Dietary Factors in Sulfur Metabolism and Pathogenesis of Ulcerative Colitis

 ,
,  ,
,
Abstract
1. Introduction
1.1. Toxic Effects of Hydrogen Sulfide
1.2. Pre-Clinical Models
1.3. Clinical Observations
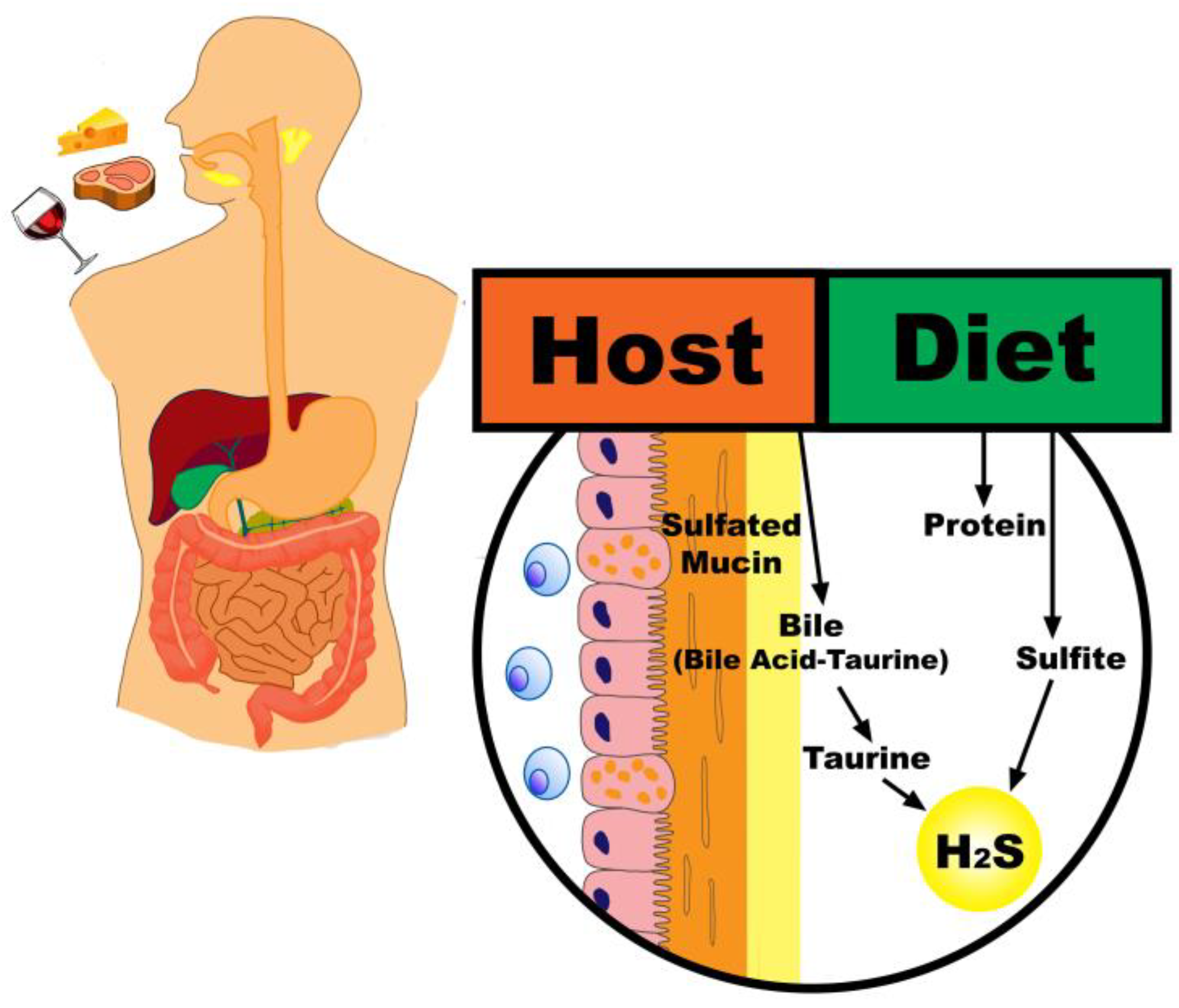
2. Sources of Sulfur in the Colon
2.1. Dietary Intake
2.2. Endogenous Sources
2.3. H2S Measurement
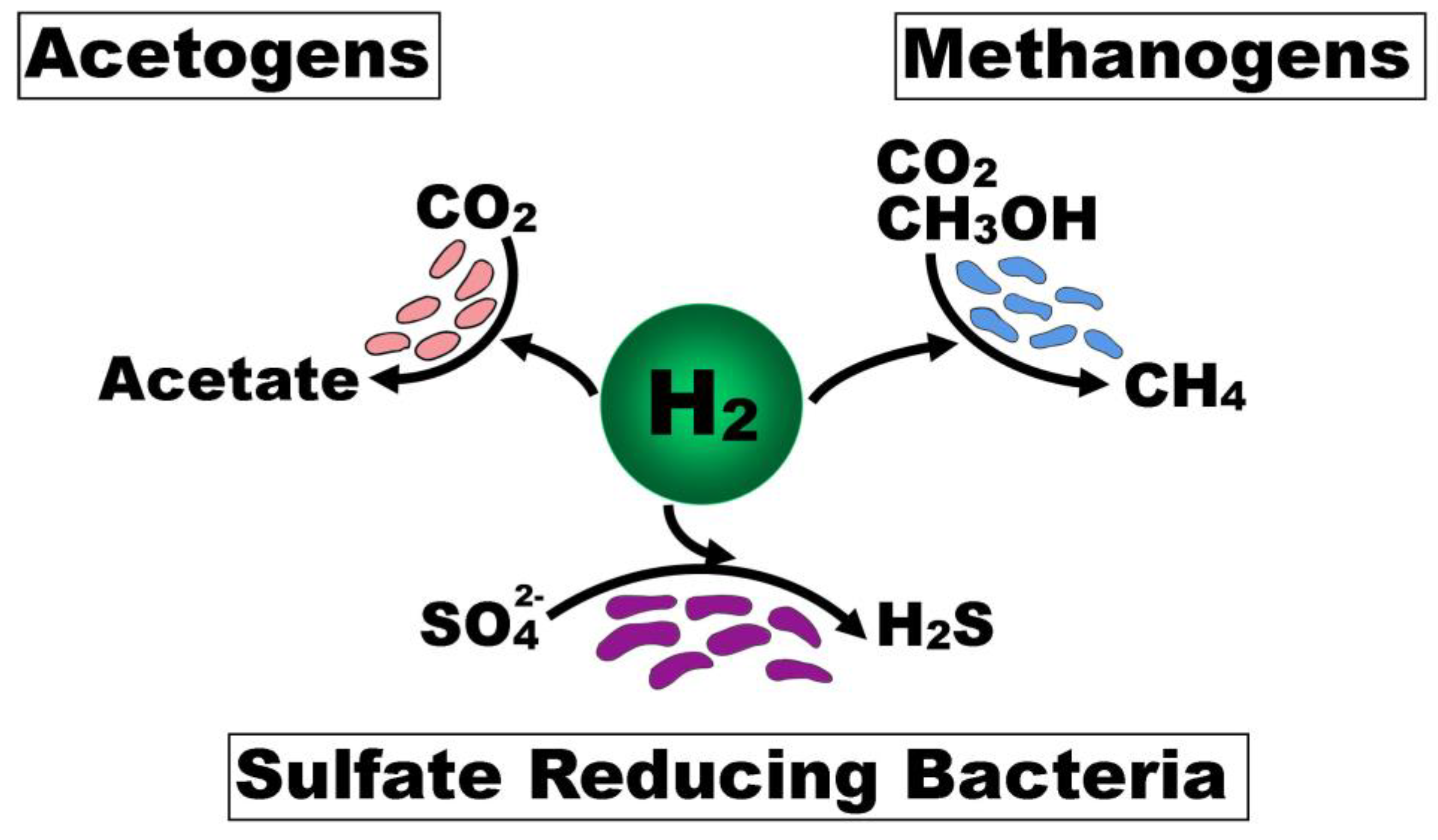
3. Colonic Microbiota and H2S Production
3.1. Sulfate-Reducing Bacteria
3.2. Cysteine Degraders
3.3. Interplay Between Functional Microbial Groups
3.4. H2S Clearance
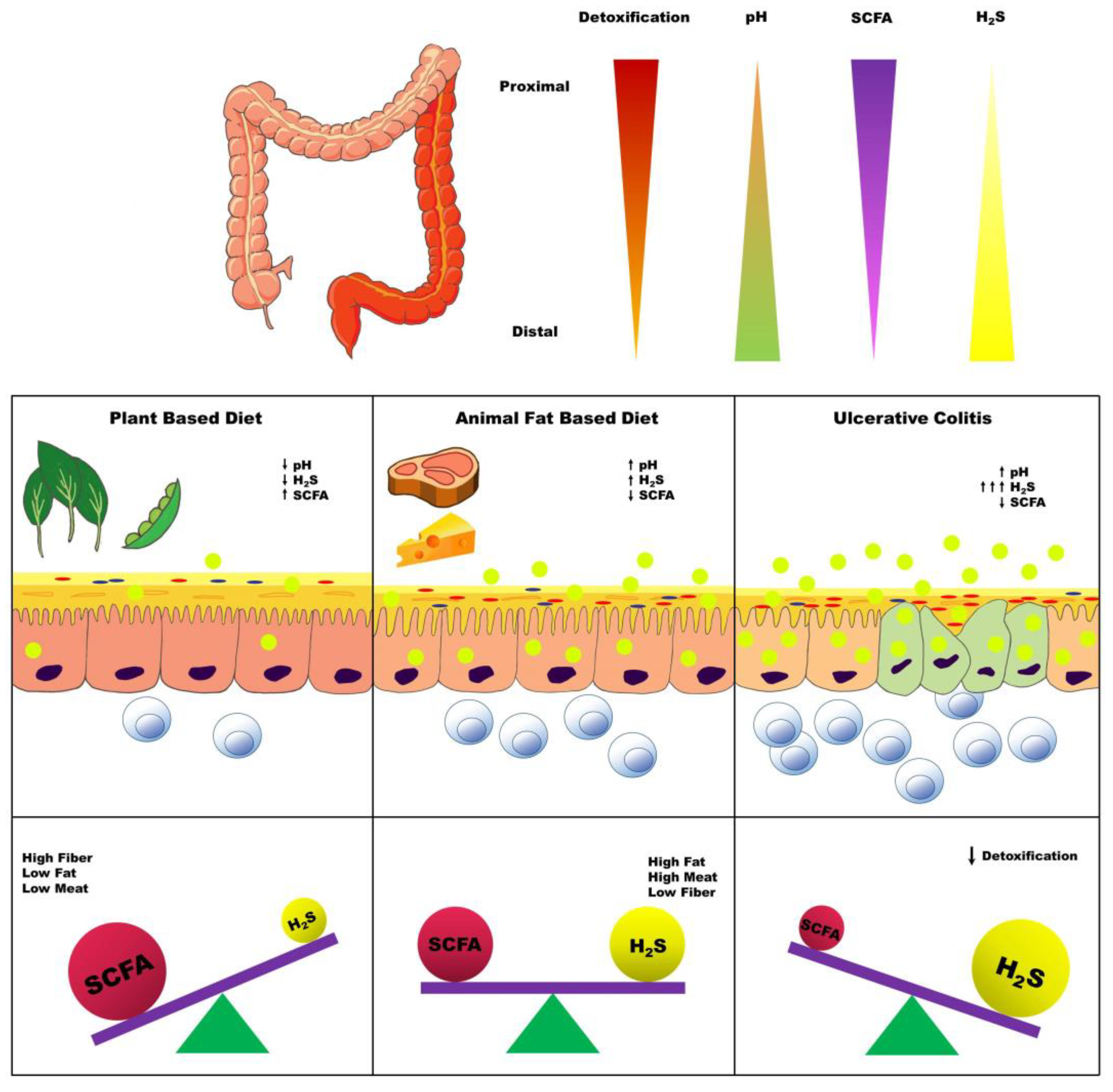
4. Dietary Factors Influencing Colonic H2S Production
4.1. Protein
4.2. Carbohydrate and Fiber
4.3. Dietary Fat
4.4. Endogenous Sulfur
4.5. Miscellaneous Dietary Factors
4.6. Inflammatory Factors
4.7. Interventions Targeting the Colon Microbiota
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kaplan, G.G. The global burden of IBD: From 2015 to 2025. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 720–727. [Google Scholar] [CrossRef]
- Ng, S.C.; Shi, H.Y.; Hamidi, N.; Underwood, F.E.; Tang, W.; Benchimol, E.I.; Panaccione, R.; Ghosh, S.; Wu, J.C.Y.; Chan, F.K.L.; et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: A systematic review of population-based studies. Lancet 2017, 390, 2769–2778. [Google Scholar] [CrossRef]
- Ng, S.C.; Kaplan, G.G.; Tang, W.; Banerjee, R.; Adigopula, B.; Underwood, F.E.; Tanyingoh, D.; Wei, S.C.; Lin, W.C.; Lin, H.H.; et al. Population Density and Risk of Inflammatory Bowel Disease: A Prospective Population-Based Study in 13 Countries or Regions in Asia-Pacific. Am. J. Gastroenterol. 2019, 114, 107–115. [Google Scholar] [CrossRef]
- Rothschild, D.; Weissbrod, O.; Barkan, E.; Kurilshikov, A.; Korem, T.; Zeevi, D.; Costea, P.I.; Godneva, A.; Kalka, I.N.; Bar, N.; et al. Environment dominates over host genetics in shaping human gut microbiota. Nature 2018, 555, 210–215. [Google Scholar] [CrossRef] [PubMed]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Benchimol, E.I.; Mack, D.R.; Guttmann, A.; Nguyen, G.C.; To, T.; Mojaverian, N.; Quach, P.; Manuel, D.G. Inflammatory bowel disease in immigrants to Canada and their children: A population-based cohort study. Am. J. Gastroenterol. 2015, 110, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Vangay, P.; Johnson, A.J.; Ward, T.L.; Al-Ghalith, G.A.; Shields-Cutler, R.R.; Hillmann, B.M.; Lucas, S.K.; Beura, L.K.; Thompson, E.A.; Till, L.M.; et al. US Immigration Westernizes the Human Gut Microbiome. Cell 2018, 175, 962–972.e10. [Google Scholar] [CrossRef]
- Hou, J.K.; Abraham, B.; El-Serag, H. Dietary intake and risk of developing inflammatory bowel disease: A systematic review of the literature. Am. J. Gastroenterol. 2011, 106, 563–573. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, G.G.; Ng, S.C. Understanding and Preventing the Global Increase of Inflammatory Bowel Disease. Gastroenterology 2017, 152, 313–321.e2. [Google Scholar] [CrossRef] [PubMed]
- Yang, G. Hydrogen sulfide in cell survival: A double-edged sword. Expert Rev. Clin. Pharmacol. 2011, 4, 33–47. [Google Scholar] [CrossRef]
- Barton, L.L.; Ritz, N.L.; Fauque, G.D.; Lin, H.C. Sulfur Cycling and the Intestinal Microbiome. Dig. Dis. Sci. 2017, 62, 2241–2257. [Google Scholar] [CrossRef]
- Pitcher, M.C.L.; Cummings, J.H. Hydrogen sulphide: A bacterial toxin in ulcerative colitis? Gut 1996, 39, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Roediger, W.E.W.; Moore, J.; Babidge, W. Colonic sulfide in pathogenesis and treatment of ulcerative colitis. Dig. Dis. Sci. 1997, 42, 1571–1579. [Google Scholar] [CrossRef] [PubMed]
