Abstract
The biogeography of inflammation in ulcerative colitis (UC) suggests a proximal to distal concentration gradient of a toxin. Hydrogen sulfide (H2S) has long been considered one such toxin candidate, and dietary sulfur along with the abundance of sulfate reducing bacteria (SRB) were considered the primary determinants of H2S production and clinical course of UC. The metabolic milieu in the lumen of the colon, however, is the result of a multitude of factors beyond dietary sulfur intake and SRB abundance. Here we present an updated formulation of the H2S toxin hypothesis for UC pathogenesis, which strives to incorporate the interdependency of diet composition and the metabolic activity of the entire colon microbial community. Specifically, we suggest that the increasing severity of inflammation along the proximal-to-distal axis in UC is due to the dilution of beneficial factors, concentration of toxic factors, and changing detoxification capacity of the host, all of which are intimately linked to the nutrient flow from the diet.
1. Introduction
Inflammatory bowel disease (IBD), which includes both Crohn’s disease and ulcerative colitis (UC), is estimated to affect ~3 million individuals in the U.S. alone and continues to increase in incidence and prevalence worldwide [1]. Epidemiological evidence implicates industrialization, an increasingly western lifestyle, and associated changes in the microbiome with the development of IBD [2,3,4]. Diet is a major determinant of intestinal microbiota composition and activity [4,5]; therefore, it is a focus of intense interest for the mechanistic understanding of IBD pathogenesis.
The risk of IBD development increases in immigrant children from developing countries [6] and substantial changes in the intestinal microbiome, including loss of diversity and displacement of Prevotella by Bacteroides strains, which can be observed following immigration from developing countries to the United States [7]. These findings lend strong support to the notion that changes in the gut microbiota, driven in part by diet, contribute to the pathogenesis of UC. Although attempts to identify consistent, specific ‘trigger’ foods that predict the development of UC have been unsuccessful, there appears to be an overall trend for an association between an animal-based diet and UC development and activity, while a plant-based diet may be protective against UC [8]. Conceptually, the epidemiological data that demonstrates an increasing prevalence of IBD with westernization lend support to this notion as westernization generally results in a transition from a plant-based to an animal-based diet [9].
The unique distribution of gut inflammation seen with UC, typically greatest in the rectum and extending continuously towards the proximal colon, supports the notion that a directionally concentrated toxic substance(s) may be involved in its pathogenesis. While the nature of this compound remains unknown, several lines of evidence, both observational and mechanistic, implicate hydrogen sulfide (H2S) as a possible candidate. The H2S toxin hypothesis, however, is complicated by the fact that H2S at low levels can be anti-inflammatory [10]. Consequently, the H2S toxin hypothesis may be better viewed in a concentration dependent manner where each individual host likely has a varied tolerance to a specific concentration of H2S above which intestinal damage occurs [11].
To date, attempts to examine the role of diet in the H2S toxin hypothesis have focused primarily on the intake of sulfur-containing foods, without regard for the overall diet composition [12,13,14,15]. It is important to consider, however, that outside of a laboratory setting diets are far more complex than can be captured by the measurement of a single or a few nutrients. Therefore, rather than focus on the specific intake of a single dietary component (e.g., sulfur) in the pathogenesis of UC, an emphasis should be placed on overall diet patterns (e.g., animal- vs plant-based diet). This approach has been the focus of a number of recent reviews [16,17], and preliminary data suggest that these types of compositional dietary changes may be beneficial in IBD [18].
The high protein content of animal-based diets provides a greater amount of sulfur that becomes available to distal gut microbiota [19]. Additionally, the higher fat content of a typical animal-based diet may lead to an increase in taurine conjugation to bile acids and a corresponding increase in the quantity of sulfur in the colonic lumen through an endogenous source [20]. Importantly, an animal-based diet pattern will also tend to be low in fiber, which may drive a metabolic shift of microbiota away from carbohydrate and towards protein fermentation and increased mucin degradation (Figure 1) [21,22].
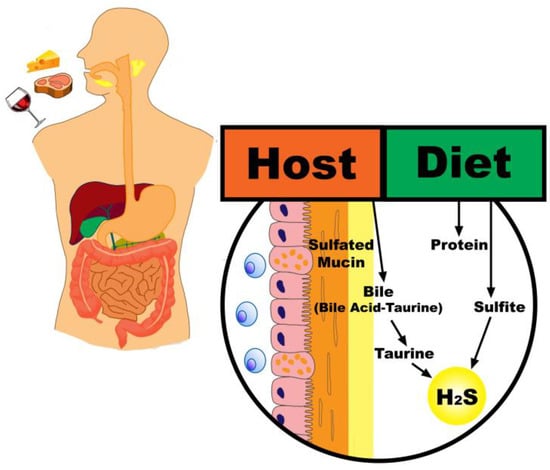
Figure 1.
Contributions of an animal-based diet to hydrogen sulfide production. Legend: An animal-based diet results in a greater amount of dietary sulfur available to distal gut microbiota, both directly through sulfur-containing amino acids and indirectly through an increase in taurine conjugated bile acids and mucin degradation.
A complimentary hypothesis to explain the directional biogeography of UC may be the dilution of beneficial microbial metabolites along the luminal flow in the intestine. One such set of products is the short-chain fatty acids (SCFA), which are generated as a result of complex polysaccharide fermentation and play essential roles in colon physiology and mucosal homeostasis. Thus, butyrate (a SCFA), in addition to being a preferred energy source for the colonocytes [23,24], also promotes the presence of regulatory T cells in the colon [25,26], strengthens the gut barrier function [27], decreases epithelial oxygenation [28,29], and inhibits excessive colonic stem cell proliferation [30]. Furthermore, the chemical milieu established by microbial metabolites impact the overall microbial community structure through feedback loops affecting the relative abundances of different microbial trophic groups. Lower pH, driven by higher concentrations of SCFA, favors the growth of methanogens over that of sulfate reducing bacteria (SRB) [31], whereas H2S may increase the oxygenation in colonocytes by inhibiting β-oxidation [32] and lead to the inhibition of obligate anaerobes that produce SCFA.
Therefore, the updated understanding of the H2S toxin hypothesis for UC pathogenesis presented here strives to account for the activity of the entire microbial community rather than individual microbial subgroups working in isolation (e.g., Desulfovibrio) and appreciates the interdependency of diet composition on both the structure and function of the microbial community. In many respects, this is a synecological approach to study of the relationship between gut microbiota and the development of UC.
1.1. Toxic Effects of Hydrogen Sulfide
Although H2S is generated by the host and is increasingly recognized to have a multitude of important beneficial physiologic functions [33], it becomes a potent toxin once its concentration exceeds the detoxifying capacity in the tissue. Specifically, higher amounts of H2S generated in the intestine have the potential to disrupt the gut barrier function, which may be an early and critical initiating event in triggering the onset of UC and the perpetuation of its activity [34]. Traditionally, the H2S toxin hypothesis has focused on the potential injurious effects of sulfide gas on the cellular metabolism of colonocytes, mainly the inhibition of cytochrome c oxidase activity in mitochondria, which induces oxidative stress in a fashion similar to cyanide [35]. Roediger and colleagues demonstrated that H2S inhibits β-oxidation of butyrate by colonocytes, their preferred energy source [36]. Notably, UC mucosa has lower rates of butyrate uptake and oxidation relative to healthy controls [37]. In summary, oxidative stress and energy starvation caused by excessive H2S concentrations may lead to colonocyte death, penetration of the epithelial barrier by the intestinal microbes and their direct interaction with the mucosal immune system. Resulting inflammation leads to further disruption of the gut barrier, decreased butyrate oxidation [37], and decreased mucosal sulfide detoxification and the subsequent perpetuation of inflammation [38].
In recent years, considerable attention has also been focused on mucus integrity in UC. The mucus layer of the colon consists of an outer (loose) and an inner (dense) layer, the latter being largely impenetrable to the resident intestinal microbiota [39,40]. The inner mucus layer in UC patients and in some animal models of IBD is more penetrable to bacteria relative to healthy controls [41]. Mucin glycoproteins form the major building blocks of mucus. The dominant mucin secreted by the goblet cells in the colon is MUC2, which contains cysteine-rich domains that mediate its multimerization through disulfide bonds. H2S can directly break the sulfide bonds in the mucus and disrupt the MUC2 network leading to a loss of mucus viscosity and greater permeability [42].
1.2. Pre-Clinical Models
Some of the strongest support for the H2S toxin hypothesis in the literature comes from pre-clinical models. The most commonly used animal model of IBD uses dextran sodium sulfate (DSS) to induce damage to the epithelium [43]. This allows for bacterial translocation from the lumen of the intestine resulting in inflammation [43]. This model is renowned for its simplicity and consistency in producing epithelial damage similar to that seen in IBD and also lends support to a role of sulfur in the pathogenesis of UC in humans.
In IL-10 knockout mice, a common murine model of IBD, a diet high in saturated fat increases the presence of taurine-conjugated bile acids, leading to an expansion of the sulphite-reducing pathobiont Bilophila wadsworthia and a greater severity of inflammation [20]. High fat diets also promote intestinal inflammation in rats as well as adenoma formation in the presence of a mutagen [44]. A high protein diet in a similar model increases SRB abundance and sulfide production and decreases the abundance of bacterial taxa associated with SCFA production and the amount of butyrate in stool [45]. Similarly, a high protein diet has been associated with post-weaning diarrhea in piglets [46]. Taken together, a high protein and saturated fat diet (characteristic of a western, animal-based diet) seems to result in an increased capacity for H2S production, decreased capacity for butyrate production, and a potential to cause intestinal inflammation.
1.3. Clinical Observations
Mesalamine, a first-line agent in treatment of mild-to-moderate ulcerative colitis, inhibits sulfide production in a dose-dependent manner when added to a fecal slurry in vitro [47]. Furthermore, UC patients taking mesalamine have reduced fecal sulfide relative to patients who do not [47]. It is noteworthy that mesalamine and butyrate are both peroxisome proliferator activated receptor gamma (PPAR-γ) agonists [48,49]. Stimulation of PPAR-γ promotes β oxidation of fatty acids and epithelial hypoxia in the colon, which favors obligate anaerobes and lowers the abundance of Proteobacteria [50]. Interestingly, some antibiotics (e.g., aminoglycosides) which target Proteobacteria (including SRB) have short-term benefits in UC patients [13,51].
Few studies have attempted to link dietary interventions targeting the updated H2S toxin hypothesis (i.e., transition from a western-style diet) with UC. The available handful of reports is limited to small case series or individual case studies (Table 1) [52,53,54,55,56]. These low dietary sulfur interventions generally emphasize transitions to a plant-based, semi-vegetarian diet. While all these experiences describe positive outcomes, conclusions are limited given the small patient numbers. The earliest case series conducted by Roediger et al. included only four patients and relied on symptom and histological criteria [52]. These patients sustained improvement in the activity of UC over 18 months of follow-up, while being maintained on the low sulfur diet. Chiba et al. reported a decreased relapse rate of UC relative to historical expectations among 60 patients instructed to follow a plant-based diet and followed for up to five years [56].

