dobersberger, T.; Kaserer, K.; Hackl, F.; Polli, C.; Steindl, P.E.; Penner, E.; Ferenci, P. The relation of iron status and hemochromatosis gene mutations in patients with chronic hepatitis C. Gastroenterology 1999, 116, 127–134. [Google Scholar] [CrossRef]
- Barton, J.C.; McLaren, C.E.; Chen, W.P.; Ramm, G.A.; Anderson, G.J.; Powell, L.W.; Subramaniam, V.N.; Adams, P.C.; Phatak, P.D.; Gurrin, L.C.; et al. Cirrhosis in Hemochromatosis: Independent Risk Factors in 368 HFE p.C282Y Homozygotes. Ann. Hepatol 2018, 17, 871–879. [Google Scholar] [CrossRef]
- Deugnier, Y.; Morcet, J.; Laine, F.; Hamdi-Roze, H.; Bollard, A.S.; Guyader, D.; Moirand, R.; Bardou-Jacquet, E. Reduced phenotypic expression in genetic hemochromatosis with time: Role of exposure to nongenetic modifiers. J. Hepatol. 2018. [Google Scholar] [CrossRef]
- Bridle, K.; Cheung, T.K.; Murphy, T.; Walters, M.; Anderson, G.; Crawford, D.G.; Fletcher, L.M. Hepcidin is down-regulated in alcoholic liver injury: Implications for the pathogenesis of alcoholic liver disease. Alcohol. Clin. Exp. Res. 2006, 30, 106–112. [Google Scholar] [CrossRef]
- Harrison-Findik, D.D.; Schafer, D.; Klein, E.; Timchenko, N.A.; Kulaksiz, H.; Clemens, D.; Fein, E.; Andriopoulos, B.; Pantopoulos, K.; Gollan, J. Alcohol metabolism-mediated oxidative stress down-regulates hepcidin transcription and leads to increased duodenal iron transporter expression. J. Biol. Chem. 2006, 281, 22974–22982. [Google Scholar] [CrossRef]
- Kaltwasser, J.P.; Werner, E.; Schalk, K.; Hansen, C.; Gottschalk, R.; Seidl, C. Clinical trial on the effect of regular tea drinking on iron accumulation in genetic haemochromatosis. Gut 1998, 43, 699–704. [Google Scholar] [CrossRef]
- Saleem, K.; Wani, W.A.; Haque, A.; Lone, M.N.; Hsieh, M.F.; Jairajpuri, M.A.; Ali, I. Synthesis, DNA binding, hemolysis assays and anticancer studies of copper(II), nickel(II) and iron(III) complexes of a pyrazoline-based ligand. Future Med. Chem. 2013, 5, 135–146. [Google Scholar] [CrossRef]
- Ko, H.; Maymani, H.; Rojas-Hernandez, C. Hemolytic uremic syndrome associated with Escherichia coli O157:H7 infection in older adults: A case report and review of the literature. J. Med. Case Rep. 2016, 10, 175. [Google Scholar] [CrossRef]
- Puchala, M.; Szweda-Lewandowska, Z.; Kiefer, J. The influence of radiation quality on radiation-induced hemolysis and hemoglobin oxidation of human erythrocytes. J. Radiat. Res. 2004, 45, 275–279. [Google Scholar] [CrossRef]
- DeLoach, J.R.; Gyongyossy-Issa, M.I.; Khachatourians, G.G. Species-specific hemolysis of erythrocytes by T-2 toxin. Toxicol. Appl. Pharmacol. 1989, 97, 107–112. [Google Scholar] [CrossRef]
- Oh, C.K.; Park, S.H.; Kim, J.; Moon, Y. Non-mutagenic Suppression of Enterocyte Ferroportin 1 by Chemical Ribosomal Inactivation via p38 Mitogen-activated Protein Kinase (MAPK)-mediated Regulation: Evidence for environmental hemochromatosis. J. Biol. Chem. 2016, 291, 19858–19872. [Google Scholar] [CrossRef]
