Non-Nutritive Sweeteners and Their Implications on the Development of Metabolic Syndrome

 , ,
, , 
Abstract
1. Introduction
2. Current Status on the Use of Non-Nutritive Sweeteners
3. Future of Artificial Sweeteners in the Food Industry
- Those that are interested in having low-sugar, low-calorie options to promote a healthy lifestyle and to avoid some of the health issues associated with consuming high amounts of sugar, such as obesity, diabetes, and heart disease.
- Those who already have with one or more of these health issues and are looking for ways to improve their diet and manage their health.
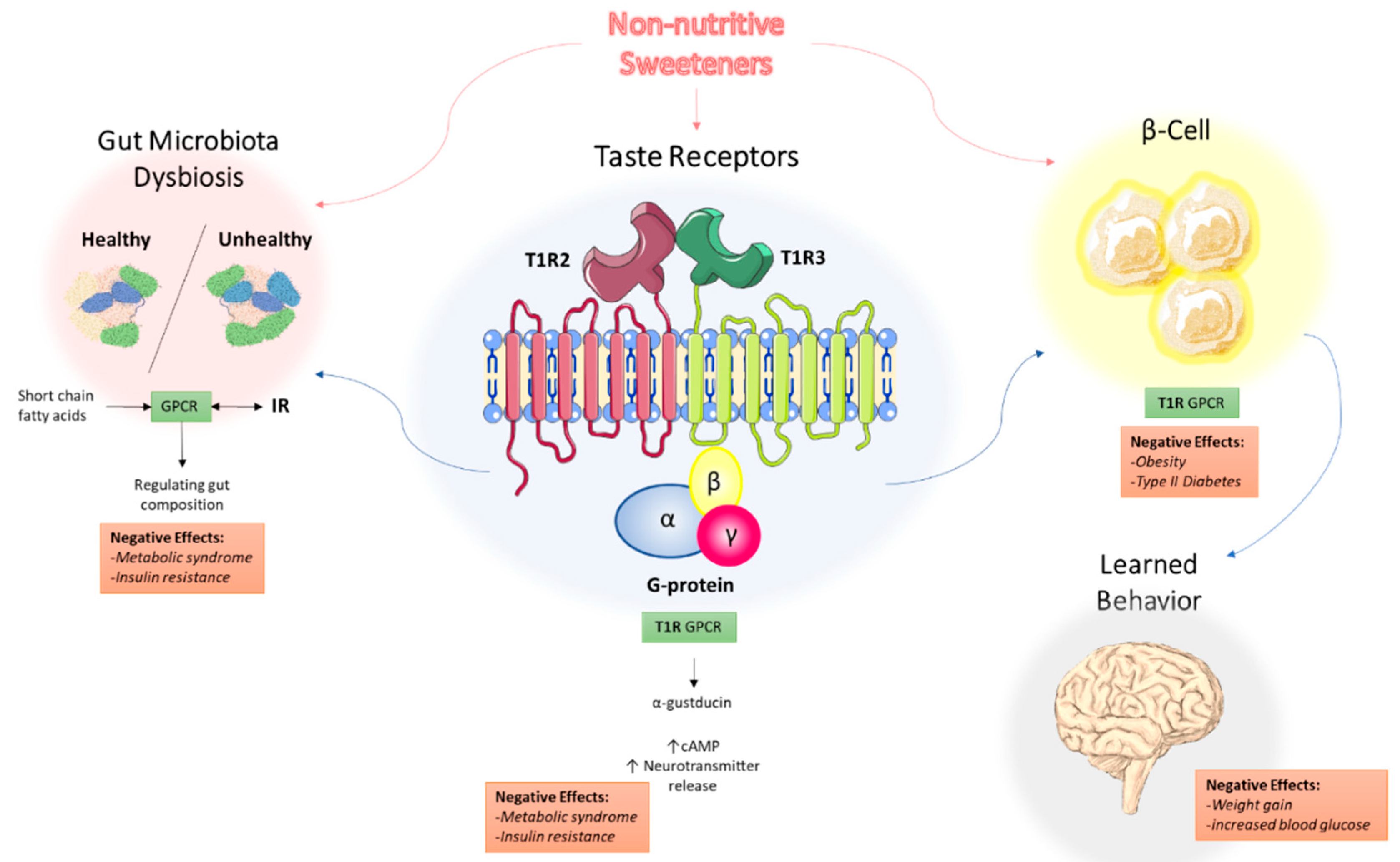
4. Physiological Effects of Non-Nutritive Sweeteners
5. Non-Nutritive Sweeteners Interact with Sweet-Taste Receptors
5.1. Sweet-Taste Receptors in the Mouth: Perception of Sweetness
5.2. Sweet-Taste Receptors in the Gut: Effect of Sweeteners on Hormone Secretion
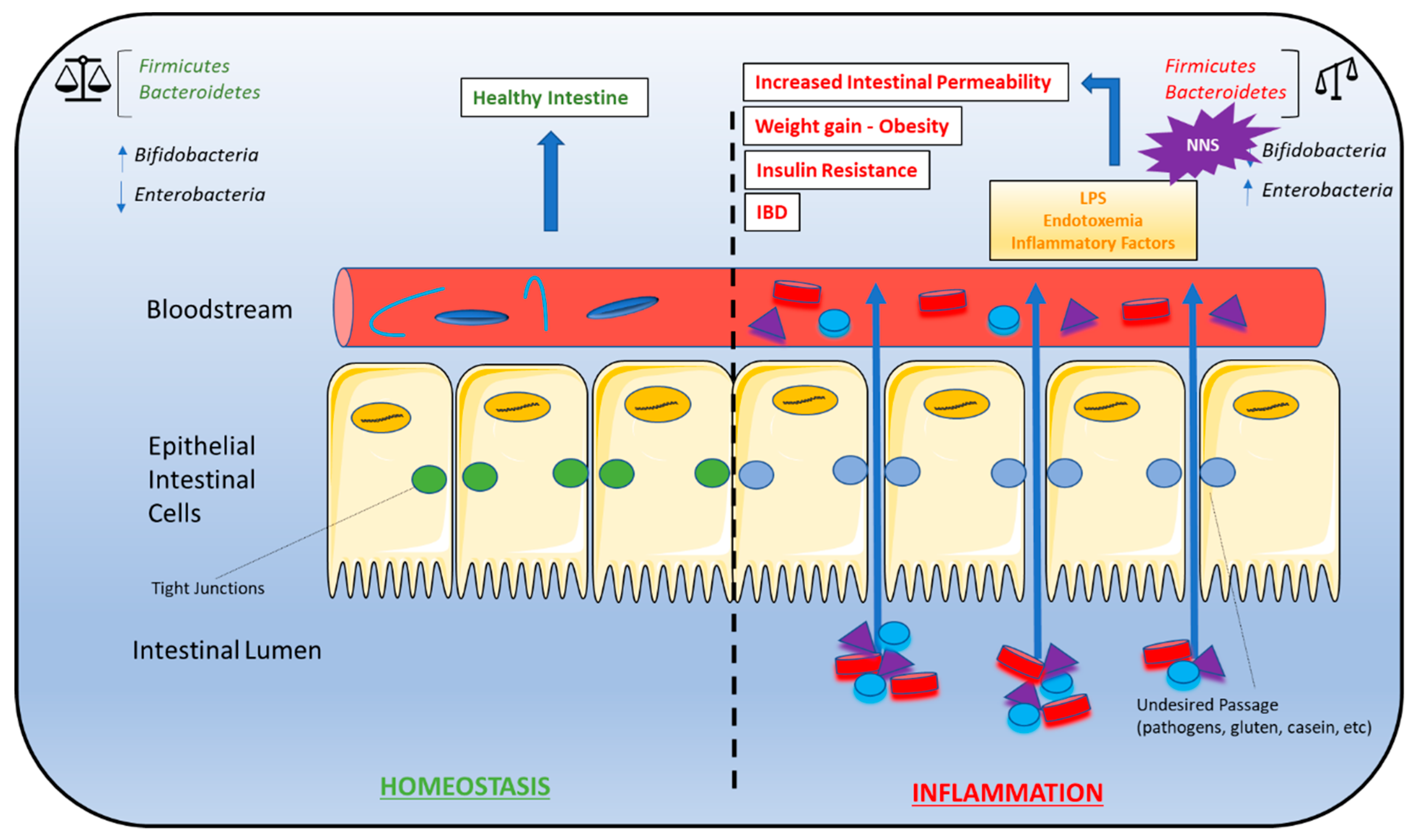
6. Non-Nutritive Sweeteners Interfere with Gut Microbiota Composition
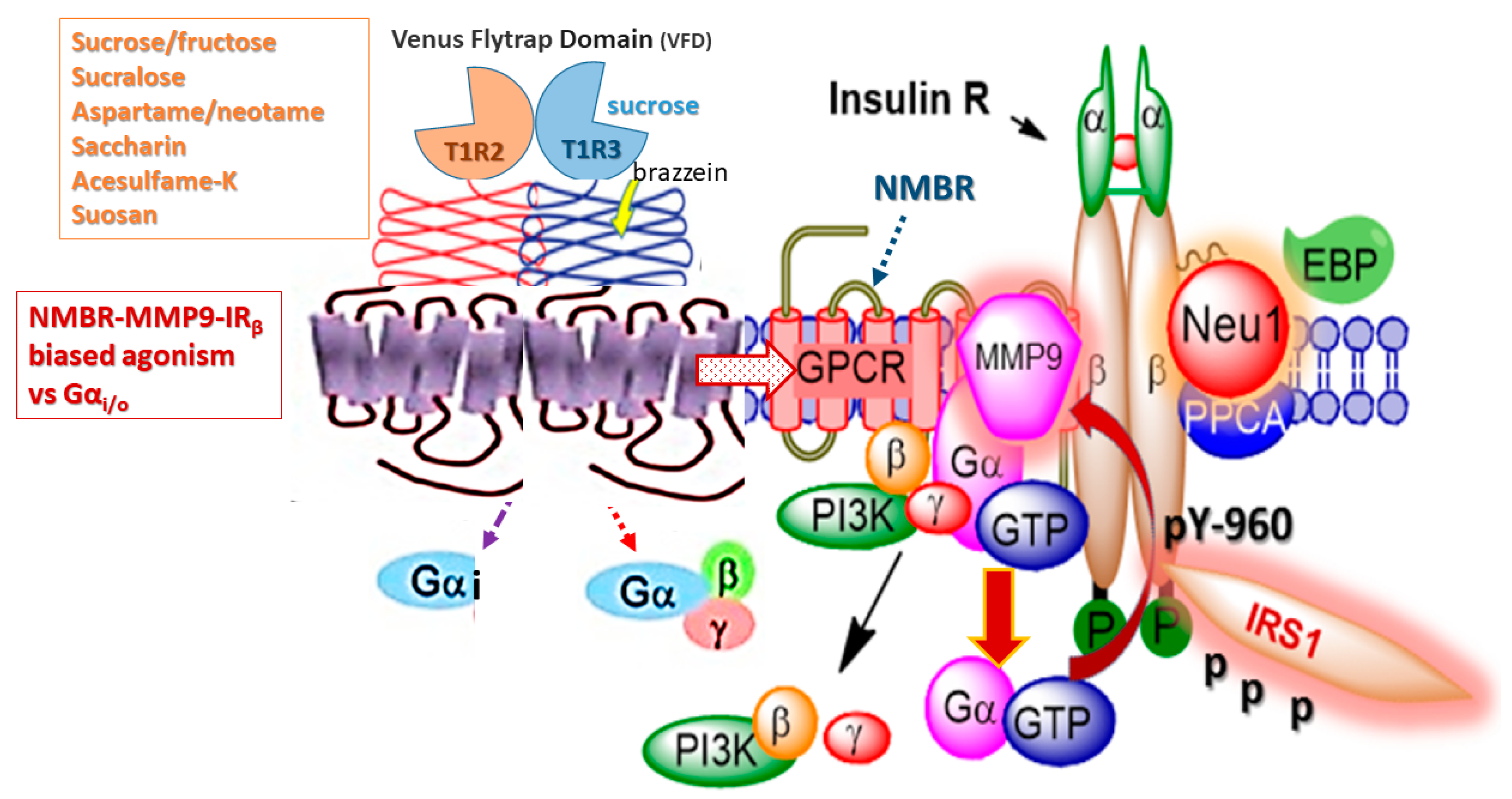
6.1. The Role of GPCR-IR Crosstalk in Metabolic Syndrome
6.2. NAS Modify miRNAs in Regulating Gut Composition
7. Non-Nutritive Sweeteners Interfere with Learned Responses to Sweetness
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Ace-K | Acesulfame-potassium |
| CCK | Cholecystokinin |
| CHD | Congenital heart defect |
| EEC | Enteroendocrine cells |
| FDA | Food and Drug Administration |
| GI | Gastrointestinal |
| GLP-1 | Glucagon-like peptide-1 |
| GLUT2 | Glucose transporter-2 |
| GPCR | G protein coupled receptor |
| GRAS | Generally recognized as safe |
| IBD | Inflammatory bowel disease |
| IEC | Intestinal epithelial cell |
| IR | insulin receptor |
| LPS | Lipopolysaccharide |
| miRNA | MicroRNA |