Table 1.
Summary of reports detailing dietary interventions in the treatment of ulcerative colitis (UC) targeting the updated H2S hypothesis.
2. Sources of Sulfur in the Colon
2.1. Dietary Intake
While sulfur is ubiquitous in the food supply [57], and the fifth most abundant element on earth, its dietary linkage is not well characterized. For example, the USDA Food Composition Database does not include sulfur as a searchable nutrient. The sulfur content of food can be estimated using the sulfur-containing amino acids (methionine and cysteine) as surrogates, but this fails to account for sulfur-containing food modifiers or additives, such as sulfiting agents (e.g., potassium bisulfate, sodium bisulfate), sulfuric acid, or carrageenan [58]. Therefore, the most robust method of estimating dietary sulfur intake, including an estimate for inorganic sulfur, is through a 24–48 hour urine collection [59,60]. Because the primary dietary sources of sulfur are the sulfur-containing amino acids, in practice, quantification of sulfur-containing amino acid content has been used to create low- and high-sulfur diets [52,61].
Dietary input and small intestinal absorption are considered the main determinants of sulfur delivery to the colon [19]. The small intestine has an efficient but saturable dietary sulfate absorptive capacity of ~5–7 mmol/day [62]. Once dietary sulfate intake exceeds ~5–7 mmol/day the amount reaching the colon increases linearly with intake. Therefore, the amount of dietary sulfur that reaches the colon, particularly with increasing levels, can be generally assumed to parallel dietary intake. A number of additional factors, however, such as food preparation (e.g., cooked versus uncooked, cooking temperature and method, ground versus whole), meal consumption habits (e.g., chewing), and transit time have been shown to influence small intestinal absorption of sulfur-containing molecules and the total amount of sulfur reaching the colon [63,64,65,66,67].
2.2. Endogenous Sources
The main endogenous sources of sulfur are taurine-conjugated bile acids and mucin. Taurine is a sulfur-containing amino acid that is synthesized in the liver from methionine and cysteine. Diet can influence the rate of taurine conjugation and subsequent presence of taurine conjugated bile acids in the colon [68]. Although the majority of bile acids are re-absorbed in the distal small intestine into the enterohepatic circulation, a small percentage will spill into the colon [69]. Deconjugation of bile acids is an early step in secondary bile metabolism and is mediated by gut microbiota.
Intestinal mucins are glycoproteins that form a mucus barrier along the epithelial surface of the gastrointestinal tract. The primary intestinal mucin MUC2 contains cysteine amino acids as core components of its structure [39,70]. Glycosylation of mucin proteins makes up a substantial portion of their size and provides protection of the peptide backbone and the gel-forming capacity [70]. The glycosylation pattern of mucin varies along the intestinal tract with increasing sulfation moving distally through the small intestine and into the colon [71,72]. The colonic mucus barrier is predominately composed of sulfomucins with a trend toward slightly diminished sulfation moving distally (100% sulfation in the right colon and 86% in the rectum) [73]. The normal sulfation pattern is altered in UC and is associated with the concentration of SRB [73].
2.3. H2S Measurement
The ideal experimental system to test the role of H2S in UC pathogenesis would be able to measure its intraluminal concentration at different segments in the intestine. Unfortunately, technology to do that does not yet exist; therefore, studies that measure H2S have focused on ex-vivo determination of its production from fecal samples. Fecal concentrations of hydrogen sulfide, however, are notoriously difficult due to the volatility of the gas, low intestinal concentrations, and instrumentation issues. Historically, the majority of work assessing stool sulfide concentrations in UC relied on colorimetric assays [62,74,75]. This method requires ‘trapping’ H2S in fresh fecal samples for measurement and is associated with a number of inherent limitations [76,77]. Ion-exchange chromatography has also been used to measure H2S in feces [78].
H2S gas measurements, done using gas chromatography (GC) with sulfur chemiluminescence detection, are considered the reference standard for H2S measurement [79]. These measures, however, cannot be obtained in real time and require in vitro incubation of the fecal sample. Only one study of UC has used GC as the measurement method and found that H2S production was elevated substantially in UC compared to controls over a 24 hour period [80]. Although this method does not capture the H2S content of fresh fecal samples, in vitro incubation may reflect in vivo H2S production capacity of the microbiota, providing a functional measure of the microbiota metabolic capacity. Furthermore, this method allows for interrogation of the capacity of various dietary components to affect H2S production (e.g., more H2S production with cysteine or mucin vs sulfate) [80,81].
3. Colonic Microbiota and H2S Production
3.1. Sulfate-Reducing Bacteria
Microbial production of H2S is generally thought to be carried out by a limited number of bacteria and archael species. Sulfate-reducing bacteria (SRB) and the dissimilatory sulfur cycle, a form of anaerobic respiration that uses sulfate as the terminal electron acceptor, have been the primary focus of the H2S toxin hypothesis in UC [14]. This pathway generates H2S as an end-product of sulfate reduction. The pathway consists of three enzymatic steps involving ATP sulfurylase, adenosine 5’-phosphosulfate reductase, and sulfite reductase [82]. The final step in the pathway, sulfite reduction, is considered the rate limiting step. The genus Desulfovibrio is regarded as the most abundant SRB in humans [83]. Despite the focus on Desulfovibrio spp. as the primary producer of H2S in the human gut, the number of microbial groups known to be capable of dissimilatory sulfate reduction continues to expand [82,84]. Many of these microbes can also reduce sulfite, dithionite, thiosulfate, elemental sulfur, and several thionates. The three sulfite reductase genes which catalyze the production of hydrogen sulfide are dissimilatory sulfite reductase (dsr), anaerobic sulfite reductase (asr), and cytochrome c sulfite reductase (mccA) [82].
In humans, the measurement of key enzymes involved in the sulfate reduction pathway is a more specific method for measurement of SRB than measurement of bacterial genera such as Desulfovibrio [85]. This approach identified a unique capacity of SRB genera in UC to generate H2S and induce epithelial apoptosis, compared with healthy controls [74,86]. Recent multiomics data in colon cancer lends additional support to the importance of accounting for the functional capacity of microbes (e.g., via measurement of key enzymes) in addition to their community composition [87].
Despite their importance and usefulness, there is a dearth of literature that relies on the quantification of sulfite reductase genes to characterize SRBs in large UC cohorts. Use of this technique in healthy subjects demonstrated that an animal-based diet increases sulfite reductase gene expression [5]. Recent work by Jia et al. used a semi-nested PCR method to detect dsrB DNA, but failed to find a difference in abundance between healthy and UC cohorts (n = 18 and n = 14, respectively) [88]. The agar shake dilution method, which measures SRB growth over a specified incubation period, has often been employed to measure SRB in individuals with UC. Studies done using this methodology found elevated SRB abundance in individuals with UC compared to those without [47,74,89].
3.2. Cysteine Degraders
Although sulfate intake and quantity of SRB has been the primary focus of the H2S toxin hypothesis, there is a growing appreciation for the contribution of protein intake to H2S production. The efficiency of protein degradation is greatest in the distal colon and at neutral to alkaline pH, which suggests a possible relationship with the pathogenesis of UC [90]. A cross-over diet study conducted by Magee and colleagues demonstrated that dietary protein intake positively correlates with fecal sulfide concentrations in healthy individuals [78].
A recent in vitro study that profiled gas production from incubated fecal samples implicated cysteine specifically as a primary driver of H2S production—conversely, the effect of sulfate was small [81]. Cysteine degradation to H2S is catalyzed by enzymes with cysteine desulfhydrase activity. Although the enzymes with cysteine desulfhydrase activity are well characterized in humans [91], identifying enzymes with this activity in microorganisms has proven far more challenging. Escherichia coli possess several enzymes with cysteine desulfhydrase activity, including O-acetylserine sulfhydrylase-A, O-acetylserine sulfhydrylase-B, MalY, tryptophanase and cystathionine β-lyase [92]. Because cysteine likely makes a substantial contribution to H2S production in the colonic lumen, there is a critical need to detail cysteine desulfhydrase activity within the colonic microbiome.
Prevention of excessive cysteine degradation to H2S may underlie the possible protective role of appendectomy for appendicitis in UC [93]. Although the mechanism underlying the purported beneficial effect of appendectomy in UC has yet to be elucidated, one possibility is related to the potential role of the appendix as a reservoir for gut microbes [93]. Specifically, the appendix contains Fusobacterium spp. in its healthy state [94] and appendicitis is associated with an abundance of this bacteria [95]. Fusobacterium spp. contain a number of proteins necessary to metabolize L-cysteine to H2S [96,97]. Therefore, removal of the appendix, and subsequently a source of H2S production, may contribute to its possible therapeutic role in the prevention of UC.
3.3. Interplay Between Functional Microbial Groups
A number of physicochemical factors shape the composition and functional output of microbial communities, which need to be integrated in order to fully account for the inter-individual differences. One such factor is the availability of hydrogen (H2), which is essential as a substrate for anaerobic respiration in the colonic lumen. The groups of bacteria that rely on and compete for H2 for anaerobic respiration are acetogens, methanogens, and SRB Figure 2) [98]. H2 production results from microbial fermentation of carbohydrates in the lumen of the intestine. Therefore, the balance between H2 production and consumption is often referred to as the ‘hydrogen economy’ [99]. An imbalance of H2 consumption and production may result in a metabolic environment that consumes NADH for H2 production at the expense of butyrate production [100]. In the context of the H2S toxin hypothesis, it is important to consider possible imbalances in H2 consumption (e.g., altered SRB concentration) or production (e.g., low dietary intake of fermentable carbohydrate) that could create a metabolic environment leading to UC—namely, excessive H2S production [80], mucin degradation [22,41], and diminished butyrate production [5].
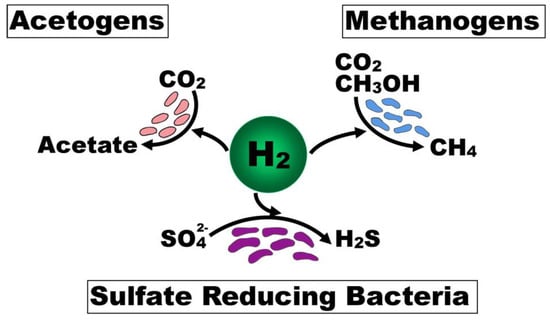
Figure 2.