- Rowan, F.E.; Docherty, N.G.; Coffey, J.C.; O’Connell, P.R. Sulphate-reducing bacteria and hydrogen sulphide in the aetiology of ulcerative colitis. Br. J. Surg. 2009, 96, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Blachier, F.; Davila, A.M.; Mimoun, S.; Benetti, P.H.; Atanasiu, C.; Andriamihaja, M.; Benamouzig, R.; Bouillaud, F.; Tome, D. Luminal sulfide and large intestine mucosa: Friend or foe? Amino Acids 2010, 39, 335–347. [Google Scholar] [CrossRef]
- Blachier, F.; Beaumont, M.; Andriamihaja, M.; Davila, A.M.; Lan, A.; Grauso, M.; Armand, L.; Benamouzig, R.; Tome, D. Changes in the Luminal Environment of the Colonic Epithelial Cells and Physiopathological Consequences. Am. J. Pathol. 2017, 187, 476–486. [Google Scholar] [CrossRef]
- Levine, A.; Sigall Boneh, R.; Wine, E. Evolving role of diet in the pathogenesis and treatment of inflammatory bowel diseases. Gut 2018, 67, 1726–1738. [Google Scholar] [CrossRef]
- Chiba, M.; Abe, T.; Tsuda, H.; Sugawara, T.; Tsuda, S.; Tozawa, H.; Fujiwara, K.; Imai, H. Lifestyle-related disease in Crohn’s disease: Relapse prevention by a semi-vegetarian diet. World J. Gastroenterol. 2010, 16, 2484–2495. [Google Scholar] [CrossRef]
- Florin, T.; Neale, G.; Gibson, G.R.; Christl, S.U.; Cummings, J.H. Metabolism of dietary sulphate: Absorption and excretion in humans. Gut 1991, 32, 766–773. [Google Scholar] [CrossRef]
- Devkota, S.; Wang, Y.; Musch, M.W.; Leone, V.; Fehlner-Peach, H.; Nadimpalli, A.; Antonopoulos, D.A.; Jabri, B.; Chang, E.B. Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10-/- mice. Nature 2012, 487, 104–108. [Google Scholar] [CrossRef]
- Holmes, A.J.; Chew, Y.V.; Colakoglu, F.; Cliff, J.B.; Klaassens, E.; Read, M.N.; Solon-Biet, S.M.; McMahon, A.C.; Cogger, V.C.; Ruohonen, K.; et al. Diet-Microbiome Interactions in Health Are Controlled by Intestinal Nitrogen Source Constraints. Cell Metab. 2017, 25, 140–151. [Google Scholar] [CrossRef] [PubMed]
- Desai, M.S.; Seekatz, A.M.; Koropatkin, N.M.; Kamada, N.; Hickey, C.A.; Wolter, M.; Pudlo, N.A.; Kitamoto, S.; Terrapon, N.; Muller, A.; et al. A Dietary Fiber-Deprived Gut Microbiota Degrades the Colonic Mucus Barrier and Enhances Pathogen Susceptibility. Cell 2016, 167, 1339–1353.e1321. [Google Scholar] [CrossRef]
- Wong, J.M.; de Souza, R.; Kendall, C.W.; Emam, A.; Jenkins, D.J. Colonic health: Fermentation and short chain fatty acids. J. Clin. Gastroenterol. 2006, 40, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Roediger, W.E. Role of anaerobic bacteria in the metabolic welfare of the colonic mucosa in man. Gut 1980, 21, 793–798. [Google Scholar] [CrossRef] [PubMed]
- Furusawa, Y.; Obata, Y.; Fukuda, S.; Endo, T.A.; Nakato, G.; Takahashi, D.; Nakanishi, Y.; Uetake, C.; Kato, K.; Kato, T.; et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 2013, 504, 446–450. [Google Scholar] [CrossRef] [PubMed]
- Arpaia, N.; Campbell, C.; Fan, X.; Dikiy, S.; van der Veeken, J.; deRoos, P.; Liu, H.; Cross, J.R.; Pfeffer, K.; Coffer, P.J.; et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 2013, 504, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Finnie, I.A.; Dwarakanath, A.D.; Taylor, B.A.; Rhodes, J.M. Colonic mucin synthesis is increased by sodium butyrate. Gut 1995, 36, 93–99. [Google Scholar] [CrossRef]
- Byndloss, M.X.; Olsan, E.E.; Rivera-Chavez, F.; Tiffany, C.R.; Cevallos, S.A.; Lokken, K.L.; Torres, T.P.; Byndloss, A.J.; Faber, F.; Gao, Y.; et al. Microbiota-activated PPAR-gamma signaling inhibits dysbiotic Enterobacteriaceae expansion. Science 2017, 357, 570–575. [Google Scholar] [CrossRef]