| MMP9 | Matrix metalloproteinase-9 |
| NAS | Noncaloric artificial sweeteners |
| Neu-1 | Neuraminidase-1 |
| NHLBI | National Heart, Lung and Blood Institute |
| NNS | Non-nutritive sweeteners |
| NOD | Nucleotide oligomerization domain |
| PDE | Phosphodiesterase |
| PLCβ2 | Phospholipase Cβ2 |
| PYY | Peptide YY |
| rEM | Rapid evidence mapping |
| SCFA | Short-chain fatty acid |
| SGLT-1 | Sodium-glucose co-transporter-1 |
| T1R | Taste 1 receptor |
| T2DM | Type 2 diabetes mellitus |
| T2R | Taste 2 receptor |
| TCR | Taste receptor cell |
| TRPM5 | Transient receptor potential cation channel subfamily M member 5 |
References
- Siervo, M.; Montagnese, C.; Mathers, J.C.; Soroka, K.R.; Stephan, B.C.; Wells, J.C. Sugar consumption and global prevalence of obesity and hypertension: An ecological analysis. Public Health Nutr. 2014, 17, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, S.; Raychaudhuri, U.; Chakraborty, R. Artificial sweeteners—A review. J. Food Sci. Technol. 2014, 51, 611–621. [Google Scholar] [CrossRef] [PubMed]
- Fitch, C.; Keim, K.S. Position of the Academy of Nutrition and Dietetics: Use of nutritive and nonnutritive sweeteners. J. Acad. Nutr. Diet. 2012, 112, 739–758. [Google Scholar] [CrossRef]
- Burke, M.V.; Small, D.M. Physiological mechanisms by which non-nutritive sweeteners may impact body weight and metabolism. Physiol. Behav. 2015, 152, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Hess, E.L.; Myers, E.A.; Swithers, S.E.; Hedrick, V.E. Associations between Nonnutritive Sweetener Intake and Metabolic Syndrome in Adults. J. Am. Coll. Nutr. 2018, 37, 487–493. [Google Scholar] [CrossRef]
- Kaur, J. A Comprehensive Review on Metabolic Syndrome. Cardiol. Res. Pract. 2014, 2014, 943162. [Google Scholar] [CrossRef] [PubMed]