Bacterial competition for hydrogen for anaerobic respiration in the lumen of the intestine. Legend: Acetogens, methanogens, and sulfate reducing bacteria are the microbial groups that compete for H2 in anaerobic respiration in the lumen of the colon. The availability of hydrogen can shape the composition and functional output of microbial communities.
Other physical (e.g., intestinal transit time) and chemical factors (e.g., pH and oxygen tension), also contribute to the microbial community structure in the colon by favoring some groups of microbes over others. Accelerated colon motility is likely to benefit fast-growing microorganisms and disfavor slow growing microorganisms, and constipation is associated with higher carriage of methanogenic archaea and increased methane production [101]. Fermentation of digestible carbohydrate results in the production of SCFA and a drop in luminal pH in the colon relative to the small intestine [102,103,104]. Increased dietary fiber also results in faster intestinal transit [105]. Indeed, the colon pH is lowest proximally and increases distally, which is likely driven by the decreasing availability of digestible carbohydrate [102]. Mildly acidic pH, characteristic of the proximal colon, is inhibitory to the growth of SRB, but favors the growth of methanogens and some butyrate producers [30,106,107]. In contrast, sulfate reduction and H2S production are optimal at an alkaline pH [108].
3.4. H2S Clearance
Clearance of hydrogen sulfide in the gut can be viewed as the final component of the H2S toxin hypothesis. Due to its potential deleterious effects, H2S is highly regulated in the cell. As H2S is freely permeable across membranes, luminal concentration in the colon would be expected to correlate with intracellular concentrations [10]. Recent work in animal models demonstrates the relationship between luminal H2S and intracellular regulation [109]. Although a small amount of H2S present in the lumen of the colon is able to be cleared through flatus [110], the primary pathway for H2S clearance is via intracellular sulfide oxidation. Interestingly, this pathway may be facilitated by cyanide, which has been proposed by Levitt and colleagues to explain the well-documented beneficial role of smoking in UC [111].
Colonic mucosa possesses a very efficient system for H2S detoxification, and defects in these pathways are hypothesized to contribute to pathogenesis of colitis [111,112]. In earlier work, thiol methyltransferase and rhodanese were thought to have a major role in the detoxification of intestinal H2S [113]. However, the investigators found conflicting results when using these enzymes to estimate the H2S detoxification capacity in UC [38,114]. Around the time this work was being conducted, however, a more detailed and comprehensive understanding of H2S detoxification was being developed that implicates sulfide quinone oxidoreductase (SQR) as the rate-limiting enzyme in sulfur oxidation [115,116].
The SQR enzyme is located within the inner mitochondrial membrane [117]. The reaction of H2S with SQR results in oxidation of H2S to a small-molecule persulfide and reduction of coenzyme Q (CoQ), which links H2S oxidation with complex III of the electron transport chain [117]. The sulfur transfer from H2S to a small molecule acceptor occurs via a cysteine intermediate within the SQR enzyme. Therefore, the availability of a small molecule acceptor is believed to be the rate-limiting step in the SQR reaction [118]. The antioxidant glutathione (GSH) is also believed to be the primary acceptor in a physiological setting, but sulfite has also been proposed as an alternative acceptor [118]. If GSH is utilized as the acceptor, the result is formation of glutathione persulfide (GSSH), which is then utilized by persulfide dioxygenase (ETHE1) or rhodanese to form sulfite or thiosulfate, respectively [117,119]. If sulfite is used as the acceptor, thiosulfate is produced, and rhodanese instead converts thiosulfate to GSSH, which is then converted to sulfite by ETHE1 [117,119]. In a steady state, GSH has been estimated to be favored ~5:1 over sulfite [118], but elevated intracellular sulfite concentrations or depletion of GSH have been suggested to inhibit SQR [116,118,119]. Furthermore, although H2S oxidation appears to be a robust pathway for intracellular control of H2S levels, an excess of H2S may paradoxically result in feedback inhibition of SQR from the electron transport chain as a result of its deleterious effect on cytochrome c oxidase activity [119].
The H2S detoxification capacity and expression of enzymes involved in the H2S detoxification pathway (SQR, ETHE1, and thiosulfate sulfurtransferase) have been shown to follow a general trend of highest in the proximal colon to lowest in the rectum [120]. This expression pattern lends further support to the H2S toxin hypothesis as the progression of UC progresses from the lowest to the highest H2S detoxification capacity and enzyme expression (Figure 3). In addition, GSH synthesis has been demonstrated to be impaired in IBD [121].
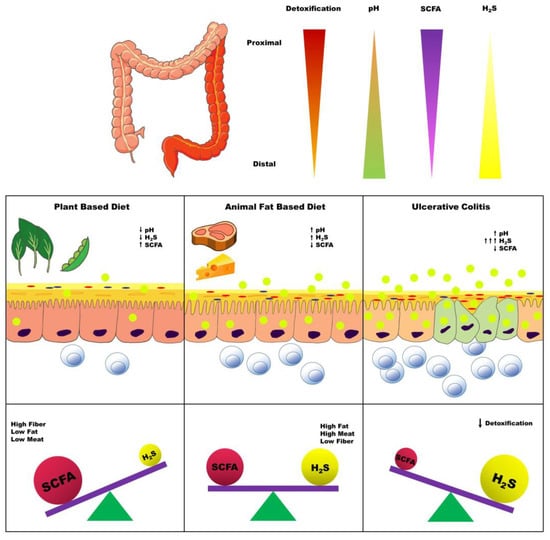
Figure 3.
Overview of metabolic milieu and influence of diet in the pathogenesis of ulcerative colitis. Legend: The distribution of inflammation seen with ulcerative colitis is typically greatest in the rectum and extends continuously towards the proximal colon with varying severity. This pattern of inflammation parallels decreasing expression of the host H2S detoxifying enzymes, rising pH, and decreasing concentrations of short-chain fatty acids (SCFA). A plant-based diet promotes greater production of SCFA, but not H2S. SCFA production decreases in an animal-based diet, while H2S production increases; however, there is a sufficient capacity for H2S detoxification to prevent epithelial cell damage. Nonetheless, a decreased capacity for H2S detoxification results in inflammation, which in turn further exacerbates the gradients of SCFA and H2S concentrations along the proximal-to-distal colon axis.
4. Dietary Factors Influencing Colonic H2S Production
4.1. Protein
Dietary protein has been shown to be a robust driver of in vitro and in vivo H2S production [78,81]. Furthermore, the efficiency of protein degradation is inverse to that of carbohydrate fermentation (and SCFA production) and parallels that of UC pathogenesis (highest in the distal colon and decreasing proximally) [90]. Dietary protein is efficiently absorbed in the small intestine, but the amount reaching the colon is thought to be positively correlated with the amount consumed [90].
In both human and animal studies, a high protein diet results in fecal microbiota changes that increase H2S production and decrease SCFA production [5,45]. Increases in SRB and Bacteroidetes sp. observed in UC also occur in a high protein diet [5,45,122] and observed decreases in specific butyrate producing species within a high protein diet (F. Prausnitzii, Akkermansia, and Ruminococcus) mirror those seen in UC [45,122,123,124].
Sulfur-containing amino acids are the primary source of dietary sulfur. Because of limited information on the sulfur content of food, outside of a metabolic unit, a ‘high’ or ‘low’ sulfur diet is defined according to cysteine and methionine concentration [52]. Therefore, a ‘low sulfur’ diet is actually a ‘sulfur-containing amino acid-controlled diet’, rather than a true ‘sulfur-controlled diet’ [52]. In general, a sulfur-containing amino acid-controlled diet results in a shift from a more traditional western diet (high in animal protein and fat, and low in fiber) to more of a plant-based diet (high in fiber, low in animal protein and fat). Recently published diet composition data from a small sulfur-containing amino acid diet intervention study (n = 4) conducted in the U.S. suggests that a typical U.S. diet is already high in sulfur-containing amino acids; a transition from a baseline diet to a ‘high sulfur’ diet results in minor diet composition changes, while a transition from a baseline to a ‘low sulfur’ diet results in a substantial transition from an animal-based to a plant-based diet (resulting in increased fiber, and decreased protein and fat intake) [61].
As described above, H2S production capacity is often characterized by quantifying SRB, but cysteine desulfhydrase activity provides another route of H2S production for the colonic microbiota that is often left unassessed. In fact, in vitro modeling using healthy human feces suggests that the contribution of cysteine to H2S production is substantially more than that from sulfate [81].
4.2. Carbohydrate and Fiber
High carbohydrate availability in the colon, as noted above, promotes microbial groups able to utilize carbohydrate substrates, but also affects other aspects of microbial metabolism and especially impacts protein degradation. The addition of fermentable fiber to healthy human feces in an in vitro setting drastically reduces H2S production from any source (e.g., cysteine, sulfate) [81]. Lower pH associated with microbial carbohydrate fermentation also leads to inhibition of dissimilatory aromatic amino acid metabolism [125]. Interestingly, stool pH is lower in vegan and vegetarian individuals relative to omnivores, consistent with greater abundance of carbohydrates in the proximal colon, faster colon transit, and higher delivery of SCFA to the distal colon [126]. This finding underscores the importance of overall diet composition when considering the relative proportions of different end-products of microbial metabolism.
4.3. Dietary Fat
Although there is little data supporting a direct role of dietary fat in the H2S toxin hypothesis, animal fat was shown in a murine model to result in increased production of taurine-conjugated bile acids and a bloom of the sulfite-reducer Bilophila wadsworthia in the colonic microenvironment [127]. A high fat diet in mice is associated with lesser production of SCFA and greater production of H2S, even when the protein content of high fat chow is lower relative to regular chow [128]. It is obvious that a relative contribution of macronutrients to H2S production is difficult to isolate in a single comparison, given that an increase in one will correspondingly necessitate reduction in at least one other to maintain the same caloric intake.
4.4. Endogenous Sulfur
The most significant source of endogenous sulfur (both as sulfate and cysteine) is intestinal mucin. The mucous layer of the colon is composed of two layers [40], the microbiota is abundant in the outer loose mucous layer, while the inner dense mucous layer—largely devoid of bacteria—maintains a barrier between the colonic microbiota and colonocytes [39,40]. The composition of the colon microbiota has been shown to influence mucin secretion, breakdown, structure, and the glycosylation pattern [22,42,129,130,131]. Conversely, mucin glycosylation patterns have been shown to influence the colonic microbiota [132]. Therefore, the H2S toxin hypothesis must consider these complex relationships between the mucin, microbiota, and diet. A characteristically high sulfur diet (high in animal protein and fat, and low in fiber) leads to nutrient deprivation for the microbiota and results in increased mucin desulfating sulfatase activity and mucin degradation [5,22,131,133]. This pattern is remarkably similar to that seen in UC [134,135,136]. The phenotypic changes resulting in increased mucin breakdown (e.g., low fiber) and H2S production (e.g., high protein/sulfur intake) [5,22,45,78] may produce H2S concentrations that overwhelm the detoxification capacity of colonocytes and contribute to H2S toxicity.