- Brandao, R.; Lara, F.S.; Pagliosa, L.B.; Soares, F.A.; Rocha, J.B.; Nogueira, C.W.; Farina, M. Hemolytic effects of sodium selenite and mercuric chloride in human blood. Drug Chem. Toxicol. 2005, 28, 397–407. [Google Scholar] [CrossRef]
- Horiguchi, H.; Oguma, E.; Kayama, F. Cadmium induces anemia through interdependent progress of hemolysis, body iron accumulation, and insufficient erythropoietin production in rats. Toxicol. Sci. 2011, 122, 198–210. [Google Scholar] [CrossRef]
- Wang, X.; Wang, L.; Liu, S. Heme-regulated eIF2alpha kinase plays a crucial role in protecting erythroid cells against Pb-induced hemolytic stress. Chem. Res. Toxicol. 2015, 28, 460–469. [Google Scholar] [CrossRef]
- Ribarov, S.R.; Benov, L.C. Relationship between the hemolytic action of heavy metals and lipid peroxidation. Biochim. Biophys. Acta 1981, 640, 721–726. [Google Scholar] [CrossRef]
- Jozefczak, M.; Remans, T.; Vangronsveld, J.; Cuypers, A. Glutathione is a key player in metal-induced oxidative stress defenses. Int. J. Mol. Sci. 2012, 13, 3145–3175. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Jomova, K.; Rhodes, C.J.; Kuca, K.; Musilek, K. Redox- and non-redox-metal-induced formation of free radicals and their role in human disease. Arch. Toxicol. 2016, 90, 1–37. [Google Scholar] [CrossRef] [PubMed]
- Beutin, L. Emerging enterohaemorrhagic Escherichia coli, causes and effects of the rise of a human pathogen. J. Vet. Med. B Infect. Dis. Vet. Public Health 2006, 53, 299–305. [Google Scholar] [CrossRef]
- Spears, K.J.; Roe, A.J.; Gally, D.L. A comparison of enteropathogenic and enterohaemorrhagic Escherichia coli pathogenesis. FEMS Microbiol. Lett. 2006, 255, 187–202. [Google Scholar] [CrossRef] [PubMed]
- Orth, D.; Wurzner, R. Complement in typical hemolytic uremic syndrome. Semin. Thromb. Hemost. 2010, 36, 620–624. [Google Scholar] [CrossRef] [PubMed]
- Stahl, A.L.; Sartz, L.; Karpman, D. Complement activation on platelet-leukocyte complexes and microparticles in enterohemorrhagic Escherichia coli-induced hemolytic uremic syndrome. Blood 2011, 117, 5503–5513. [Google Scholar] [CrossRef]
- Friedman, D.I.; Court, D.L. Bacteriophage lambda: Alive and well and still doing its thing. Curr. Opin. Microbiol. 2001, 4, 201–207. [Google Scholar] [CrossRef]
- Mayer, C.L.; Leibowitz, C.S.; Kurosawa, S.; Stearns-Kurosawa, D.J. Shiga toxins and the pathophysiology of hemolytic uremic syndrome in humans and animals. Toxins 2012, 4, 1261–1287. [Google Scholar] [CrossRef]
- Moon, Y. Cellular alterations of mucosal integrity by ribotoxins: Mechanistic implications of environmentally-linked epithelial inflammatory diseases. Toxicon 2012, 59, 192–204. [Google Scholar] [CrossRef]
- Park, S.H.; Moon, Y. Integrated stress response-altered pro-inflammatory signals in mucosal immune-related cells. Immunopharmacol. Immunotoxicol. 2013, 35, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Gyongyossy-Issa, M.I.; Khanna, V.; Khachatourians, G.G. Characterisation of hemolysis induced by T-2 toxin. Biochim. Biophys. Acta 1985, 838, 252–256. [Google Scholar] [CrossRef]