- Litvak, Y.; Byndloss, M.X.; Tsolis, R.M.; Baumler, A.J. Dysbiotic Proteobacteria expansion: A microbial signature of epithelial dysfunction. Curr. Opin. Microbiol. 2017, 39, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Christl, S.U.; Eisner, H.D.; Dusel, G.; Kasper, H.; Scheppach, W. Antagonistic effects of sulfide and butyrate on proliferation of colonic mucosa: A potential role for these agents in the pathogenesis of ulcerative colitis. Dig. Dis. Sci. 1996, 41, 2477–2481. [Google Scholar] [CrossRef]
- O’Flaherty, V.; Mahony, T.; O’Kennedy, R.; Colleran, E. Effect of pH on growth kinetics and sulphide toxicity thresholds of a range of methanogeic, syntrophic and sulphate-reducing bacteria. Process Biochem. 1998, 33, 555–569. [Google Scholar] [CrossRef]
- Babidge, W.; Millard, S.; Roediger, W. Sulfides impair short chain fatty acid beta-oxidation at acyl-CoA dehydrogenase level in colonocytes: Implications for ulcerative colitis. Mol. Cell. Biochem. 1998, 181, 117–124. [Google Scholar] [CrossRef]
- Wallace, J.L.; Blackler, R.W.; Chan, M.V.; Da Silva, G.J.; Elsheikh, W.; Flannigan, K.L.; Gamaniek, I.; Manko, A.; Wang, L.; Motta, J.P.; et al. Anti-inflammatory and cytoprotective actions of hydrogen sulfide: Translation to therapeutics. Antioxid. Redox Signal. 2015, 22, 398–410. [Google Scholar] [CrossRef] [PubMed]
- Odenwald, M.A.; Turner, J.R. The intestinal epithelial barrier: A therapeutic target? Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 9–21. [Google Scholar] [CrossRef]
- Jiang, J.; Chan, A.; Ali, S.; Saha, A.; Haushalter, K.J.; Lam, W.L.; Glasheen, M.; Parker, J.; Brenner, M.; Mahon, S.B.; et al. Hydrogen Sulfide--Mechanisms of Toxicity and Development of an Antidote. Sci. Rep. 2016, 6, 20831. [Google Scholar] [CrossRef]
- Roediger, W.E.; Duncan, A.; Kapaniris, O.; Millard, S. Reducing sulfur compounds of the colon impair colonocyte nutrition: Implications for ulcerative colitis. Gastroenterology 1993, 104, 802–809. [Google Scholar] [CrossRef]
- De Preter, V.; Arijs, I.; Windey, K.; Vanhove, W.; Vermeire, S.; Schuit, F.; Rutgeerts, P.; Verbeke, K. Impaired butyrate oxidation in ulcerative colitis is due to decreased butyrate uptake and a defect in the oxidation pathway. Inflamm. Bowel Dis. 2012, 18, 1127–1136. [Google Scholar] [CrossRef] [PubMed]
- De Preter, V.; Arijs, I.; Windey, K.; Vanhove, W.; Vermeire, S.; Schuit, F.; Rutgeerts, P.; Verbeke, K. Decreased mucosal sulfide detoxification is related to an impaired butyrate oxidation in ulcerative colitis. Inflamm. Bowel Dis. 2012, 18, 2371–2380. [Google Scholar] [CrossRef] [PubMed]
- Johansson, M.E.; Hansson, G.C. Immunological aspects of intestinal mucus and mucins. Nat. Rev. Immunol. 2016, 16, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Johansson, M.E.; Phillipson, M.; Petersson, J.; Velcich, A.; Holm, L.; Hansson, G.C. The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Proc. Natl. Acad. Sci. USA 2008, 105, 15064–15069. [Google Scholar] [CrossRef] [PubMed]
- Johansson, M.E.; Gustafsson, J.K.; Holmen-Larsson, J.; Jabbar, K.S.; Xia, L.; Xu, H.; Ghishan, F.K.; Carvalho, F.A.; Gewirtz, A.T.; Sjovall, H.; et al. Bacteria penetrate the normally impenetrable inner colon mucus layer in both murine colitis models and patients with ulcerative colitis. Gut 2014, 63, 281–291. [Google Scholar] [CrossRef]
- Ijssennagger, N.; Belzer, C.; Hooiveld, G.J.; Dekker, J.; van Mil, S.W.; Muller, M.; Kleerebezem, M.; van der Meer, R. Gut microbiota facilitates dietary heme-induced epithelial hyperproliferation by opening the mucus barrier in colon. Proc. Natl. Acad. Sci. USA 2015, 112, 10038–10043. [Google Scholar] [CrossRef]
- Eichele, D.D.; Kharbanda, K.K. Dextran sodium sulfate colitis murine model: An indispensable tool for advancing our understanding of inflammatory bowel diseases pathogenesis. World J. Gastroenterol. 2017, 23, 6016–6029. [Google Scholar] [CrossRef]