4.5. Miscellaneous Dietary Factors
Finally, it is also important to consider factors that may correlate with sulfur content in the diet but are not related to sulfur metabolism. An increased intake of heme, present in its highest concentrations in red meat, can have cytotoxic effects on colonocytes [42]. Consumption of processed foods expose individuals to additives such as phosphates, nitrates, and emulsifiers, which have been shown to influence the composition of microbiota, mucin layer thickness, and intestinal inflammation [137,138,139,140]. Even specific cooking methods may alter the potential of diet to impact the intestinal microbiome and microbiota-host interactions. Both enzymatic and non-enzymatic mechanisms of protein modification may also influence intestinal H2S production. Thus, glycation of dietary protein can to shift the colonic microbiota towards greater abundance of SRB and lesser production of butyrate [64]. Conversely, there are likely undiscovered or underappreciated components of a plant-based diet that may be protective in UC [141].
4.6. Inflammatory Factors
Onset of inflammation triggers a number of positive feedback loops that amplify the detrimental effects of H2S on the homeostasis of colonic mucosa. Increased synthesis of inducible nitric oxide synthetase (iNOS) results in increased release of nitrate, which contributes to dysbiosis by encouraging the expansion of facultative anaerobic Proteobacteria [28]. Nitric oxide also impairs H2S detoxification [109], thus lowering the threshold concentration where H2S becomes toxic to epithelial cells. Epithelial injury results in crypt hyperplasia and loss of epithelial hypoxia, which are detrimental to butyrate-producing obligate anaerobes. In animal models and human studies, SCFA (and specifically butyrate) production is inhibited in the presence of inflammation [122,123,124,142]. Inflammation is also correlated with a lower BcoAT gene content of fecal microbiota, consistent with lower butyrate production capacity [143]. Collectively, these changes hasten the collapse of the gut barrier to intestinal microbes and inhibit regulatory immune circuits, such as regulatory T cells, which further augment the inflammatory response.
4.7. Interventions Targeting the Colon Microbiota
The underlying mechanisms that make up the H2S toxin hypothesis are supported by changes produced with fermentable fiber supplementation in individuals with UC (increased butyrate production) [144] and findings from a recent fecal microbial transplantation (FMT) trial that demonstrated a correlation between sustained remission and increased butyrate production, while increased abundances of Bacteroidetes and Proteobacteria correlated with no response or relapse [122]. Notably, members of the Bacteroidetes phylum contain mucin desulfating sulfatase enzymes [122,145,146]. The FMT in UC findings have also been recently expanded upon by Wei et al. who demonstrated a role for fiber in maintaining the gut microbiota composition in individuals with UC following FMT [147].
The role of carbohydrate, and more specifically fermentable carbohydrate, intake within the H2S toxin hypothesis may be its overall impact on protein degradation by colonic bacteria. These findings are supported by a diet study in healthy volunteers [148], where administration of metronidazole effectively reduced SRB counts, but fecal concentration of H2S remained unchanged. In contrast, consumption of fermentable fiber (oligofructose) resulted in a decrease in fecal H2S, concomitant increase in SCFA concentrations, but failure to influence SRB counts. The impact of fiber on microbial H2S production is further supported by recent data demonstrating a decrease in SRB with a high brassica diet (sulfur-containing vegetables) [149]. Although brassica vegetables are technically considered to be high sulfate foods, they also have a high fiber content. These findings underscore the beneficial role for the latter within the H2S toxin hypothesis and support a clinical approach focusing on plant versus animal-based diets rather than high- and low-sulfur diets.
It is important to consider that while short-term changes in diet can affect activities of different microbial groups in the intestine, they do not change the overall individual-specific taxonomic composition of microbiota [61,150]. Short-term low carbohydrate, high protein diets are among the most popular approaches used to achieve weight loss. Such diets do result in decreases in relative abundance of some butyrate producing bacteria, as well as altered fecal metabolite profiles, including lower content of SCFA and fiber-derived antioxidant phenolic acids [151]. It is possible that such dietary changes can even trigger the onset of UC, which may be reversed with increasing plant-derived dietary components [54,56]. However, long-term diets low in microbiota-accessible carbohydrates can lead to complete extinctions of microbial taxa [152]. Therefore, it is possible that most consistent benefits of dietary therapies will also require transplantation of relevant microbiota, optimized in capacity for the individual patients.
5. Conclusions
There is an intriguing body of literature supporting a relationship between dietary sulfur intake and the H2S toxin hypothesis in the development of UC. To date, the focus of the H2S toxin hypothesis in UC has primarily been focused on the abundance and activity of SRB (e.g., Desulfovibrio) and dietary sulfur intake. However, this provides only a partial window on the total activity of the microbiota that is relevant to UC pathogenesis. An updated approach to the H2S hypothesis has to incorporate interactions between different microbial groups as well as multiple components of the diet. A compelling case does exist that a typical western diet, which tends to be high in protein and low in fermentable fiber, may promote the accumulation of harmful products such as H2S and even increase their toxicity, and is also associated with a lower production of beneficial products such as butyrate. Patients are intensely interested in adjunctive dietary therapies for UC. Multiple technological developments that allow compositional and functional measurements of entire microbial communities should facilitate the development and testing of novel microbiota-targeting interventions that include the dietary management of IBD.
Author Contributions
Conceptulization, L.M.T, A.K.; writing—original draft preparation, L.M.T.; writing—review and editing, L.M.T, M.J.S., B.P.V., M.J.H., Z.G., A.K.; supervision, A.K.
Funding
This research received no external funding.
Acknowledgments
This work was partially supported by grants from the Healthy Foods Healthy Lives Institute, the Allen Foundation, and Achieving Cures Together.
Conflicts of Interest
B.P.V. has received compensation from speaking and/or ad boards from Abbvie and Janssen and has received grant support from Roche, Takeda, Celgene and Diasorin.
References
- Kaplan, G.G. The global burden of IBD: From 2015 to 2025. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 720–727. [Google Scholar] [CrossRef]
- Ng, S.C.; Shi, H.Y.; Hamidi, N.; Underwood, F.E.; Tang, W.; Benchimol, E.I.; Panaccione, R.; Ghosh, S.; Wu, J.C.Y.; Chan, F.K.L.; et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: A systematic review of population-based studies. Lancet 2017, 390, 2769–2778. [Google Scholar] [CrossRef]
- Ng, S.C.; Kaplan, G.G.; Tang, W.; Banerjee, R.; Adigopula, B.; Underwood, F.E.; Tanyingoh, D.; Wei, S.C.; Lin, W.C.; Lin, H.H.; et al. Population Density and Risk of Inflammatory Bowel Disease: A Prospective Population-Based Study in 13 Countries or Regions in Asia-Pacific. Am. J. Gastroenterol. 2019, 114, 107–115. [Google Scholar] [CrossRef]
- Rothschild, D.; Weissbrod, O.; Barkan, E.; Kurilshikov, A.; Korem, T.; Zeevi, D.; Costea, P.I.; Godneva, A.; Kalka, I.N.; Bar, N.; et al. Environment dominates over host genetics in shaping human gut microbiota. Nature 2018, 555, 210–215. [Google Scholar] [CrossRef] [PubMed]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Benchimol, E.I.; Mack, D.R.; Guttmann, A.; Nguyen, G.C.; To, T.; Mojaverian, N.; Quach, P.; Manuel, D.G. Inflammatory bowel disease in immigrants to Canada and their children: A population-based cohort study. Am. J. Gastroenterol. 2015, 110, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Vangay, P.; Johnson, A.J.; Ward, T.L.; Al-Ghalith, G.A.; Shields-Cutler, R.R.; Hillmann, B.M.; Lucas, S.K.; Beura, L.K.; Thompson, E.A.; Till, L.M.; et al. US Immigration Westernizes the Human Gut Microbiome. Cell 2018, 175, 962–972.e10. [Google Scholar] [CrossRef]
- Hou, J.K.; Abraham, B.; El-Serag, H. Dietary intake and risk of developing inflammatory bowel disease: A systematic review of the literature. Am. J. Gastroenterol. 2011, 106, 563–573. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, G.G.; Ng, S.C. Understanding and Preventing the Global Increase of Inflammatory Bowel Disease. Gastroenterology 2017, 152, 313–321.e2. [Google Scholar] [CrossRef] [PubMed]
- Yang, G. Hydrogen sulfide in cell survival: A double-edged sword. Expert Rev. Clin. Pharmacol. 2011, 4, 33–47. [Google Scholar] [CrossRef]
- Barton, L.L.; Ritz, N.L.; Fauque, G.D.; Lin, H.C. Sulfur Cycling and the Intestinal Microbiome. Dig. Dis. Sci. 2017, 62, 2241–2257. [Google Scholar] [CrossRef]
- Pitcher, M.C.L.; Cummings, J.H. Hydrogen sulphide: A bacterial toxin in ulcerative colitis? Gut 1996, 39, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Roediger, W.E.W.; Moore, J.; Babidge, W. Colonic sulfide in pathogenesis and treatment of ulcerative colitis. Dig. Dis. Sci. 1997, 42, 1571–1579. [Google Scholar] [CrossRef] [PubMed]
- Rowan, F.E.; Docherty, N.G.; Coffey, J.C.; O’Connell, P.R. Sulphate-reducing bacteria and hydrogen sulphide in the aetiology of ulcerative colitis. Br. J. Surg. 2009, 96, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Blachier, F.; Davila, A.M.; Mimoun, S.; Benetti, P.H.; Atanasiu, C.; Andriamihaja, M.; Benamouzig, R.; Bouillaud, F.; Tome, D. Luminal sulfide and large intestine mucosa: Friend or foe? Amino Acids 2010, 39, 335–347. [Google Scholar] [CrossRef]
- Blachier, F.; Beaumont, M.; Andriamihaja, M.; Davila, A.M.; Lan, A.; Grauso, M.; Armand, L.; Benamouzig, R.; Tome, D. Changes in the Luminal Environment of the Colonic Epithelial Cells and Physiopathological Consequences. Am. J. Pathol. 2017, 187, 476–486. [Google Scholar] [CrossRef]