- Segal, R.; Milo-Goldzweig, I.; Joffe, A.Z.; Yagen, B. Trichothecene-induced hemolysis. I. The hemolytic activity of T-2 toxin. Toxicol. Appl. Pharmacol. 1983, 70, 343–349. [Google Scholar] [CrossRef]
- Rizzo, A.F.; Atroshi, F.; Hirvi, T.; Saloniemi, H. The hemolytic activity of deoxynivalenol and T-2 toxin. Nat. Toxins 1992, 1, 106–110. [Google Scholar] [CrossRef]
- Gyongyossy-Issa, M.I.; Khachatourians, G.G. Interaction of T-2 toxin and murine lymphocytes and the demonstration of a threshold effect on macromolecular synthesis. Biochim. Biophys. Acta 1985, 844, 167–173. [Google Scholar] [CrossRef]
- Schiefer, H.B.; Hancock, D.S. Systemic effects of topical application of T-2 toxin in mice. Toxicol. Appl. Pharmacol. 1984, 76, 464–472. [Google Scholar] [CrossRef]
- Ward, D.M.; Kaplan, J. Ferroportin-mediated iron transport: Expression and regulation. Biochim. Biophys. Acta 2012, 1823, 1426–1433. [Google Scholar] [CrossRef]
- Nemeth, E.; Tuttle, M.S.; Powelson, J.; Vaughn, M.B.; Donovan, A.; Ward, D.M.; Ganz, T.; Kaplan, J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 2004, 306, 2090–2093. [Google Scholar] [CrossRef]
- De Domenico, I.; Ward, D.M.; Langelier, C.; Vaughn, M.B.; Nemeth, E.; Sundquist, W.I.; Ganz, T.; Musci, G.; Kaplan, J. The molecular mechanism of hepcidin-mediated ferroportin down-regulation. Mol. Biol. Cell 2007, 18, 2569–2578. [Google Scholar] [CrossRef]
- Bridle, K.R.; Frazer, D.M.; Wilkins, S.J.; Dixon, J.L.; Purdie, D.M.; Crawford, D.H.; Subramaniam, V.N.; Powell, L.W.; Anderson, G.J.; Ramm, G.A. Disrupted hepcidin regulation in HFE-associated haemochromatosis and the liver as a regulator of body iron homoeostasis. Lancet 2003, 361, 669–673. [Google Scholar] [CrossRef]
- Nemeth, E.; Rivera, S.; Gabayan, V.; Keller, C.; Taudorf, S.; Pedersen, B.K.; Ganz, T. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J. Clin. Investig. 2004, 113, 1271–1276. [Google Scholar] [CrossRef]
- Ganz, T. Hepcidin, a key regulator of iron metabolism and mediator of anemia of inflammation. Blood 2003, 102, 783–788. [Google Scholar] [CrossRef]
- Rodrigues, P.N.; Vazquez-Dorado, S.; Neves, J.V.; Wilson, J.M. Dual function of fish hepcidin: Response to experimental iron overload and bacterial infection in sea bass (Dicentrarchus labrax). Dev. Comp. Immunol. 2006, 30, 1156–1167. [Google Scholar] [CrossRef]
- Chen, J.; Shi, Y.H.; Li, M.Y. Changes in transferrin and hepcidin genes expression in the liver of the fish Pseudosciaena crocea following exposure to cadmium. Arch. Toxicol. 2008, 82, 525–530. [Google Scholar] [CrossRef]
- Ilback, N.G.; Frisk, P.; Tallkvist, J.; Gadhasson, I.L.; Blomberg, J.; Friman, G. Gastrointestinal uptake of trace elements are changed during the course of a common human viral (Coxsackievirus B3) infection in mice. J. Trace Elem. Med. Biol. 2008, 22, 120–130. [Google Scholar] [CrossRef]