- Zhu, Q.C.; Gao, R.Y.; Wu, W.; Guo, B.M.; Peng, J.Y.; Qin, H.L. Effect of a high-fat diet in development of colonic adenoma in an animal model. World J. Gastroenterol. 2014, 20, 8119–8129. [Google Scholar] [CrossRef]
- Mu, C.; Yang, Y.; Luo, Z.; Guan, L.; Zhu, W. The Colonic Microbiome and Epithelial Transcriptome Are Altered in Rats Fed a High-Protein Diet Compared with a Normal-Protein Diet. J. Nutr. 2016, 146, 474–483. [Google Scholar] [CrossRef]
- Heo, J.M.; Kim, J.C.; Hansen, C.F.; Mullan, B.P.; Hampson, D.J.; Pluske, J.R. Effects of feeding low protein diets to piglets on plasma urea nitrogen, faecal ammonia nitrogen, the incidence of diarrhoea and performance after weaning. Arch. Anim. Nutr. 2008, 62, 343–358. [Google Scholar] [CrossRef]
- Pitcher, M.C.; Beatty, E.R.; Cummings, J.H. The contribution of sulphate reducing bacteria and 5-aminosalicylic acid to faecal sulphide in patients with ulcerative colitis. Gut 2000, 46, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Dubuquoy, L.; Rousseaux, C.; Thuru, X.; Peyrin-Biroulet, L.; Romano, O.; Chavatte, P.; Chamaillard, M.; Desreumaux, P. PPARgamma as a new therapeutic target in inflammatory bowel diseases. Gut 2006, 55, 1341–1349. [Google Scholar] [CrossRef]
- Alex, S.; Lange, K.; Amolo, T.; Grinstead, J.S.; Haakonsson, A.K.; Szalowska, E.; Koppen, A.; Mudde, K.; Haenen, D.; Al-Lahham, S.; et al. Short-chain fatty acids stimulate angiopoietin-like 4 synthesis in human colon adenocarcinoma cells by activating peroxisome proliferator-activated receptor gamma. Mol. Cell. Biol. 2013, 33, 1303–1316. [Google Scholar] [CrossRef]
- Xu, J.; Chen, N.; Wu, Z.; Song, Y.; Zhang, Y.; Wu, N.; Zhang, F.; Ren, X.; Liu, Y. 5-Aminosalicylic Acid Alters the Gut Bacterial Microbiota in Patients with Ulcerative Colitis. Front. Microbiol. 2018, 9, 1274. [Google Scholar] [CrossRef]
- Burke, D.A.; Axon, A.T.; Clayden, S.A.; Dixon, M.F.; Johnston, D.; Lacey, R.W. The efficacy of tobramycin in the treatment of ulcerative colitis. Aliment. Pharmacol. Ther. 1990, 4, 123–129. [Google Scholar] [CrossRef]
- Roediger, W.E.W. Decreased sulphur aminoacid intake in ulcerative colitis. Lancet 1998, 351. [Google Scholar] [CrossRef]
- Kashyap, P.C.; Reigstad, C.S.; Loftus, E.V., Jr. Role of diet and gut microbiota in management of inflammatory bowel disease in an Asian migrant. J. Allergy Clin. Immunol. 2013, 132, 250–250.e255. [Google Scholar] [CrossRef] [PubMed]
- Chiba, M.; Tsuda, S.; Komatsu, M.; Tozawa, H.; Takayama, Y. Onset of Ulcerative Colitis during a Low-Carbohydrate Weight-Loss Diet and Treatment with a Plant-Based Diet: A Case Report. Perm. J. 2016, 20, 80–84. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chiba, M.; Sugawara, T.; Komatsu, M.; Tozawa, H. Onset of Ulcerative Colitis in the Second Trimester after Emesis Gravidarum: Treatment with Plant-based Diet. Inflamm. Bowel Dis. 2018, 24, e8–e9. [Google Scholar] [CrossRef]
- Chiba, M.; Nakane, K.; Tsuji, T.; Tsuda, S.; Ishii, H.; Ohno, H.; Watanabe, K.; Ito, M.; Komatsu, M.; Yamada, K.; et al. Relapse Prevention in Ulcerative Colitis by Plant-Based Diet Through Educational Hospitalization: A Single-Group Trial. Perm. J. 2018, 22. [Google Scholar] [CrossRef] [PubMed]
- Florin, T.H.J.; Neale, G.; Goretski, S.; Cummings, J.H. The sulfate content of foods and beverages. J. Food Compos. Anal. 1993, 6, 140–151. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. Food Additive Status List. Available online: https://www.fda.gov/food/ingredientspackaginglabeling/foodadditivesingredients/ucm091048.htm (accessed on 15 August 2018).