- Levine, A.; Sigall Boneh, R.; Wine, E. Evolving role of diet in the pathogenesis and treatment of inflammatory bowel diseases. Gut 2018, 67, 1726–1738. [Google Scholar] [CrossRef]
- Chiba, M.; Abe, T.; Tsuda, H.; Sugawara, T.; Tsuda, S.; Tozawa, H.; Fujiwara, K.; Imai, H. Lifestyle-related disease in Crohn’s disease: Relapse prevention by a semi-vegetarian diet. World J. Gastroenterol. 2010, 16, 2484–2495. [Google Scholar] [CrossRef]
- Florin, T.; Neale, G.; Gibson, G.R.; Christl, S.U.; Cummings, J.H. Metabolism of dietary sulphate: Absorption and excretion in humans. Gut 1991, 32, 766–773. [Google Scholar] [CrossRef]
- Devkota, S.; Wang, Y.; Musch, M.W.; Leone, V.; Fehlner-Peach, H.; Nadimpalli, A.; Antonopoulos, D.A.; Jabri, B.; Chang, E.B. Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10-/- mice. Nature 2012, 487, 104–108. [Google Scholar] [CrossRef]
- Holmes, A.J.; Chew, Y.V.; Colakoglu, F.; Cliff, J.B.; Klaassens, E.; Read, M.N.; Solon-Biet, S.M.; McMahon, A.C.; Cogger, V.C.; Ruohonen, K.; et al. Diet-Microbiome Interactions in Health Are Controlled by Intestinal Nitrogen Source Constraints. Cell Metab. 2017, 25, 140–151. [Google Scholar] [CrossRef] [PubMed]
- Desai, M.S.; Seekatz, A.M.; Koropatkin, N.M.; Kamada, N.; Hickey, C.A.; Wolter, M.; Pudlo, N.A.; Kitamoto, S.; Terrapon, N.; Muller, A.; et al. A Dietary Fiber-Deprived Gut Microbiota Degrades the Colonic Mucus Barrier and Enhances Pathogen Susceptibility. Cell 2016, 167, 1339–1353.e1321. [Google Scholar] [CrossRef]
- Wong, J.M.; de Souza, R.; Kendall, C.W.; Emam, A.; Jenkins, D.J. Colonic health: Fermentation and short chain fatty acids. J. Clin. Gastroenterol. 2006, 40, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Roediger, W.E. Role of anaerobic bacteria in the metabolic welfare of the colonic mucosa in man. Gut 1980, 21, 793–798. [Google Scholar] [CrossRef] [PubMed]
- Furusawa, Y.; Obata, Y.; Fukuda, S.; Endo, T.A.; Nakato, G.; Takahashi, D.; Nakanishi, Y.; Uetake, C.; Kato, K.; Kato, T.; et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 2013, 504, 446–450. [Google Scholar] [CrossRef] [PubMed]
- Arpaia, N.; Campbell, C.; Fan, X.; Dikiy, S.; van der Veeken, J.; deRoos, P.; Liu, H.; Cross, J.R.; Pfeffer, K.; Coffer, P.J.; et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 2013, 504, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Finnie, I.A.; Dwarakanath, A.D.; Taylor, B.A.; Rhodes, J.M. Colonic mucin synthesis is increased by sodium butyrate. Gut 1995, 36, 93–99. [Google Scholar] [CrossRef]
- Byndloss, M.X.; Olsan, E.E.; Rivera-Chavez, F.; Tiffany, C.R.; Cevallos, S.A.; Lokken, K.L.; Torres, T.P.; Byndloss, A.J.; Faber, F.; Gao, Y.; et al. Microbiota-activated PPAR-gamma signaling inhibits dysbiotic Enterobacteriaceae expansion. Science 2017, 357, 570–575. [Google Scholar] [CrossRef]
- Litvak, Y.; Byndloss, M.X.; Tsolis, R.M.; Baumler, A.J. Dysbiotic Proteobacteria expansion: A microbial signature of epithelial dysfunction. Curr. Opin. Microbiol. 2017, 39, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Christl, S.U.; Eisner, H.D.; Dusel, G.; Kasper, H.; Scheppach, W. Antagonistic effects of sulfide and butyrate on proliferation of colonic mucosa: A potential role for these agents in the pathogenesis of ulcerative colitis. Dig. Dis. Sci. 1996, 41, 2477–2481. [Google Scholar] [CrossRef]
- O’Flaherty, V.; Mahony, T.; O’Kennedy, R.; Colleran, E. Effect of pH on growth kinetics and sulphide toxicity thresholds of a range of methanogeic, syntrophic and sulphate-reducing bacteria. Process Biochem. 1998, 33, 555–569. [Google Scholar] [CrossRef]
- Babidge, W.; Millard, S.; Roediger, W. Sulfides impair short chain fatty acid beta-oxidation at acyl-CoA dehydrogenase level in colonocytes: Implications for ulcerative colitis. Mol. Cell. Biochem. 1998, 181, 117–124. [Google Scholar] [CrossRef]
- Wallace, J.L.; Blackler, R.W.; Chan, M.V.; Da Silva, G.J.; Elsheikh, W.; Flannigan, K.L.; Gamaniek, I.; Manko, A.; Wang, L.; Motta, J.P.; et al. Anti-inflammatory and cytoprotective actions of hydrogen sulfide: Translation to therapeutics. Antioxid. Redox Signal. 2015, 22, 398–410. [Google Scholar] [CrossRef] [PubMed]
- Odenwald, M.A.; Turner, J.R. The intestinal epithelial barrier: A therapeutic target? Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 9–21. [Google Scholar] [CrossRef]
- Jiang, J.; Chan, A.; Ali, S.; Saha, A.; Haushalter, K.J.; Lam, W.L.; Glasheen, M.; Parker, J.; Brenner, M.; Mahon, S.B.; et al. Hydrogen Sulfide--Mechanisms of Toxicity and Development of an Antidote. Sci. Rep. 2016, 6, 20831. [Google Scholar] [CrossRef]
- Roediger, W.E.; Duncan, A.; Kapaniris, O.; Millard, S. Reducing sulfur compounds of the colon impair colonocyte nutrition: Implications for ulcerative colitis. Gastroenterology 1993, 104, 802–809. [Google Scholar] [CrossRef]
- De Preter, V.; Arijs, I.; Windey, K.; Vanhove, W.; Vermeire, S.; Schuit, F.; Rutgeerts, P.; Verbeke, K. Impaired butyrate oxidation in ulcerative colitis is due to decreased butyrate uptake and a defect in the oxidation pathway. Inflamm. Bowel Dis. 2012, 18, 1127–1136. [Google Scholar] [CrossRef] [PubMed]
- De Preter, V.; Arijs, I.; Windey, K.; Vanhove, W.; Vermeire, S.; Schuit, F.; Rutgeerts, P.; Verbeke, K. Decreased mucosal sulfide detoxification is related to an impaired butyrate oxidation in ulcerative colitis. Inflamm. Bowel Dis. 2012, 18, 2371–2380. [Google Scholar] [CrossRef] [PubMed]
- Johansson, M.E.; Hansson, G.C. Immunological aspects of intestinal mucus and mucins. Nat. Rev. Immunol. 2016, 16, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Johansson, M.E.; Phillipson, M.; Petersson, J.; Velcich, A.; Holm, L.; Hansson, G.C. The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Proc. Natl. Acad. Sci. USA 2008, 105, 15064–15069. [Google Scholar] [CrossRef] [PubMed]
- Johansson, M.E.; Gustafsson, J.K.; Holmen-Larsson, J.; Jabbar, K.S.; Xia, L.; Xu, H.; Ghishan, F.K.; Carvalho, F.A.; Gewirtz, A.T.; Sjovall, H.; et al. Bacteria penetrate the normally impenetrable inner colon mucus layer in both murine colitis models and patients with ulcerative colitis. Gut 2014, 63, 281–291. [Google Scholar] [CrossRef]
- Ijssennagger, N.; Belzer, C.; Hooiveld, G.J.; Dekker, J.; van Mil, S.W.; Muller, M.; Kleerebezem, M.; van der Meer, R. Gut microbiota facilitates dietary heme-induced epithelial hyperproliferation by opening the mucus barrier in colon. Proc. Natl. Acad. Sci. USA 2015, 112, 10038–10043. [Google Scholar] [CrossRef]
- Eichele, D.D.; Kharbanda, K.K. Dextran sodium sulfate colitis murine model: An indispensable tool for advancing our understanding of inflammatory bowel diseases pathogenesis. World J. Gastroenterol. 2017, 23, 6016–6029. [Google Scholar] [CrossRef]
- Zhu, Q.C.; Gao, R.Y.; Wu, W.; Guo, B.M.; Peng, J.Y.; Qin, H.L. Effect of a high-fat diet in development of colonic adenoma in an animal model. World J. Gastroenterol. 2014, 20, 8119–8129. [Google Scholar] [CrossRef]
- Mu, C.; Yang, Y.; Luo, Z.; Guan, L.; Zhu, W. The Colonic Microbiome and Epithelial Transcriptome Are Altered in Rats Fed a High-Protein Diet Compared with a Normal-Protein Diet. J. Nutr. 2016, 146, 474–483. [Google Scholar] [CrossRef]
- Heo, J.M.; Kim, J.C.; Hansen, C.F.; Mullan, B.P.; Hampson, D.J.; Pluske, J.R. Effects of feeding low protein diets to piglets on plasma urea nitrogen, faecal ammonia nitrogen, the incidence of diarrhoea and performance after weaning. Arch. Anim. Nutr. 2008, 62, 343–358. [Google Scholar] [CrossRef]
- Pitcher, M.C.; Beatty, E.R.; Cummings, J.H. The contribution of sulphate reducing bacteria and 5-aminosalicylic acid to faecal sulphide in patients with ulcerative colitis. Gut 2000, 46, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Dubuquoy, L.; Rousseaux, C.; Thuru, X.; Peyrin-Biroulet, L.; Romano, O.; Chavatte, P.; Chamaillard, M.; Desreumaux, P. PPARgamma as a new therapeutic target in inflammatory bowel diseases. Gut 2006, 55, 1341–1349. [Google Scholar] [CrossRef]
- Alex, S.; Lange, K.; Amolo, T.; Grinstead, J.S.; Haakonsson, A.K.; Szalowska, E.; Koppen, A.; Mudde, K.; Haenen, D.; Al-Lahham, S.; et al. Short-chain fatty acids stimulate angiopoietin-like 4 synthesis in human colon adenocarcinoma cells by activating peroxisome proliferator-activated receptor gamma. Mol. Cell. Biol. 2013, 33, 1303–1316. [Google Scholar] [CrossRef]
- Xu, J.; Chen, N.; Wu, Z.; Song, Y.; Zhang, Y.; Wu, N.; Zhang, F.; Ren, X.; Liu, Y. 5-Aminosalicylic Acid Alters the Gut Bacterial Microbiota in Patients with Ulcerative Colitis. Front. Microbiol. 2018, 9, 1274. [Google Scholar] [CrossRef]
- Burke, D.A.; Axon, A.T.; Clayden, S.A.; Dixon, M.F.; Johnston, D.; Lacey, R.W. The efficacy of tobramycin in the treatment of ulcerative colitis. Aliment. Pharmacol. Ther. 1990, 4, 123–129. [Google Scholar] [CrossRef]
- Roediger, W.E.W. Decreased sulphur aminoacid intake in ulcerative colitis. Lancet 1998, 351. [Google Scholar] [CrossRef]
- Kashyap, P.C.; Reigstad, C.S.; Loftus, E.V., Jr. Role of diet and gut microbiota in management of inflammatory bowel disease in an Asian migrant. J. Allergy Clin. Immunol. 2013, 132, 250–250.e255. [Google Scholar] [CrossRef] [PubMed]
- Chiba, M.; Tsuda, S.; Komatsu, M.; Tozawa, H.; Takayama, Y. Onset of Ulcerative Colitis during a Low-Carbohydrate Weight-Loss Diet and Treatment with a Plant-Based Diet: A Case Report. Perm. J. 2016, 20, 80–84. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chiba, M.; Sugawara, T.; Komatsu, M.; Tozawa, H. Onset of Ulcerative Colitis in the Second Trimester after Emesis Gravidarum: Treatment with Plant-based Diet. Inflamm. Bowel Dis. 2018, 24, e8–e9. [Google Scholar] [CrossRef]
- Chiba, M.; Nakane, K.; Tsuji, T.; Tsuda, S.; Ishii, H.; Ohno, H.; Watanabe, K.; Ito, M.; Komatsu, M.; Yamada, K.; et al. Relapse Prevention in Ulcerative Colitis by Plant-Based Diet Through Educational Hospitalization: A Single-Group Trial. Perm. J. 2018, 22. [Google Scholar] [CrossRef] [PubMed]
- Florin, T.H.J.; Neale, G.; Goretski, S.; Cummings, J.H. The sulfate content of foods and beverages. J. Food Compos. Anal. 1993, 6, 140–151. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. Food Additive Status List. Available online: https://www.fda.gov/food/ingredientspackaginglabeling/foodadditivesingredients/ucm091048.htm (accessed on 15 August 2018).