- Troadec, M.B.; Ward, D.M.; Lo, E.; Kaplan, J.; De Domenico, I. Induction of FPN1 transcription by MTF-1 reveals a role for ferroportin in transition metal efflux. Blood 2010, 116, 4657–4664. [Google Scholar] [CrossRef]
- Ross, S.L.; Tran, L.; Winters, A.; Lee, K.J.; Plewa, C.; Foltz, I.; King, C.; Miranda, L.P.; Allen, J.; Beckman, H.; et al. Molecular mechanism of hepcidin-mediated ferroportin internalization requires ferroportin lysines, not tyrosines or JAK-STAT. Cell Metab. 2012, 15, 905–917. [Google Scholar] [CrossRef]
- Qiao, B.; Sugianto, P.; Fung, E.; Del-Castillo-Rueda, A.; Moran-Jimenez, M.J.; Ganz, T.; Nemeth, E. Hepcidin-induced endocytosis of ferroportin is dependent on ferroportin ubiquitination. Cell Metab. 2012, 15, 918–924. [Google Scholar] [CrossRef]
- Masaldan, S.; Bush, A.I.; Devos, D.; Rolland, A.S.; Moreau, C. Striking while the iron is hot: Iron metabolism and Ferroptosis in neurodegeneration. Free Radic. Biol. Med. 2018. [Google Scholar] [CrossRef]
- Wang, Q.; Du, F.; Qian, Z.M.; Ge, X.H.; Zhu, L.; Yung, W.H.; Yang, L.; Ke, Y. Lipopolysaccharide induces a significant increase in expression of iron regulatory hormone hepcidin in the cortex and substantia nigra in rat brain. Endocrinology 2008, 149, 3920–3925. [Google Scholar] [CrossRef]
- Bayeva, M.; Chang, H.C.; Wu, R.; Ardehali, H. When less is more: Novel mechanisms of iron conservation. Trends Endocrinol. Metab. 2013, 24, 569–577. [Google Scholar] [CrossRef]
- Zhou, Z.D.; Tan, E.K. Iron regulatory protein (IRP)-iron responsive element (IRE) signaling pathway in human neurodegenerative diseases. Mol. Neurodegener. 2017, 12, 75. [Google Scholar] [CrossRef]
- Kim, S.; Wing, S.S.; Ponka, P. S-nitrosylation of IRP2 regulates its stability via the ubiquitin-proteasome pathway. Mol. Cell Biol. 2004, 24, 330–337. [Google Scholar] [CrossRef]
- Wang, J.; Chen, G.; Muckenthaler, M.; Galy, B.; Hentze, M.W.; Pantopoulos, K. Iron-mediated degradation of IRP2, an unexpected pathway involving a 2-oxoglutarate-dependent oxygenase activity. Mol. Cell Biol. 2004, 24, 954–965. [Google Scholar] [CrossRef]
- Cazzola, M.; Skoda, R.C. Translational pathophysiology: A novel molecular mechanism of human disease. Blood 2000, 95, 3280–3288. [Google Scholar]
- Birgegard, G.; Hallgren, R.; Killander, A.; Stromberg, A.; Venge, P.; Wide, L. Serum ferritin during infection. A longitudinal study. Scand. J. Haematol. 1978, 21, 333–340. [Google Scholar] [CrossRef]
- Friis, H.; Range, N.; Braendgaard Kristensen, C.; Kaestel, P.; Changalucha, J.; Malenganisho, W.; Krarup, H.; Magnussen, P.; Bengaard Andersen, A. Acute- phase response and iron status markers among pulmonary tuberculosis patients: A cross-sectional study in Mwanza, Tanzania. Br. J. Nutr. 2009, 102, 310–317. [Google Scholar] [CrossRef]
- Suarez-Ortegon, M.F.; Blanco, E.; McLachlan, S.; Fernandez-Real, J.M.; Burrows, R.; Wild, S.H.; Lozoff, B.; Gahagan, S. Ferritin levels throughout childhood and metabolic syndrome in adolescent stage. Nutr. Metab. Cardiovasc. Dis. 2018. [Google Scholar] [CrossRef]