- Grundy, S.M.; Cleeman, J.I.; Daniels, S.R.; Donato, K.A.; Eckel, R.H.; Franklin, B.A.; Gordon, D.J.; Krauss, R.M.; Savage, P.J.; Smith, S.C., Jr.; et al. Diagnosis and management of the metabolic syndrome: An American Heart Association/National Heart, Lung, and Blood Institute scientific statement. Circulation 2005, 112, 2735–2752. [Google Scholar] [CrossRef] [PubMed]
- Rebholz, C.M.; Young, B.A.; Katz, R.; Tucker, K.L.; Carithers, T.C.; Norwood, A.F.; Correa, A. Patterns of Beverages Consumed and Risk of Incident Kidney Disease. Clin. J. Am. Soc. Nephrol. 2019, 14, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Praveena, S.M.; Cheema, M.S.; Guo, H.-R. Non-nutritive artificial sweeteners as an emerging contaminant in environment: A global review and risks perspectives. Ecotoxicol. Environ. Saf. 2019, 170, 699–707. [Google Scholar] [CrossRef] [PubMed]
- Dale, M.T.G.; Magnus, P.; Leirgul, E.; Holmstrøm, H.; Gjessing, H.K.; Brodwall, K.; Haugen, M.; Stoltenberg, C.; Øyen, N. Intake of sucrose-sweetened soft beverages during pregnancy and risk of congenital heart defects (CHD) in offspring: A Norwegian pregnancy cohort study. Eur. J. Epidemiol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Shwide-Slavin, C.; Swift, C.; Ross, T. Nonnutritive sweeteners: Where are we today? Diabetes Spectr. 2012, 25, 104–110. [Google Scholar] [CrossRef]
- Neil, M.J. The Merck Index: An Encyclopedia of Chemicals, Drugs and Biologicals; Merck Research Laboratories, Division of Merck and Co. Inc.: Whitehouse Station, NJ, USA, 2006. [Google Scholar]