- Curno, R.; Magee, E.A.; Edmond, L.M.; Cummings, J.H. Studies of a urinary biomarker of dietary inorganic sulphur in subjects on diets containing 1–38 mmol sulphur/day and of the half-life of ingested 34SO4(2-). Eur. J. Clin. Nutr. 2008, 62, 1106–1115. [Google Scholar] [CrossRef]
- Magee, E.A.; Curno, R.; Edmond, L.M.; Cummings, J.H. Contribution of dietary protein and inorganic sulfur to urinary sulfate: Toward a biomarker of inorganic sulfur intake. Am. J. Clin. Nutr. 2004, 80, 137–142. [Google Scholar] [CrossRef]
- Dostal Webster, A.; Staley, C.; Hamilton, M.J.; Huang, M.; Fryxell, K.; Erickson, R.; Kabage, A.J.; Sadowsky, M.J.; Khoruts, A. Influence of short-term changes in dietary sulfur on the relative abundances of intestinal sulfate-reducing bacteria. Gut Microbes 2019, 1–11. [Google Scholar] [CrossRef]
- Florin, T.H. Hydrogen sulphide and total acid-volatile sulphide in faeces, determined with a direct spectrophotometric method. Clin. Chim. Acta 1991, 196, 127–134. [Google Scholar] [CrossRef]
- Buffiere, C.; Gaudichon, C.; Hafnaoui, N.; Migne, C.; Scislowsky, V.; Khodorova, N.; Mosoni, L.; Blot, A.; Boirie, Y.; Dardevet, D.; et al. In the elderly, meat protein assimilation from rare meat is lower than that from meat that is well done. Am. J. Clin. Nutr. 2017, 106, 1257–1266. [Google Scholar] [CrossRef]
- Mills, D.J.; Tuohy, K.M.; Booth, J.; Buck, M.; Crabbe, M.J.; Gibson, G.R.; Ames, J.M. Dietary glycated protein modulates the colonic microbiota towards a more detrimental composition in ulcerative colitis patients and non-ulcerative colitis subjects. J. Appl. Microbiol. 2008, 105, 706–714. [Google Scholar] [CrossRef] [PubMed]
- Oberli, M.; Marsset-Baglieri, A.; Airinei, G.; Sante-Lhoutellier, V.; Khodorova, N.; Remond, D.; Foucault-Simonin, A.; Piedcoq, J.; Tome, D.; Fromentin, G.; et al. High True Ileal Digestibility but Not Postprandial Utilization of Nitrogen from Bovine Meat Protein in Humans Is Moderately Decreased by High-Temperature, Long-Duration Cooking. J. Nutr. 2015, 145, 2221–2228. [Google Scholar] [CrossRef]
- Pennings, B.; Groen, B.B.; van Dijk, J.W.; de Lange, A.; Kiskini, A.; Kuklinski, M.; Senden, J.M.; van Loon, L.J. Minced beef is more rapidly digested and absorbed than beef steak, resulting in greater postprandial protein retention in older men. Am. J. Clin. Nutr. 2013, 98, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Remond, D.; Machebeuf, M.; Yven, C.; Buffiere, C.; Mioche, L.; Mosoni, L.; Mirand, P.P. Postprandial whole-body protein metabolism after a meat meal is influenced by chewing efficiency in elderly subjects. Am. J. Clin. Nutr. 2007, 85, 1286–1292. [Google Scholar] [CrossRef] [PubMed]
- Ridlon, J.M.; Wolf, P.G.; Gaskins, H.R. Taurocholic acid metabolism by gut microbes and colon cancer. Gut Microbes 2016, 7, 201–215. [Google Scholar] [CrossRef] [PubMed]
- Di Ciaula, A.; Garruti, G.; Lunardi Baccetto, R.; Molina-Molina, E.; Bonfrate, L.; Wang, D.Q.; Portincasa, P. Bile Acid Physiology. Ann. Hepatol. 2017, 16, s4–s14. [Google Scholar] [CrossRef]
- Johansson, M.E.; Larsson, J.M.; Hansson, G.C. The two mucus layers of colon are organized by the MUC2 mucin, whereas the outer layer is a legislator of host-microbial interactions. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. 1), 4659–4665. [Google Scholar] [CrossRef]
- Holmen Larsson, J.M.; Thomsson, K.A.; Rodriguez-Pineiro, A.M.; Karlsson, H.; Hansson, G.C. Studies of mucus in mouse stomach, small intestine, and colon. III. Gastrointestinal Muc5ac and Muc2 mucin O-glycan patterns reveal a regiospecific distribution. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 305, G357–G363. [Google Scholar] [CrossRef] [PubMed]
- Robbe, C.; Capon, C.; Coddeville, B.; Michalski, J.-C. Structural diversity and specific distribution of O-glycans in normal human mucins along the intestinal tract. Biochem. J. 2004, 384, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Lennon, G.; Balfe, A.; Bambury, N.; Lavelle, A.; Maguire, A.; Docherty, N.G.; Coffey, J.C.; Winter, D.C.; Sheahan, K.; O’Connell, P.R. Correlations between colonic crypt mucin chemotype, inflammatory grade and Desulfovibrio species in ulcerative colitis. Colorectal Dis. 2014, 16, O161–O169. [Google Scholar] [CrossRef] [PubMed]
- Gibson, G.R.; Cummings, J.H.; Macfarlane, G.T. Growth and activities of sulphate-reducing bacteria in gut contents of healthy subjects and patients with ulcerative colitis. FEMS Microbiol. Ecol. 1991, 86, 103–112. [Google Scholar] [CrossRef]
- Moore, J.; Babidge, W.; Millard, S.; Roediger, W. Colonic luminal hydrogen sulfide is not elevated in ulcerative colitis. Dig. Dis. Sci. 1998, 43, 162–165. [Google Scholar] [CrossRef]
- Hughes, M.N.; Centelles, M.N.; Moore, K.P. Making and working with hydrogen sulfide: The chemistry and generation of hydrogen sulfide in vitro and its measurement in vivo: A review. Free Radic. Biol. Med. 2009, 47, 1346–1353. [Google Scholar] [CrossRef]
- Kolluru, G.K.; Shen, X.; Bir, S.C.; Kevil, C.G. Hydrogen sulfide chemical biology: Pathophysiological roles and detection. Nitric Oxide 2013, 35, 5–20. [Google Scholar] [CrossRef] [PubMed]
- Magee, E.A.; Richardson, C.J.; Hughes, R.; Cummings, J.H. Contribution of dietary protein to sulfide production in the large intestine: An in vitro and a controlled feeding study in humans. Am. J. Clin. Nutr. 2000, 72, 1488–1494. [Google Scholar] [CrossRef] [PubMed]
- Vitvitsky, V.; Banerjee, R. H2S analysis in biological samples using gas chromatography with sulfur chemiluminescence detection. Methods Enzymol. 2015, 554, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Levine, J.; Ellis, C.J.; Furne, J.K.; Springfield, J.; Levitt, M.D. Fecal hydrogen sulfide production in ulcerative colitis. Am. J. Gastroenterol. 1998, 93, 83–87. [Google Scholar] [CrossRef]
- Yao, C.K.; Rotbart, A.; Ou, J.Z.; Kalantar-Zadeh, K.; Muir, J.G.; Gibson, P.R. Modulation of colonic hydrogen sulfide production by diet and mesalazine utilizing a novel gas-profiling technology. Gut Microbes 2018, 1–13. [Google Scholar] [CrossRef]
- Anantharaman, K.; Hausmann, B.; Jungbluth, S.P.; Kantor, R.S.; Lavy, A.; Warren, L.A.; Rappe, M.S.; Pester, M.; Loy, A.; Thomas, B.C.; et al. Expanded diversity of microbial groups that shape the dissimilatory sulfur cycle. ISME J. 2018, 12, 1715–1728. [Google Scholar] [CrossRef]
- Scanlan, P.D.; Shanahan, F.; Marchesi, J.R. Culture-independent analysis of desulfovibrios in the human distal colon of healthy, colorectal cancer and polypectomized individuals. FEMS Microbiol. Ecol. 2009, 69, 213–221. [Google Scholar] [CrossRef]
- Muller, A.L.; Kjeldsen, K.U.; Rattei, T.; Pester, M.; Loy, A. Phylogenetic and environmental diversity of DsrAB-type dissimilatory (bi)sulfite reductases. ISME J. 2015, 9, 1152–1165. [Google Scholar] [CrossRef] [PubMed]
- Christophersen, C.T.; Morrison, M.; Conlon, M.A. Overestimation of the abundance of sulfate-reducing bacteria in human feces by quantitative PCR targeting the Desulfovibrio 16S rRNA gene. Appl. Environ. Microbiol. 2011, 77, 3544–3546. [Google Scholar] [CrossRef] [PubMed]
- Coutinho, C.; Coutinho-Silva, R.; Zinkevich, V.; Pearce, C.B.; Ojcius, D.M.; Beech, I. Sulphate-reducing bacteria from ulcerative colitis patients induce apoptosis of gastrointestinal epithelial cells. Microb. Pathog. 2017, 112, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Hale, V.L.; Jeraldo, P.; Mundy, M.; Yao, J.; Keeney, G.; Scott, N.; Cheek, E.H.; Davidson, J.; Green, M.; Martinez, C.; et al. Synthesis of multi-omic data and community metabolic models reveals insights into the role of hydrogen sulfide in colon cancer. Methods 2018. [Google Scholar] [CrossRef] [PubMed]
- Jia, W.; Whitehead, R.N.; Griffiths, L.; Dawson, C.; Bai, H.; Waring, R.H.; Ramsden, D.B.; Hunter, J.O.; Cauchi, M.; Bessant, C.; et al. Diversity and distribution of sulphate-reducing bacteria in human faeces from healthy subjects and patients with inflammatory bowel disease. FEMS Immunol. Med. Microbiol. 2012, 65, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Duffy, M.; O’Mahony, L.; Coffey, J.C.; Collins, J.K.; Shanahan, F.; Redmond, H.P.; Kirwan, W.O. Sulfate-Reducing Bacteria Colonize Pouches Formed for Ulcerative Colitis but Not for Familial Adenomatous Polyposis. Dis. Colon Rectum 2002, 45, 384–388. [Google Scholar] [CrossRef]