- Curno, R.; Magee, E.A.; Edmond, L.M.; Cummings, J.H. Studies of a urinary biomarker of dietary inorganic sulphur in subjects on diets containing 1–38 mmol sulphur/day and of the half-life of ingested 34SO4(2-). Eur. J. Clin. Nutr. 2008, 62, 1106–1115. [Google Scholar] [CrossRef]
- Magee, E.A.; Curno, R.; Edmond, L.M.; Cummings, J.H. Contribution of dietary protein and inorganic sulfur to urinary sulfate: Toward a biomarker of inorganic sulfur intake. Am. J. Clin. Nutr. 2004, 80, 137–142. [Google Scholar] [CrossRef]
- Dostal Webster, A.; Staley, C.; Hamilton, M.J.; Huang, M.; Fryxell, K.; Erickson, R.; Kabage, A.J.; Sadowsky, M.J.; Khoruts, A. Influence of short-term changes in dietary sulfur on the relative abundances of intestinal sulfate-reducing bacteria. Gut Microbes 2019, 1–11. [Google Scholar] [CrossRef]
- Florin, T.H. Hydrogen sulphide and total acid-volatile sulphide in faeces, determined with a direct spectrophotometric method. Clin. Chim. Acta 1991, 196, 127–134. [Google Scholar] [CrossRef]
- Buffiere, C.; Gaudichon, C.; Hafnaoui, N.; Migne, C.; Scislowsky, V.; Khodorova, N.; Mosoni, L.; Blot, A.; Boirie, Y.; Dardevet, D.; et al. In the elderly, meat protein assimilation from rare meat is lower than that from meat that is well done. Am. J. Clin. Nutr. 2017, 106, 1257–1266. [Google Scholar] [CrossRef]
- Mills, D.J.; Tuohy, K.M.; Booth, J.; Buck, M.; Crabbe, M.J.; Gibson, G.R.; Ames, J.M. Dietary glycated protein modulates the colonic microbiota towards a more detrimental composition in ulcerative colitis patients and non-ulcerative colitis subjects. J. Appl. Microbiol. 2008, 105, 706–714. [Google Scholar] [CrossRef] [PubMed]
- Oberli, M.; Marsset-Baglieri, A.; Airinei, G.; Sante-Lhoutellier, V.; Khodorova, N.; Remond, D.; Foucault-Simonin, A.; Piedcoq, J.; Tome, D.; Fromentin, G.; et al. High True Ileal Digestibility but Not Postprandial Utilization of Nitrogen from Bovine Meat Protein in Humans Is Moderately Decreased by High-Temperature, Long-Duration Cooking. J. Nutr. 2015, 145, 2221–2228. [Google Scholar] [CrossRef]
- Pennings, B.; Groen, B.B.; van Dijk, J.W.; de Lange, A.; Kiskini, A.; Kuklinski, M.; Senden, J.M.; van Loon, L.J. Minced beef is more rapidly digested and absorbed than beef steak, resulting in greater postprandial protein retention in older men. Am. J. Clin. Nutr. 2013, 98, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Remond, D.; Machebeuf, M.; Yven, C.; Buffiere, C.; Mioche, L.; Mosoni, L.; Mirand, P.P. Postprandial whole-body protein metabolism after a meat meal is influenced by chewing efficiency in elderly subjects. Am. J. Clin. Nutr. 2007, 85, 1286–1292. [Google Scholar] [CrossRef] [PubMed]
- Ridlon, J.M.; Wolf, P.G.; Gaskins, H.R. Taurocholic acid metabolism by gut microbes and colon cancer. Gut Microbes 2016, 7, 201–215. [Google Scholar] [CrossRef] [PubMed]
- Di Ciaula, A.; Garruti, G.; Lunardi Baccetto, R.; Molina-Molina, E.; Bonfrate, L.; Wang, D.Q.; Portincasa, P. Bile Acid Physiology. Ann. Hepatol. 2017, 16, s4–s14. [Google Scholar] [CrossRef]
- Johansson, M.E.; Larsson, J.M.; Hansson, G.C. The two mucus layers of colon are organized by the MUC2 mucin, whereas the outer layer is a legislator of host-microbial interactions. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. 1), 4659–4665. [Google Scholar] [CrossRef]
- Holmen Larsson, J.M.; Thomsson, K.A.; Rodriguez-Pineiro, A.M.; Karlsson, H.; Hansson, G.C. Studies of mucus in mouse stomach, small intestine, and colon. III. Gastrointestinal Muc5ac and Muc2 mucin O-glycan patterns reveal a regiospecific distribution. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 305, G357–G363. [Google Scholar] [CrossRef] [PubMed]
- Robbe, C.; Capon, C.; Coddeville, B.; Michalski, J.-C. Structural diversity and specific distribution of O-glycans in normal human mucins along the intestinal tract. Biochem. J. 2004, 384, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Lennon, G.; Balfe, A.; Bambury, N.; Lavelle, A.; Maguire, A.; Docherty, N.G.; Coffey, J.C.; Winter, D.C.; Sheahan, K.; O’Connell, P.R. Correlations between colonic crypt mucin chemotype, inflammatory grade and Desulfovibrio species in ulcerative colitis. Colorectal Dis. 2014, 16, O161–O169. [Google Scholar] [CrossRef] [PubMed]
- Gibson, G.R.; Cummings, J.H.; Macfarlane, G.T. Growth and activities of sulphate-reducing bacteria in gut contents of healthy subjects and patients with ulcerative colitis. FEMS Microbiol. Ecol. 1991, 86, 103–112. [Google Scholar] [CrossRef]
- Moore, J.; Babidge, W.; Millard, S.; Roediger, W. Colonic luminal hydrogen sulfide is not elevated in ulcerative colitis. Dig. Dis. Sci. 1998, 43, 162–165. [Google Scholar] [CrossRef]
- Hughes, M.N.; Centelles, M.N.; Moore, K.P. Making and working with hydrogen sulfide: The chemistry and generation of hydrogen sulfide in vitro and its measurement in vivo: A review. Free Radic. Biol. Med. 2009, 47, 1346–1353. [Google Scholar] [CrossRef]
- Kolluru, G.K.; Shen, X.; Bir, S.C.; Kevil, C.G. Hydrogen sulfide chemical biology: Pathophysiological roles and detection. Nitric Oxide 2013, 35, 5–20. [Google Scholar] [CrossRef] [PubMed]
- Magee, E.A.; Richardson, C.J.; Hughes, R.; Cummings, J.H. Contribution of dietary protein to sulfide production in the large intestine: An in vitro and a controlled feeding study in humans. Am. J. Clin. Nutr. 2000, 72, 1488–1494. [Google Scholar] [CrossRef] [PubMed]
- Vitvitsky, V.; Banerjee, R. H2S analysis in biological samples using gas chromatography with sulfur chemiluminescence detection. Methods Enzymol. 2015, 554, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Levine, J.; Ellis, C.J.; Furne, J.K.; Springfield, J.; Levitt, M.D. Fecal hydrogen sulfide production in ulcerative colitis. Am. J. Gastroenterol. 1998, 93, 83–87. [Google Scholar] [CrossRef]
- Yao, C.K.; Rotbart, A.; Ou, J.Z.; Kalantar-Zadeh, K.; Muir, J.G.; Gibson, P.R. Modulation of colonic hydrogen sulfide production by diet and mesalazine utilizing a novel gas-profiling technology. Gut Microbes 2018, 1–13. [Google Scholar] [CrossRef]
- Anantharaman, K.; Hausmann, B.; Jungbluth, S.P.; Kantor, R.S.; Lavy, A.; Warren, L.A.; Rappe, M.S.; Pester, M.; Loy, A.; Thomas, B.C.; et al. Expanded diversity of microbial groups that shape the dissimilatory sulfur cycle. ISME J. 2018, 12, 1715–1728. [Google Scholar] [CrossRef]
- Scanlan, P.D.; Shanahan, F.; Marchesi, J.R. Culture-independent analysis of desulfovibrios in the human distal colon of healthy, colorectal cancer and polypectomized individuals. FEMS Microbiol. Ecol. 2009, 69, 213–221. [Google Scholar] [CrossRef]
- Muller, A.L.; Kjeldsen, K.U.; Rattei, T.; Pester, M.; Loy, A. Phylogenetic and environmental diversity of DsrAB-type dissimilatory (bi)sulfite reductases. ISME J. 2015, 9, 1152–1165. [Google Scholar] [CrossRef] [PubMed]