- Peters John, C.; Beck, J.; Cardel, M.; Wyatt Holly, R.; Foster Gary, D.; Pan, Z.; Wojtanowski Alexis, C.; Vander Veur Stephanie, S.; Herring Sharon, J.; Brill, C.; et al. The effects of water and non-nutritive sweetened beverages on weight loss and weight maintenance: A randomized clinical trial. Obesity 2015, 24, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Sylvetsky, A.C.; Jin, Y.; Clark, E.J.; Welsh, J.A.; Rother, K.I.; Talegawkar, S.A. Consumption of Low-Calorie Sweeteners among Children and Adults in the United States. J. Acad. Nutr. Diet. 2017, 117, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Arnold, D.L.; Krewski, D.; Munro, I.C. Saccharin: A toxicological and historical perspective. Toxicology 1983, 27, 179–256. [Google Scholar] [CrossRef]
- Purohit, V.; Mishra, S. The truth about artificial sweeteners – Are they good for diabetics? Indian Heart J. 2018, 70, 197–199. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Amarnath, S.; Thulasimani, M.; Ramaswamy, S. Artificial sweeteners as a sugar substitute: Are they really safe? Indian J. Pharmacol. 2016, 48, 237–240. [Google Scholar] [CrossRef] [PubMed]
- Lam, J.; Howard, B.E.; Thayer, K.; Shah, R.R. Low-calorie sweeteners and health outcomes: A demonstration of rapid evidence mapping (rEM). Environ. Int. 2019, 123, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.J.; De Banate, M.A.; Rother, K.I. Artificial sweeteners: A systematic review of metabolic effects in youth. Int. J. Pediatr. Obes. 2010, 5, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Sylvetsky, A.C.; Rother, K.I. Trends in the consumption of low-calorie sweeteners. Physiol. Behav. 2016, 164, 446–450. [Google Scholar] [CrossRef]
- Philliphs, M.L. Gut Reaction: Environmental Effects on the Human Microbiota. Environ. Health Perspect. 2009, 117, A198–A205. [Google Scholar]
- Heidari-Beni, M.; Kelishadi, R. The role of dietary sugars and sweeteners in metabolic disorders and diabetes. In Sweeteners: Pharmacology, Biotechnology, and Applications; Merillon, J.-M., Ramawat, K.G., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 1–19. [Google Scholar]