- Windey, K.; De Preter, V.; Verbeke, K. Relevance of protein fermentation to gut health. Mol. Nutr. Food Res. 2012, 56, 184–196. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H. Metabolic turnover of hydrogen sulfide. Front. Physiol. 2012, 3, 101. [Google Scholar] [CrossRef]
- Awano, N.; Wada, M.; Mori, H.; Nakamori, S.; Takagi, H. Identification and functional analysis of Escherichia coli cysteine desulfhydrases. Appl. Environ. Microbiol. 2005, 71, 4149–4152. [Google Scholar] [CrossRef]
- Sahami, S.; Kooij, I.A.; Meijer, S.L.; Van den Brink, G.R.; Buskens, C.J.; Te Velde, A.A. The Link between the Appendix and Ulcerative Colitis: Clinical Relevance and Potential Immunological Mechanisms. Am. J. Gastroenterol. 2016, 111, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Rogers, M.B.; Brower-Sinning, R.; Firek, B.; Zhong, D.; Morowitz, M.J. Acute Appendicitis in Children Is Associated with a Local Expansion of Fusobacteria. Clin. Infect. Dis. 2016, 63, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Zhong, D.; Brower-Sinning, R.; Firek, B.; Morowitz, M.J. Acute appendicitis in children is associated with an abundance of bacteria from the phylum Fusobacteria. J. Pediatr. Surg. 2014, 49, 441–446. [Google Scholar] [CrossRef]
- Basic, A.; Blomqvist, M.; Dahlen, G.; Svensater, G. The proteins of Fusobacterium spp. involved in hydrogen sulfide production from L-cysteine. BMC Microbiol. 2017, 17, 61. [Google Scholar] [CrossRef] [PubMed]
- Basic, A.; Blomqvist, S.; Carlen, A.; Dahlen, G. Estimation of bacterial hydrogen sulfide production in vitro. J. Oral Microbiol. 2015, 7, 28166. [Google Scholar] [CrossRef] [PubMed]
- Wolf, P.G.; Biswas, A.; Morales, S.E.; Greening, C.; Gaskins, H.R. H2 metabolism is widespread and diverse among human colonic microbes. Gut Microbes 2016, 7, 235–245. [Google Scholar] [CrossRef]
- Carbonero, F.; Benefiel, A.C.; Gaskins, H.R. Contributions of the microbial hydrogen economy to colonic homeostasis. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 504–518. [Google Scholar] [CrossRef]
- Macfarlane, S.; Macfarlane, G.T. Regulation of short-chain fatty acid production. Proc. Nutr. Soc. 2003, 62, 67–72. [Google Scholar] [CrossRef]
- Kunkel, D.; Basseri, R.J.; Makhani, M.D.; Chong, K.; Chang, C.; Pimentel, M. Methane on breath testing is associated with constipation: A systematic review and meta-analysis. Dig. Dis. Sci. 2011, 56, 1612–1618. [Google Scholar] [CrossRef]
- Macfarlane, G.T.; Gibson, G.R.; Cummings, J.H. Comparison of fermentation reactions in different regions of the human colon. J. Appl. Bacteriol. 1992, 72, 57–64. [Google Scholar]
- Rao, S.S.; Kuo, B.; McCallum, R.W.; Chey, W.D.; DiBaise, J.K.; Hasler, W.L.; Koch, K.L.; Lackner, J.M.; Miller, C.; Saad, R.; et al. Investigation of colonic and whole-gut transit with wireless motility capsule and radiopaque markers in constipation. Clin. Gastroenterol. Hepatol. 2009, 7, 537–544. [Google Scholar] [CrossRef]
- Videla, S.; Vilaseca, J.; Antolin, M.; Garcia-Lafuente, A.; Guarner, F.; Crespo, E.; Casalots, J.; Salas, A.; Malagelada, J.R. Dietary inulin improves distal colitis induced by dextran sodium sulfate in the rat. Am. J. Gastroenterol. 2001, 96, 1486–1493. [Google Scholar] [CrossRef] [PubMed]
- de Vries, J.; Miller, P.E.; Verbeke, K. Effects of cereal fiber on bowel function: A systematic review of intervention trials. World J. Gastroenterol. 2015, 21, 8952–8963. [Google Scholar] [CrossRef]
- Walker, A.W.; Duncan, S.H.; McWilliam Leitch, E.C.; Child, M.W.; Flint, H.J. pH and peptide supply can radically alter bacterial populations and short-chain fatty acid ratios within microbial communities from the human colon. Appl. Environ. Microbiol. 2005, 71, 3692–3700. [Google Scholar] [CrossRef] [PubMed]
- Duncan, S.H.; Louis, P.; Thomson, J.M.; Flint, H.J. The role of pH in determining the species composition of the human colonic microbiota. Environ. Microbiol. 2009, 11, 2112–2122. [Google Scholar] [CrossRef]
- Gibson, G.R.; Cummings, J.H.; Macfarlane, G.T.; Allison, C.; Segal, I.; Vorster, H.H.; Walker, A.R.P. Alternative pathways for hydrogen disposal during fermentation in the human colon. Gut 1990, 31, 679–683. [Google Scholar] [CrossRef]
- Beaumont, M.; Andriamihaja, M.; Lan, A.; Khodorova, N.; Audebert, M.; Blouin, J.M.; Grauso, M.; Lancha, L.; Benetti, P.H.; Benamouzig, R.; et al. Detrimental effects for colonocytes of an increased exposure to luminal hydrogen sulfide: The adaptive response. Free Radic. Biol. Med. 2016, 93, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Suarez, F.; Furne, J.; Springfield, J.; Levitt, M. Insights into human colonic physiology obtained from the study of flatus composition. Am. Physiol. Soc. 1997, 272, G1028–G1033. [Google Scholar] [CrossRef] [PubMed]
- Levitt, M.D.; Furne, J.; Springfield, J.; Suarez, F.; DeMaster, E. Detoxification of hydrogen sulfide and methanethiol in the cecal mucosa. J. Clin. Investig. 1999, 104, 1107–1114. [Google Scholar] [CrossRef] [PubMed]
- Furne, J.; Springfield, J.; Koenig, T.; DeMaster, E.; Levitt, M.D. Oxidation of hydrogen sulfide and methanethiol to thiosulfate by rat tissues: A specialized function of the colonic mucosa. Biochem. Pharmacol. 2001, 62, 255–259. [Google Scholar] [CrossRef]
- Picton, R.; Eggo, M.C.; Merrill, G.A.; Langman, M.J.S.; Singh, S. Mucosal protection against sulphide: Importance of the enzyme rhodanese. Gut 2002, 50, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Picton, R.; Eggo, M.C.; Langman, M.J.; Singh, S. Impaired detoxication of hydrogen sulfide in ulcerative colitis? Dig. Dis. Sci. 2007, 52, 373–378. [Google Scholar] [CrossRef]
- Wilson, K.; Mudra, M.; Furne, J.; Levitt, M. Differentiation of the roles of sulfide oxidase and rhodanese in the detoxification of sulfide by the colonic mucosa. Dig. Dis. Sci. 2008, 53, 277–283. [Google Scholar] [CrossRef]
- Landry, A.P.; Ballou, D.P.; Banerjee, R. Modulation of Catalytic Promiscuity during Hydrogen Sulfide Oxidation. ACS Chem. Biol. 2018, 13, 1651–1658. [Google Scholar] [CrossRef]
- Filipovic, M.R.; Zivanovic, J.; Alvarez, B.; Banerjee, R. Chemical Biology of H2S Signaling through Persulfidation. Chem. Rev. 2018, 118, 1253–1337. [Google Scholar] [CrossRef]
- Landry, A.P.; Ballou, D.P.; Banerjee, R. H2S oxidation by nanodisc-embedded human sulfide quinone oxidoreductase. J. Biol. Chem. 2017, 292, 11641–11649. [Google Scholar] [CrossRef] [PubMed]
- Libiad, M.; Yadav, P.K.; Vitvitsky, V.; Martinov, M.; Banerjee, R. Organization of the human mitochondrial hydrogen sulfide oxidation pathway. J. Biol. Chem. 2014, 289, 30901–30910. [Google Scholar] [CrossRef] [PubMed]
- Mimoun, S.; Andriamihaja, M.; Chaumontet, C.; Atanasiu, C.; Benamouzig, R.; Blouin, J.M.; Tome, D.; Bouillaud, F.; Blachier, F. Detoxification of H(2)S by differentiated colonic epithelial cells: Implication of the sulfide oxidizing unit and of the cell respiratory capacity. Antioxid. Redox Signal. 2012, 17, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Sido, B.; Hack, V.; Hochlehnert, A.; Lipps, H.; Herfarth, C.; Droge, W. Impairment of intestinal glutathione synthesis in patients with inflammatory bowel disease. Gut 1998, 42, 485–492. [Google Scholar] [CrossRef]
- Fuentes, S.; Rossen, N.G.; van der Spek, M.J.; Hartman, J.H.; Huuskonen, L.; Korpela, K.; Salojarvi, J.; Aalvink, S.; de Vos, W.M.; D’Haens, G.R.; et al. Microbial shifts and signatures of long-term remission in ulcerative colitis after faecal microbiota transplantation. ISME J. 2017, 11, 1877–1889. [Google Scholar] [CrossRef]
- Bajer, L.; Kverka, M.; Kostovcik, M.; Macinga, P.; Dvorak, J.; Stehlikova, Z.; Brezina, J.; Wohl, P.; Spicak, J.; Drastich, P. Distinct gut microbiota profiles in patients with primary sclerosing cholangitis and ulcerative colitis. World J. Gastroenterol. 2017, 23, 4548–4558. [Google Scholar] [CrossRef]