- Christophersen, C.T.; Morrison, M.; Conlon, M.A. Overestimation of the abundance of sulfate-reducing bacteria in human feces by quantitative PCR targeting the Desulfovibrio 16S rRNA gene. Appl. Environ. Microbiol. 2011, 77, 3544–3546. [Google Scholar] [CrossRef] [PubMed]
- Coutinho, C.; Coutinho-Silva, R.; Zinkevich, V.; Pearce, C.B.; Ojcius, D.M.; Beech, I. Sulphate-reducing bacteria from ulcerative colitis patients induce apoptosis of gastrointestinal epithelial cells. Microb. Pathog. 2017, 112, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Hale, V.L.; Jeraldo, P.; Mundy, M.; Yao, J.; Keeney, G.; Scott, N.; Cheek, E.H.; Davidson, J.; Green, M.; Martinez, C.; et al. Synthesis of multi-omic data and community metabolic models reveals insights into the role of hydrogen sulfide in colon cancer. Methods 2018. [Google Scholar] [CrossRef] [PubMed]
- Jia, W.; Whitehead, R.N.; Griffiths, L.; Dawson, C.; Bai, H.; Waring, R.H.; Ramsden, D.B.; Hunter, J.O.; Cauchi, M.; Bessant, C.; et al. Diversity and distribution of sulphate-reducing bacteria in human faeces from healthy subjects and patients with inflammatory bowel disease. FEMS Immunol. Med. Microbiol. 2012, 65, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Duffy, M.; O’Mahony, L.; Coffey, J.C.; Collins, J.K.; Shanahan, F.; Redmond, H.P.; Kirwan, W.O. Sulfate-Reducing Bacteria Colonize Pouches Formed for Ulcerative Colitis but Not for Familial Adenomatous Polyposis. Dis. Colon Rectum 2002, 45, 384–388. [Google Scholar] [CrossRef]
- Windey, K.; De Preter, V.; Verbeke, K. Relevance of protein fermentation to gut health. Mol. Nutr. Food Res. 2012, 56, 184–196. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H. Metabolic turnover of hydrogen sulfide. Front. Physiol. 2012, 3, 101. [Google Scholar] [CrossRef]
- Awano, N.; Wada, M.; Mori, H.; Nakamori, S.; Takagi, H. Identification and functional analysis of Escherichia coli cysteine desulfhydrases. Appl. Environ. Microbiol. 2005, 71, 4149–4152. [Google Scholar] [CrossRef]
- Sahami, S.; Kooij, I.A.; Meijer, S.L.; Van den Brink, G.R.; Buskens, C.J.; Te Velde, A.A. The Link between the Appendix and Ulcerative Colitis: Clinical Relevance and Potential Immunological Mechanisms. Am. J. Gastroenterol. 2016, 111, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Rogers, M.B.; Brower-Sinning, R.; Firek, B.; Zhong, D.; Morowitz, M.J. Acute Appendicitis in Children Is Associated with a Local Expansion of Fusobacteria. Clin. Infect. Dis. 2016, 63, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Zhong, D.; Brower-Sinning, R.; Firek, B.; Morowitz, M.J. Acute appendicitis in children is associated with an abundance of bacteria from the phylum Fusobacteria. J. Pediatr. Surg. 2014, 49, 441–446. [Google Scholar] [CrossRef]
- Basic, A.; Blomqvist, M.; Dahlen, G.; Svensater, G. The proteins of Fusobacterium spp. involved in hydrogen sulfide production from L-cysteine. BMC Microbiol. 2017, 17, 61. [Google Scholar] [CrossRef] [PubMed]
- Basic, A.; Blomqvist, S.; Carlen, A.; Dahlen, G. Estimation of bacterial hydrogen sulfide production in vitro. J. Oral Microbiol. 2015, 7, 28166. [Google Scholar] [CrossRef] [PubMed]
- Wolf, P.G.; Biswas, A.; Morales, S.E.; Greening, C.; Gaskins, H.R. H2 metabolism is widespread and diverse among human colonic microbes. Gut Microbes 2016, 7, 235–245. [Google Scholar] [CrossRef]
- Carbonero, F.; Benefiel, A.C.; Gaskins, H.R. Contributions of the microbial hydrogen economy to colonic homeostasis. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 504–518. [Google Scholar] [CrossRef]
- Macfarlane, S.; Macfarlane, G.T. Regulation of short-chain fatty acid production. Proc. Nutr. Soc. 2003, 62, 67–72. [Google Scholar] [CrossRef]
- Kunkel, D.; Basseri, R.J.; Makhani, M.D.; Chong, K.; Chang, C.; Pimentel, M. Methane on breath testing is associated with constipation: A systematic review and meta-analysis. Dig. Dis. Sci. 2011, 56, 1612–1618. [Google Scholar] [CrossRef]
- Macfarlane, G.T.; Gibson, G.R.; Cummings, J.H. Comparison of fermentation reactions in different regions of the human colon. J. Appl. Bacteriol. 1992, 72, 57–64. [Google Scholar]
- Rao, S.S.; Kuo, B.; McCallum, R.W.; Chey, W.D.; DiBaise, J.K.; Hasler, W.L.; Koch, K.L.; Lackner, J.M.; Miller, C.; Saad, R.; et al. Investigation of colonic and whole-gut transit with wireless motility capsule and radiopaque markers in constipation. Clin. Gastroenterol. Hepatol. 2009, 7, 537–544. [Google Scholar] [CrossRef]
- Videla, S.; Vilaseca, J.; Antolin, M.; Garcia-Lafuente, A.; Guarner, F.; Crespo, E.; Casalots, J.; Salas, A.; Malagelada, J.R. Dietary inulin improves distal colitis induced by dextran sodium sulfate in the rat. Am. J. Gastroenterol. 2001, 96, 1486–1493. [Google Scholar] [CrossRef] [PubMed]
- de Vries, J.; Miller, P.E.; Verbeke, K. Effects of cereal fiber on bowel function: A systematic review of intervention trials. World J. Gastroenterol. 2015, 21, 8952–8963. [Google Scholar] [CrossRef]
- Walker, A.W.; Duncan, S.H.; McWilliam Leitch, E.C.; Child, M.W.; Flint, H.J. pH and peptide supply can radically alter bacterial populations and short-chain fatty acid ratios within microbial communities from the human colon. Appl. Environ. Microbiol. 2005, 71, 3692–3700. [Google Scholar] [CrossRef] [PubMed]
- Duncan, S.H.; Louis, P.; Thomson, J.M.; Flint, H.J. The role of pH in determining the species composition of the human colonic microbiota. Environ. Microbiol. 2009, 11, 2112–2122. [Google Scholar] [CrossRef]
- Gibson, G.R.; Cummings, J.H.; Macfarlane, G.T.; Allison, C.; Segal, I.; Vorster, H.H.; Walker, A.R.P. Alternative pathways for hydrogen disposal during fermentation in the human colon. Gut 1990, 31, 679–683. [Google Scholar] [CrossRef]
- Beaumont, M.; Andriamihaja, M.; Lan, A.; Khodorova, N.; Audebert, M.; Blouin, J.M.; Grauso, M.; Lancha, L.; Benetti, P.H.; Benamouzig, R.; et al. Detrimental effects for colonocytes of an increased exposure to luminal hydrogen sulfide: The adaptive response. Free Radic. Biol. Med. 2016, 93, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Suarez, F.; Furne, J.; Springfield, J.; Levitt, M. Insights into human colonic physiology obtained from the study of flatus composition. Am. Physiol. Soc. 1997, 272, G1028–G1033. [Google Scholar] [CrossRef] [PubMed]
- Levitt, M.D.; Furne, J.; Springfield, J.; Suarez, F.; DeMaster, E. Detoxification of hydrogen sulfide and methanethiol in the cecal mucosa. J. Clin. Investig. 1999, 104, 1107–1114. [Google Scholar] [CrossRef] [PubMed]
- Furne, J.; Springfield, J.; Koenig, T.; DeMaster, E.; Levitt, M.D. Oxidation of hydrogen sulfide and methanethiol to thiosulfate by rat tissues: A specialized function of the colonic mucosa. Biochem. Pharmacol. 2001, 62, 255–259. [Google Scholar] [CrossRef]