- Fernaeus, S.; Halldin, J.; Bedecs, K.; Land, T. Changed iron regulation in scrapie-infected neuroblastoma cells. Brain Res. Mol. Brain Res. 2005, 133, 266–273. [Google Scholar] [CrossRef]
- Kim, B.H.; Jun, Y.C.; Jin, J.K.; Kim, J.I.; Kim, N.H.; Leibold, E.A.; Connor, J.R.; Choi, E.K.; Carp, R.I.; Kim, Y.S. Alteration of iron regulatory proteins (IRP1 and IRP2) and ferritin in the brains of scrapie-infected mice. Neurosci. Lett. 2007, 422, 158–163. [Google Scholar] [CrossRef]
- Nairz, M.; Ferring-Appel, D.; Casarrubea, D.; Sonnweber, T.; Viatte, L.; Schroll, A.; Haschka, D.; Fang, F.C.; Hentze, M.W.; Weiss, G.; et al. Iron Regulatory Proteins Mediate Host Resistance to Salmonella Infection. Cell Host. Microbe 2015, 18, 254–261. [Google Scholar] [CrossRef]
- Jiao, Y.; Wilkinson, J.T.; Di, X.; Wang, W.; Hatcher, H.; Kock, N.D.; D’Agostino, R., Jr.; Knovich, M.A.; Torti, F.M.; Torti, S.V. Curcumin, a cancer chemopreventive and chemotherapeutic agent, is a biologically active iron chelator. Blood 2009, 113, 462–469. [Google Scholar] [CrossRef]
- Jiao, Y.; Wilkinson, J.t.; Christine Pietsch, E.; Buss, J.L.; Wang, W.; Planalp, R.; Torti, F.M.; Torti, S.V. Iron chelation in the biological activity of curcumin. Free Radic. Biol. Med. 2006, 40, 1152–1160. [Google Scholar] [CrossRef]
- Pietsch, E.C.; Chan, J.Y.; Torti, F.M.; Torti, S.V. Nrf2 mediates the induction of ferritin H in response to xenobiotics and cancer chemopreventive dithiolethiones. J. Biol Chem. 2003, 278, 2361–2369. [Google Scholar] [CrossRef]
- Yang, C.; Zhang, X.; Fan, H.; Liu, Y. Curcumin upregulates transcription factor Nrf2, HO-1 expression and protects rat brains against focal ischemia. Brain Res. 2009, 1282, 133–141. [Google Scholar] [CrossRef]
- Tanigawa, S.; Fujii, M.; Hou, D.X. Action of Nrf2 and Keap1 in ARE-mediated NQO1 expression by quercetin. Free Radic. Biol. Med. 2007, 42, 1690–1703. [Google Scholar] [CrossRef]
- Afonyushkin, T.; Oskolkova, O.V.; Philippova, M.; Resink, T.J.; Erne, P.; Binder, B.R.; Bochkov, V.N. Oxidized phospholipids regulate expression of ATF4 and VEGF in endothelial cells via NRF2-dependent mechanism: Novel point of convergence between electrophilic and unfolded protein stress pathways. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1007–1013. [Google Scholar] [CrossRef]
- Motohashi, H.; Yamamoto, M. Nrf2-Keap1 defines a physiologically important stress response mechanism. Trends Mol. Med. 2004, 10, 549–557. [Google Scholar] [CrossRef]
- Boesch-Saadatmandi, C.; Wagner, A.E.; Graeser, A.C.; Hundhausen, C.; Wolffram, S.; Rimbach, G. Ochratoxin A impairs Nrf2-dependent gene expression in porcine kidney tubulus cells. J. Anim. Physiol. Anim. Nutr. 2009, 93, 547–554. [Google Scholar] [CrossRef]
- Limonciel, A.; Jennings, P. A review of the evidence that ochratoxin A is an Nrf2 inhibitor: Implications for nephrotoxicity and renal carcinogenicity. Toxins 2014, 6, 371–379. [Google Scholar] [CrossRef]