- Vermeire, S.; Joossens, M.; Verbeke, K.; Wang, J.; Machiels, K.; Sabino, J.; Ferrante, M.; Van Assche, G.; Rutgeerts, P.; Raes, J. Donor Species Richness Determines Faecal Microbiota Transplantation Success in Inflammatory Bowel Disease. J. Crohn’s Colitis 2016, 10, 387–394. [Google Scholar] [CrossRef]
- Smith, E.A.; Macfarlane, G.T. Enumeration of human colonic bacteria producing phenolic and indolic compounds: Effects of pH, carbohydrate availability and retention time on dissimilatory aromatic amino acid metabolism. J. Appl. Bacteriol. 1996, 81, 288–302. [Google Scholar] [CrossRef]
- Zimmer, J.; Lange, B.; Frick, J.S.; Sauer, H.; Zimmermann, K.; Schwiertz, A.; Rusch, K.; Klosterhalfen, S.; Enck, P. A vegan or vegetarian diet substantially alters the human colonic faecal microbiota. Eur. J. Clin. Nutr. 2012, 66, 53–60. [Google Scholar] [CrossRef]
- Devkota, S.; Chang, E.B. Interactions between Diet, Bile Acid Metabolism, Gut Microbiota, and Inflammatory Bowel Diseases. Dig. Dis. 2015, 33, 351–356. [Google Scholar] [CrossRef]
- Leone, V.; Gibbons, S.M.; Martinez, K.; Hutchison, A.L.; Huang, E.Y.; Cham, C.M.; Pierre, J.F.; Heneghan, A.F.; Nadimpalli, A.; Hubert, N.; et al. Effects of diurnal variation of gut microbes and high-fat feeding on host circadian clock function and metabolism. Cell Host Microbe 2015, 17, 681–689. [Google Scholar] [CrossRef]
- Barcelo, A.; Claustre, J.; Moro, F.; Chayvialle, J.-A.; Cuber, J.-C. Plaisancia. Mucin secretion is modulated by luminal factors in the isolated vascularly perfused rat colon. Gut 2000, 46, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Deplancke, B.; Finster, K.; Vallen Graham, W.; Collier, C.T.; Thurmond, J.E.; Gaskins, H.R. Gastrointestinal and Microbial Responses to Sulfate-Supplemented Drinking Water in Mice. Exp. Biol. Med. 2003, 228, 424–433. [Google Scholar] [CrossRef]
- Schroeder, B.O.; Birchenough, G.M.H.; Stahlman, M.; Arike, L.; Johansson, M.E.V.; Hansson, G.C.; Backhed, F. Bifidobacteria or Fiber Protects against Diet-Induced Microbiota-Mediated Colonic Mucus Deterioration. Cell Host Microbe 2018, 23, 27–40.e27. [Google Scholar] [CrossRef] [PubMed]
- Croix, J.A.; Carbonero, F.; Nava, G.M.; Russell, M.; Greenberg, E.; Gaskins, H.R. On the relationship between sialomucin and sulfomucin expression and hydrogenotrophic microbes in the human colonic mucosa. PLoS ONE 2011, 6, e24447. [Google Scholar] [CrossRef] [PubMed]
- Rey, F.E.; Gonzalez, M.D.; Cheng, J.; Wu, M.; Ahern, P.P.; Gordon, J.I. Metabolic niche of a prominent sulfate-reducing human gut bacterium. Proc. Natl. Acad. Sci. USA 2013, 110, 13582–13587. [Google Scholar] [CrossRef]
- Dwarakanath, A.D.; Campbell, B.J.; Tsai, H.H.; Sunderland, D.; Hart, C.A.; Rhodes, J.M. Faecal mucinase activity assessed in inflammatory bowel disease using 14C threonine labelled mucin substrate. Gut 1995, 37, 58–62. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Press, A.G.; Hauptmann, I.A.; Hauptmann, L.; Fuchs, B.; Fuchs, M.; Ewe, K.; Ramadori, G. Gastrointestinal pH profiles in patients with inflammatory bowel disease. Aliment Pharmacol. Ther. 1998, 12, 673–678. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.H.; Dwarakanath, A.D.; Hart, C.A.; Milton, J.D.; Rhodes, J.M. Increased faecal mucin sulphatase activity in ulcerative colitis: A potential target for treatment. Gut 1995, 36, 570–576. [Google Scholar] [CrossRef] [PubMed]
- Chassaing, B.; Koren, O.; Goodrich, J.K.; Poole, A.C.; Srinivasan, S.; Ley, R.E.; Gewirtz, A.T. Dietary emulsifiers impact the mouse gut microbiota promoting colitis and metabolic syndrome. Nature 2015, 519, 92–96. [Google Scholar] [CrossRef]
- Roberts, C.L.; Keita, A.V.; Duncan, S.H.; O’Kennedy, N.; Soderholm, J.D.; Rhodes, J.M.; Campbell, B.J. Translocation of Crohn’s disease Escherichia coli across M-cells: Contrasting effects of soluble plant fibres and emulsifiers. Gut 2010, 59, 1331–1339. [Google Scholar] [CrossRef] [PubMed]
- Sugihara, K.; Masuda, M.; Nakao, M.; Abuduli, M.; Imi, Y.; Oda, N.; Okahisa, T.; Yamamoto, H.; Takeda, E.; Taketani, Y. Dietary phosphate exacerbates intestinal inflammation in experimental colitis. J. Clin. Biochem. Nutr. 2017, 61, 91–99. [Google Scholar] [CrossRef]
- Swidsinski, A.; Ung, V.; Sydora, B.C.; Loening-Baucke, V.; Doerffel, Y.; Verstraelen, H.; Fedorak, R.N. Bacterial overgrowth and inflammation of small intestine after carboxymethylcellulose ingestion in genetically susceptible mice. Inflamm. Bowel Dis. 2009, 15, 359–364. [Google Scholar] [CrossRef]
- Sadar, S.S.; Vyawahare, N.S.; Bodhankar, S.L. Ferulic acid ameliorates TNBS-induced ulcerative colitis through modulation of cytokines, oxidative stress, iNOs, COX-2, and apoptosis in laboratory rats. EXCLI J. 2016, 15, 482–499. [Google Scholar] [CrossRef]
- Zheng, H.; Chen, M.; Li, Y.; Wang, Y.; Wei, L.; Liao, Z.; Wang, M.; Ma, F.; Liao, Q.; Xie, Z. Modulation of Gut Microbiome Composition and Function in Experimental Colitis Treated with Sulfasalazine. Front. Microbiol. 2017, 8, 1703. [Google Scholar] [CrossRef] [PubMed]
- Laserna-Mendieta, E.J.; Clooney, A.G.; Carretero-Gomez, J.F.; Moran, C.; Sheehan, D.; Nolan, J.A.; Hill, C.; Gahan, C.G.M.; Joyce, S.A.; Shanahan, F.; et al. Determinants of Reduced Genetic Capacity for Butyrate Synthesis by the Gut Microbiome in Crohn’s Disease and Ulcerative Colitis. J. Crohn’s Colitis 2018, 12, 204–216. [Google Scholar] [CrossRef] [PubMed]
- Valcheva, R.; Koleva, P.; Martinez, I.; Walter, J.; Ganzle, M.G.; Dieleman, L.A. Inulin-type fructans improve active ulcerative colitis associated with microbiota changes and increased short-chain fatty acids levels. Gut Microbes 2018, 1–24. [Google Scholar] [CrossRef]
- Earley, H.; Lennon, G.; Balfe, A.; Kilcoyne, M.; Clyne, M.; Joshi, L.; Carrington, S.; Martin, S.T.; Coffey, J.C.; Winter, D.C.; et al. A Preliminary Study Examining the Binding Capacity of Akkermansia muciniphila and Desulfovibrio spp., to Colonic Mucin in Health and Ulcerative Colitis. PLoS ONE 2015, 10, e0135280. [Google Scholar] [CrossRef]
- Tsai, H.H.; Sunderland, D.; Gibson, G.R.; Hart, C.A.; Rhodes, J.M. A novel mucin sulphatase from human faeces: Its identification, purification and characterization. Clin. Sci. 1992, 82, 447–454. [Google Scholar] [CrossRef]
- Wei, Y.; Gong, J.; Zhu, W.; Tian, H.; Ding, C.; Gu, L.; Li, N.; Li, J. Pectin enhances the effect of fecal microbiota transplantation in ulcerative colitis by delaying the loss of diversity of gut flora. BMC Microbiol. 2016, 16, 255. [Google Scholar] [CrossRef]
- Lewis, S.; Brazier, J.; Beard, D.; Nazem, N.; Proctor, D. Effects of metronidazole and oligofructose on faecal concentrations of sulphate-reducing bacteria and their activity in human volunteers. Scand. J. Gastroenterol. 2009, 40, 1296–1303. [Google Scholar] [CrossRef]
- Kellingray, L.; Tapp, H.S.; Saha, S.; Doleman, J.F.; Narbad, A.; Mithen, R.F. Consumption of a diet rich in Brassica vegetables is associated with a reduced abundance of sulphate-reducing bacteria: A randomised crossover study. Mol. Nutr. Food Res. 2017, 61. [Google Scholar] [CrossRef]
- Wu, G.D.; Chen, J.; Hoffmann, C.; Bittinger, K.; Chen, Y.Y.; Keilbaugh, S.A.; Bewtra, M.; Knights, D.; Walters, W.A.; Knight, R.; et al. Linking long-term dietary patterns with gut microbial enterotypes. Science 2011, 334, 105–108. [Google Scholar] [CrossRef]
- Russell, W.R.; Gratz, S.W.; Duncan, S.H.; Holtrop, G.; Ince, J.; Scobbie, L.; Duncan, G.; Johnstone, A.M.; Lobley, G.E.; Wallace, R.J.; et al. High-protein, reduced-carbohydrate weight-loss diets promote metabolite profiles likely to be detrimental to colonic health. Am. J. Clin. Nutr. 2011, 93, 1062–1072. [Google Scholar] [CrossRef]
- Sonnenburg, E.D.; Smits, S.A.; Tikhonov, M.; Higginbottom, S.K.; Wingreen, N.S.; Sonnenburg, J.L. Diet-induced extinctions in the gut microbiota compound over generations. Nature 2016, 529, 212–215. [Google Scholar] [CrossRef] [PubMed]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).