- Picton, R.; Eggo, M.C.; Merrill, G.A.; Langman, M.J.S.; Singh, S. Mucosal protection against sulphide: Importance of the enzyme rhodanese. Gut 2002, 50, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Picton, R.; Eggo, M.C.; Langman, M.J.; Singh, S. Impaired detoxication of hydrogen sulfide in ulcerative colitis? Dig. Dis. Sci. 2007, 52, 373–378. [Google Scholar] [CrossRef]
- Wilson, K.; Mudra, M.; Furne, J.; Levitt, M. Differentiation of the roles of sulfide oxidase and rhodanese in the detoxification of sulfide by the colonic mucosa. Dig. Dis. Sci. 2008, 53, 277–283. [Google Scholar] [CrossRef]
- Landry, A.P.; Ballou, D.P.; Banerjee, R. Modulation of Catalytic Promiscuity during Hydrogen Sulfide Oxidation. ACS Chem. Biol. 2018, 13, 1651–1658. [Google Scholar] [CrossRef]
- Filipovic, M.R.; Zivanovic, J.; Alvarez, B.; Banerjee, R. Chemical Biology of H2S Signaling through Persulfidation. Chem. Rev. 2018, 118, 1253–1337. [Google Scholar] [CrossRef]
- Landry, A.P.; Ballou, D.P.; Banerjee, R. H2S oxidation by nanodisc-embedded human sulfide quinone oxidoreductase. J. Biol. Chem. 2017, 292, 11641–11649. [Google Scholar] [CrossRef] [PubMed]
- Libiad, M.; Yadav, P.K.; Vitvitsky, V.; Martinov, M.; Banerjee, R. Organization of the human mitochondrial hydrogen sulfide oxidation pathway. J. Biol. Chem. 2014, 289, 30901–30910. [Google Scholar] [CrossRef] [PubMed]
- Mimoun, S.; Andriamihaja, M.; Chaumontet, C.; Atanasiu, C.; Benamouzig, R.; Blouin, J.M.; Tome, D.; Bouillaud, F.; Blachier, F. Detoxification of H(2)S by differentiated colonic epithelial cells: Implication of the sulfide oxidizing unit and of the cell respiratory capacity. Antioxid. Redox Signal. 2012, 17, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Sido, B.; Hack, V.; Hochlehnert, A.; Lipps, H.; Herfarth, C.; Droge, W. Impairment of intestinal glutathione synthesis in patients with inflammatory bowel disease. Gut 1998, 42, 485–492. [Google Scholar] [CrossRef]
- Fuentes, S.; Rossen, N.G.; van der Spek, M.J.; Hartman, J.H.; Huuskonen, L.; Korpela, K.; Salojarvi, J.; Aalvink, S.; de Vos, W.M.; D’Haens, G.R.; et al. Microbial shifts and signatures of long-term remission in ulcerative colitis after faecal microbiota transplantation. ISME J. 2017, 11, 1877–1889. [Google Scholar] [CrossRef]
- Bajer, L.; Kverka, M.; Kostovcik, M.; Macinga, P.; Dvorak, J.; Stehlikova, Z.; Brezina, J.; Wohl, P.; Spicak, J.; Drastich, P. Distinct gut microbiota profiles in patients with primary sclerosing cholangitis and ulcerative colitis. World J. Gastroenterol. 2017, 23, 4548–4558. [Google Scholar] [CrossRef]
- Vermeire, S.; Joossens, M.; Verbeke, K.; Wang, J.; Machiels, K.; Sabino, J.; Ferrante, M.; Van Assche, G.; Rutgeerts, P.; Raes, J. Donor Species Richness Determines Faecal Microbiota Transplantation Success in Inflammatory Bowel Disease. J. Crohn’s Colitis 2016, 10, 387–394. [Google Scholar] [CrossRef]
- Smith, E.A.; Macfarlane, G.T. Enumeration of human colonic bacteria producing phenolic and indolic compounds: Effects of pH, carbohydrate availability and retention time on dissimilatory aromatic amino acid metabolism. J. Appl. Bacteriol. 1996, 81, 288–302. [Google Scholar] [CrossRef]
- Zimmer, J.; Lange, B.; Frick, J.S.; Sauer, H.; Zimmermann, K.; Schwiertz, A.; Rusch, K.; Klosterhalfen, S.; Enck, P. A vegan or vegetarian diet substantially alters the human colonic faecal microbiota. Eur. J. Clin. Nutr. 2012, 66, 53–60. [Google Scholar] [CrossRef]
- Devkota, S.; Chang, E.B. Interactions between Diet, Bile Acid Metabolism, Gut Microbiota, and Inflammatory Bowel Diseases. Dig. Dis. 2015, 33, 351–356. [Google Scholar] [CrossRef]
- Leone, V.; Gibbons, S.M.; Martinez, K.; Hutchison, A.L.; Huang, E.Y.; Cham, C.M.; Pierre, J.F.; Heneghan, A.F.; Nadimpalli, A.; Hubert, N.; et al. Effects of diurnal variation of gut microbes and high-fat feeding on host circadian clock function and metabolism. Cell Host Microbe 2015, 17, 681–689. [Google Scholar] [CrossRef]
- Barcelo, A.; Claustre, J.; Moro, F.; Chayvialle, J.-A.; Cuber, J.-C. Plaisancia. Mucin secretion is modulated by luminal factors in the isolated vascularly perfused rat colon. Gut 2000, 46, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Deplancke, B.; Finster, K.; Vallen Graham, W.; Collier, C.T.; Thurmond, J.E.; Gaskins, H.R. Gastrointestinal and Microbial Responses to Sulfate-Supplemented Drinking Water in Mice. Exp. Biol. Med. 2003, 228, 424–433. [Google Scholar] [CrossRef]
- Schroeder, B.O.; Birchenough, G.M.H.; Stahlman, M.; Arike, L.; Johansson, M.E.V.; Hansson, G.C.; Backhed, F. Bifidobacteria or Fiber Protects against Diet-Induced Microbiota-Mediated Colonic Mucus Deterioration. Cell Host Microbe 2018, 23, 27–40.e27. [Google Scholar] [CrossRef] [PubMed]
- Croix, J.A.; Carbonero, F.; Nava, G.M.; Russell, M.; Greenberg, E.; Gaskins, H.R. On the relationship between sialomucin and sulfomucin expression and hydrogenotrophic microbes in the human colonic mucosa. PLoS ONE 2011, 6, e24447. [Google Scholar] [CrossRef] [PubMed]
- Rey, F.E.; Gonzalez, M.D.; Cheng, J.; Wu, M.; Ahern, P.P.; Gordon, J.I. Metabolic niche of a prominent sulfate-reducing human gut bacterium. Proc. Natl. Acad. Sci. USA 2013, 110, 13582–13587. [Google Scholar] [CrossRef]
- Dwarakanath, A.D.; Campbell, B.J.; Tsai, H.H.; Sunderland, D.; Hart, C.A.; Rhodes, J.M. Faecal mucinase activity assessed in inflammatory bowel disease using 14C threonine labelled mucin substrate. Gut 1995, 37, 58–62. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Press, A.G.; Hauptmann, I.A.; Hauptmann, L.; Fuchs, B.; Fuchs, M.; Ewe, K.; Ramadori, G. Gastrointestinal pH profiles in patients with inflammatory bowel disease. Aliment Pharmacol. Ther. 1998, 12, 673–678. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.H.; Dwarakanath, A.D.; Hart, C.A.; Milton, J.D.; Rhodes, J.M. Increased faecal mucin sulphatase activity in ulcerative colitis: A potential target for treatment. Gut 1995, 36, 570–576. [Google Scholar] [CrossRef] [PubMed]
- Chassaing, B.; Koren, O.; Goodrich, J.K.; Poole, A.C.; Srinivasan, S.; Ley, R.E.; Gewirtz, A.T. Dietary emulsifiers impact the mouse gut microbiota promoting colitis and metabolic syndrome. Nature 2015, 519, 92–96. [Google Scholar] [CrossRef]
- Roberts, C.L.; Keita, A.V.; Duncan, S.H.; O’Kennedy, N.; Soderholm, J.D.; Rhodes, J.M.; Campbell, B.J. Translocation of Crohn’s disease Escherichia coli across M-cells: Contrasting effects of soluble plant fibres and emulsifiers. Gut 2010, 59, 1331–1339. [Google Scholar] [CrossRef] [PubMed]