- Stachurska, A.; Ciesla, M.; Kozakowska, M.; Wolffram, S.; Boesch-Saadatmandi, C.; Rimbach, G.; Jozkowicz, A.; Dulak, J.; Loboda, A. Cross-talk between microRNAs, nuclear factor E2-related factor 2, and heme oxygenase-1 in ochratoxin A-induced toxic effects in renal proximal tubular epithelial cells. Mol. Nutr. Food Res. 2013, 57, 504–515. [Google Scholar] [CrossRef] [PubMed]
- Ramyaa, P.; Krishnaswamy, R.; Padma, V.V. Quercetin modulates OTA-induced oxidative stress and redox signalling in HepG2 cells—Up regulation of Nrf2 expression and down regulation of NF-kappaB and COX-2. Biochim. Biophys. Acta 2014, 1840, 681–692. [Google Scholar] [CrossRef] [PubMed]
- Cavin, C.; Delatour, T.; Marin-Kuan, M.; Holzhauser, D.; Higgins, L.; Bezencon, C.; Guignard, G.; Junod, S.; Richoz-Payot, J.; Gremaud, E.; et al. Reduction in antioxidant defenses may contribute to ochratoxin A toxicity and carcinogenicity. Toxicol. Sci. 2007, 96, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Harada, N.; Kanayama, M.; Maruyama, A.; Yoshida, A.; Tazumi, K.; Hosoya, T.; Mimura, J.; Toki, T.; Maher, J.M.; Yamamoto, M.; et al. Nrf2 regulates ferroportin 1-mediated iron efflux and counteracts lipopolysaccharide-induced ferroportin 1 mRNA suppression in macrophages. Arch. Biochem. Biophys. 2011, 508, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Ikeda, T.; Yamamoto, K.; Ogawa, H.; Kamisako, T. Dysregulated expression of fatty acid oxidation enzymes and iron-regulatory genes in livers of Nrf2-null mice. J. Gastroenterol. Hepatol. 2012, 27, 1711–1717. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Chen, L.; Peng, Z.; Wang, D.; Song, Y.; Wang, H.; Yao, P.; Yan, H.; Nussler, A.K.; Liu, L.; et al. Embryotoxicity Caused by DON-Induced Oxidative Stress Mediated by Nrf2/HO-1 Pathway. Toxins 2017, 9, 188. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Peng, Z.; Liao, Y.; Wang, L.; Li, D.; Qin, C.; Hu, J.; Wang, Z.; Cai, M.; Cai, Q.; et al. Deoxynivalenol-induced oxidative stress and Nrf2 translocation in maternal liver on gestation day 12.5d and 18.5d. Toxicon 2019, 161, 17–22. [Google Scholar] [CrossRef]
- Jarolim, K.; Del Favero, G.; Pahlke, G.; Dostal, V.; Zimmermann, K.; Heiss, E.; Ellmer, D.; Stark, T.D.; Hofmann, T.; Marko, D. Activation of the Nrf2-ARE pathway by the Alternaria alternata mycotoxins altertoxin I and II. Arch. Toxicol. 2017, 91, 203–216. [Google Scholar] [CrossRef]
- Qin, S.; Hou, D.X. Multiple regulations of Keap1/Nrf2 system by dietary phytochemicals. Mol. Nutr. Food Res. 2016, 60, 1731–1755. [Google Scholar] [CrossRef]
- Zimmermann, M.B.; Chassard, C.; Rohner, F.; N’Goran, E.K.; Nindjin, C.; Dostal, A.; Utzinger, J.; Ghattas, H.; Lacroix, C.; Hurrell, R.F. The effects of iron fortification on the gut microbiota in African children: A randomized controlled trial in Cote d’Ivoire. Am. J. Clin. Nutr 2010, 92, 1406–1415. [Google Scholar] [CrossRef]
- Buhnik-Rosenblau, K.; Moshe-Belizowski, S.; Danin-Poleg, Y.; Meyron-Holtz, E.G. Genetic modification of iron metabolism in mice affects the gut microbiota. Biometals 2012, 25, 883–892. [Google Scholar] [CrossRef]