- Sugihara, K.; Masuda, M.; Nakao, M.; Abuduli, M.; Imi, Y.; Oda, N.; Okahisa, T.; Yamamoto, H.; Takeda, E.; Taketani, Y. Dietary phosphate exacerbates intestinal inflammation in experimental colitis. J. Clin. Biochem. Nutr. 2017, 61, 91–99. [Google Scholar] [CrossRef]
- Swidsinski, A.; Ung, V.; Sydora, B.C.; Loening-Baucke, V.; Doerffel, Y.; Verstraelen, H.; Fedorak, R.N. Bacterial overgrowth and inflammation of small intestine after carboxymethylcellulose ingestion in genetically susceptible mice. Inflamm. Bowel Dis. 2009, 15, 359–364. [Google Scholar] [CrossRef]
- Sadar, S.S.; Vyawahare, N.S.; Bodhankar, S.L. Ferulic acid ameliorates TNBS-induced ulcerative colitis through modulation of cytokines, oxidative stress, iNOs, COX-2, and apoptosis in laboratory rats. EXCLI J. 2016, 15, 482–499. [Google Scholar] [CrossRef]
- Zheng, H.; Chen, M.; Li, Y.; Wang, Y.; Wei, L.; Liao, Z.; Wang, M.; Ma, F.; Liao, Q.; Xie, Z. Modulation of Gut Microbiome Composition and Function in Experimental Colitis Treated with Sulfasalazine. Front. Microbiol. 2017, 8, 1703. [Google Scholar] [CrossRef] [PubMed]
- Laserna-Mendieta, E.J.; Clooney, A.G.; Carretero-Gomez, J.F.; Moran, C.; Sheehan, D.; Nolan, J.A.; Hill, C.; Gahan, C.G.M.; Joyce, S.A.; Shanahan, F.; et al. Determinants of Reduced Genetic Capacity for Butyrate Synthesis by the Gut Microbiome in Crohn’s Disease and Ulcerative Colitis. J. Crohn’s Colitis 2018, 12, 204–216. [Google Scholar] [CrossRef] [PubMed]
- Valcheva, R.; Koleva, P.; Martinez, I.; Walter, J.; Ganzle, M.G.; Dieleman, L.A. Inulin-type fructans improve active ulcerative colitis associated with microbiota changes and increased short-chain fatty acids levels. Gut Microbes 2018, 1–24. [Google Scholar] [CrossRef]
- Earley, H.; Lennon, G.; Balfe, A.; Kilcoyne, M.; Clyne, M.; Joshi, L.; Carrington, S.; Martin, S.T.; Coffey, J.C.; Winter, D.C.; et al. A Preliminary Study Examining the Binding Capacity of Akkermansia muciniphila and Desulfovibrio spp., to Colonic Mucin in Health and Ulcerative Colitis. PLoS ONE 2015, 10, e0135280. [Google Scholar] [CrossRef]
- Tsai, H.H.; Sunderland, D.; Gibson, G.R.; Hart, C.A.; Rhodes, J.M. A novel mucin sulphatase from human faeces: Its identification, purification and characterization. Clin. Sci. 1992, 82, 447–454. [Google Scholar] [CrossRef]
- Wei, Y.; Gong, J.; Zhu, W.; Tian, H.; Ding, C.; Gu, L.; Li, N.; Li, J. Pectin enhances the effect of fecal microbiota transplantation in ulcerative colitis by delaying the loss of diversity of gut flora. BMC Microbiol. 2016, 16, 255. [Google Scholar] [CrossRef]
- Lewis, S.; Brazier, J.; Beard, D.; Nazem, N.; Proctor, D. Effects of metronidazole and oligofructose on faecal concentrations of sulphate-reducing bacteria and their activity in human volunteers. Scand. J. Gastroenterol. 2009, 40, 1296–1303. [Google Scholar] [CrossRef]
- Kellingray, L.; Tapp, H.S.; Saha, S.; Doleman, J.F.; Narbad, A.; Mithen, R.F. Consumption of a diet rich in Brassica vegetables is associated with a reduced abundance of sulphate-reducing bacteria: A randomised crossover study. Mol. Nutr. Food Res. 2017, 61. [Google Scholar] [CrossRef]
- Wu, G.D.; Chen, J.; Hoffmann, C.; Bittinger, K.; Chen, Y.Y.; Keilbaugh, S.A.; Bewtra, M.; Knights, D.; Walters, W.A.; Knight, R.; et al. Linking long-term dietary patterns with gut microbial enterotypes. Science 2011, 334, 105–108. [Google Scholar] [CrossRef]
- Russell, W.R.; Gratz, S.W.; Duncan, S.H.; Holtrop, G.; Ince, J.; Scobbie, L.; Duncan, G.; Johnstone, A.M.; Lobley, G.E.; Wallace, R.J.; et al. High-protein, reduced-carbohydrate weight-loss diets promote metabolite profiles likely to be detrimental to colonic health. Am. J. Clin. Nutr. 2011, 93, 1062–1072. [Google Scholar] [CrossRef]
- Sonnenburg, E.D.; Smits, S.A.; Tikhonov, M.; Higginbottom, S.K.; Wingreen, N.S.; Sonnenburg, J.L. Diet-induced extinctions in the gut microbiota compound over generations. Nature 2016, 529, 212–215. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Author Year | Location | Study Design | Intervention | Outcomes |
|---|---|---|---|---|
| Roediger 1998 [52] | Australia | Prospective pilot, Single-arm, 12 months duration (n = 4) | Reduction of sulfur amino acid intake for 12 months following an acute attack of UC | Histological improvement at 12 months. Decreased number of daily bowel movements and stool more formed. Two patients noted worsening symptoms when off diet. |
| Kashyap et al. 2013 [53] | United States | Case Report 26-year-old male with notable history of migration to Canada at the age of 10 from Southeast Asia and transition to western diet | Eliminated dairy, refined sugar, pork, beef, and fried food. Emphasized vegetables, fruit, chicken, and fish. | Majority of UC symptoms alleviated at 12 months. Prospectively, symptoms worsened when off diet and improved when returned to diet. |
| Chiba et al. 2016 [54] | Japan | Case Report 38-year-old male who began an Atkins-type diet that emphasized meat and animal fat 2-years prior to UC diagnosis | Transitioned to plant-based, semi-vegetarian diet | Achieved remission without medication. Qualitative comment that symptoms returned with worsening diet compliance over time. |
| Chiba et al. 2018 [55] | Japan | Case Report 36-year-old female diagnosed with UC at 21 weeks gestation who experienced notable diet changes with onset of emesis | Transitioned to a plant-based, lacto-ovo-vegetarian diet | Remission achieved and maintained through pregnancy |
| Chiba et al. 2018 [56] | Japan | Prospective, single-arm (n = 60) | Individuals with mild UC or UC in remission were provided with plant-based diet education | Relapse rates extending to follow-up at 5 years ≤19%, which the authors purport is better than those previously reported. Diet adherence decreased over time. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Teigen, L.M.; Geng, Z.; Sadowsky, M.J.; Vaughn, B.P.; Hamilton, M.J.; Khoruts, A. Dietary Factors in Sulfur Metabolism and Pathogenesis of Ulcerative Colitis. Nutrients 2019, 11, 931. https://doi.org/10.3390/nu11040931
Teigen LM, Geng Z, Sadowsky MJ, Vaughn BP, Hamilton MJ, Khoruts A. Dietary Factors in Sulfur Metabolism and Pathogenesis of Ulcerative Colitis. Nutrients. 2019; 11(4):931. https://doi.org/10.3390/nu11040931
Chicago/Turabian StyleTeigen, Levi M., Zhuo Geng, Michael J. Sadowsky, Byron P. Vaughn, Matthew J. Hamilton, and Alexander Khoruts. 2019. "Dietary Factors in Sulfur Metabolism and Pathogenesis of Ulcerative Colitis" Nutrients 11, no. 4: 931. https://doi.org/10.3390/nu11040931
APA StyleTeigen, L. M., Geng, Z., Sadowsky, M. J., Vaughn, B. P., Hamilton, M. J., & Khoruts, A. (2019). Dietary Factors in Sulfur Metabolism and Pathogenesis of Ulcerative Colitis. Nutrients, 11(4), 931. https://doi.org/10.3390/nu11040931