- Bailey, J.R.; Probert, C.S.; Cogan, T.A. Identification and characterisation of an iron-responsive candidate probiotic. PLoS ONE 2011, 6, e26507. [Google Scholar] [CrossRef]
- Funakoshi, N.; Chaze, I.; Alary, A.S.; Tachon, G.; Cunat, S.; Giansily-Blaizot, M.; Bismuth, M.; Larrey, D.; Pageaux, G.P.; Schved, J.F.; et al. The role of genetic factors in patients with hepatocellular carcinoma and iron overload—A prospective series of 234 patients. Liver Int. 2016, 36, 746–754. [Google Scholar] [CrossRef]
- Hino, K.; Nishina, S.; Sasaki, K.; Hara, Y. Mitochondrial damage and iron metabolic dysregulation in hepatitis C virus infection. Free Radic. Biol. Med. 2018. [Google Scholar] [CrossRef]
- Jiang, J.W.; Chen, X.H.; Ren, Z.G.; Zheng, S.S. Gut microbial dysbiosis associates hepatocellular carcinoma via the gut-liver axis. Hepatobiliary Pancreat. Dis. Int. 2018. [Google Scholar] [CrossRef]
- Nanba, S.; Ikeda, F.; Baba, N.; Takaguchi, K.; Senoh, T.; Nagano, T.; Seki, H.; Takeuchi, Y.; Moritou, Y.; Yasunaka, T.; et al. Association of hepatic oxidative stress and iron dysregulation with HCC development after interferon therapy in chronic hepatitis C. J. Clin. Pathol. 2016, 69, 226–233. [Google Scholar] [CrossRef]
- Harrison, S.A.; Bacon, B.R. Relation of hemochromatosis with hepatocellular carcinoma: Epidemiology, natural history, pathophysiology, screening, treatment, and prevention. Med. Clin. N. Am. 2005, 89, 391–409. [Google Scholar] [CrossRef]
- Mallory, M.A.; Kowdley, K.V. Hereditary hemochromatosis and cancer risk: More fuel to the fire? Gastroenterology 2001, 121, 1253–1254. [Google Scholar] [CrossRef]
- Toyokuni, S. Mysterious link between iron overload and CDKN2A/2B. J. Clin. Biochem. Nutr. 2011, 48, 46–49. [Google Scholar] [CrossRef]
- Utzschneider, K.M.; Kowdley, K.V. Hereditary hemochromatosis and diabetes mellitus: Implications for clinical practice. Nat. Rev. Endocrinol. 2010, 6, 26–33. [Google Scholar] [CrossRef]
- Cooksey, R.C.; Jouihan, H.A.; Ajioka, R.S.; Hazel, M.W.; Jones, D.L.; Kushner, J.P.; McClain, D.A. Oxidative stress, beta-cell apoptosis, and decreased insulin secretory capacity in mouse models of hemochromatosis. Endocrinology 2004, 145, 5305–5312. [Google Scholar] [CrossRef]
- Ammann, R.W.; Muller, E.; Bansky, J.; Schuler, G.; Hacki, W.H. High incidence of extrahepatic carcinomas in idiopathic hemochromatosis. Scand. J. Gastroenterol. 1980, 15, 733–736. [Google Scholar] [CrossRef]
- Elmberg, M.; Hultcrantz, R.; Ekbom, A.; Brandt, L.; Olsson, S.; Olsson, R.; Lindgren, S.; Loof, L.; Stal, P.; Wallerstedt, S.; et al. Cancer risk in patients with hereditary hemochromatosis and in their first-degree relatives. Gastroenterology 2003, 125, 1733–1741. [Google Scholar] [CrossRef]
- Hsing, A.W.; McLaughlin, J.K.; Olsen, J.H.; Mellemkjar, L.; Wacholder, S.; Fraumeni, J.F., Jr. Cancer risk following primary hemochromatosis: A population-based cohort study in Denmark. Int. J. Cancer 1995, 60, 160–162. [Google Scholar] [CrossRef]
- Shaheen, N.J.; Silverman, L.M.; Keku, T.; Lawrence, L.B.; Rohlfs, E.M.; Martin, C.F.; Galanko, J.; Sandler, R.S. Association between hemochromatosis (HFE) gene mutation carrier status and the risk of colon cancer. J. Natl. Cancer Inst. 2003, 95, 154–159. [Google Scholar] [CrossRef]
- Tiniakos, G.; Williams, R. Cirrhotic process, liver cell carcinoma and extrahepatic malignant tumors in idiopathic haemochromatosis. Study of 71 patients treated with venesection therapy. Appl. Pathol. 1988, 6, 128–138. [Google Scholar]
- Zacharski, L.R.; Chow, B.K.; Howes, P.S.; Shamayeva, G.; Baron, J.A.; Dalman, R.L.; Malenka, D.J.; Ozaki, C.K.; Lavori, P.W. Decreased cancer risk after iron reduction in patients with peripheral arterial disease: Results from a randomized trial. J. Natl. Cancer Inst. 2008, 100, 996–1002. [Google Scholar] [CrossRef]
- Mandishona, E.; MacPhail, A.P.; Gordeuk, V.R.; Kedda, M.A.; Paterson, A.C.; Rouault, T.A.; Kew, M.C. Dietary iron overload as a risk factor for hepatocellular carcinoma in Black Africans. Hepatology 1998, 27, 1563–1566. [Google Scholar] [CrossRef]
- Gordeuk, V.R.; McLaren, C.E.; MacPhail, A.P.; Deichsel, G.; Bothwell, T.H. Associations of iron overload in Africa with hepatocellular carcinoma and tuberculosis: Strachan’s 1929 thesis revisited. Blood 1996, 87, 3470–3476. [Google Scholar]
- Moyo, V.M.; Makunike, R.; Gangaidzo, I.T.; Gordeuk, V.R.; McLaren, C.E.; Khumalo, H.; Saungweme, T.; Rouault, T.; Kiire, C.F. African iron overload and hepatocellular carcinoma (HA-7-0-080). Eur. J. Haematol. 1998, 60, 28–34. [Google Scholar] [CrossRef]
- Pinnix, Z.K.; Miller, L.D.; Wang, W.; D’Agostino, R., Jr.; Kute, T.; Willingham, M.C.; Hatcher, H.; Tesfay, L.; Sui, G.; Di, X.; et al. Ferroportin and iron regulation in breast cancer progression and prognosis. Sci. Transl. Med. 2010, 2, 43ra56. [Google Scholar] [CrossRef]
- Gutteridge, J.M.; Fu, X.C. Enhancement of bleomycin-iron free radical damage to DNA by antioxidants and their inhibition of lipid peroxidation. FEBS Lett. 1981, 123, 71–74. [Google Scholar] [CrossRef]
- Gutteridge, J.M. Iron promoters of the Fenton reaction and lipid peroxidation can be released from haemoglobin by peroxides. FEBS Lett. 1986, 201, 291–295. [Google Scholar] [CrossRef]
- Kershenobich Stalnikowitz, D.; Weissbrod, A.B. Liver fibrosis and inflammation. A review. Ann. Hepatol. 2003, 2, 159–163. [Google Scholar]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oh, C.-K.; Moon, Y. Dietary and Sentinel Factors Leading to Hemochromatosis. Nutrients 2019, 11, 1047. https://doi.org/10.3390/nu11051047
Oh C-K, Moon Y. Dietary and Sentinel Factors Leading to Hemochromatosis. Nutrients. 2019; 11(5):1047. https://doi.org/10.3390/nu11051047
Chicago/Turabian StyleOh, Chang-Kyu, and Yuseok Moon. 2019. "Dietary and Sentinel Factors Leading to Hemochromatosis" Nutrients 11, no. 5: 1047. https://doi.org/10.3390/nu11051047
APA StyleOh, C.-K., & Moon, Y. (2019). Dietary and Sentinel Factors Leading to Hemochromatosis. Nutrients, 11(5), 1047. https://doi.org/10.3390/nu11051047