- Seetharaman, S. Chapter 24—The influences of dietary sugar and related metabolic disorders on cognitive aging and dementia. In Molecular Basis of Nutrition and Aging; Mocchegiani, E., Ed.; Academic Press: San Diego, CA, USA, 2016; pp. 331–344. [Google Scholar]
- Stanhope, K.L. Sugar consumption, metabolic disease and obesity: The state of the controversy. Crit. Rev. Clin. Lab. Sci. 2016, 53, 52–67. [Google Scholar] [CrossRef] [PubMed]
- Pepino, M.Y. Metabolic effects of non-nutritive sweeteners. Physiol. Behav. 2015, 152, 450–455. [Google Scholar] [CrossRef] [PubMed]
- Fernstrom, J.D. Non-nutritive sweeteners. In Fructose, High Fructose Corn Syrup, Sucrose and Health; Springer: New York, NY, USA, 2014; pp. 63–84. [Google Scholar]
- Janssen, S.; Depoortere, I. Nutrient sensing in the gut: New roads to therapeutics? Trends Endocrinol. Metab. 2013, 24, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Chaudhari, N.; Roper, S.D. The cell biology of taste. J. Cell Biol. 2010, 190, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Laffitte, A.; Neiers, F.; Briand, L. Functional roles of the sweet taste receptor in oral and extraoral tissues. Curr. Opin. Clin. Nutr. Metab. Care 2014, 17, 379. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Staszewski, L.; Tang, H.; Adler, E.; Zoller, M.; Li, X. Different functional roles of T1R subunits in the heteromeric taste receptors. Proc. Natl. Acad. Sci. USA 2004, 101, 14258–14263. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.J.; Rother, K.I. Non-nutritive sweeteners and their role in the gastrointestinal tract. J. Clin. Endocrinol. Metab. 2012, 97, 2597–2605. [Google Scholar] [CrossRef] [PubMed]
- Chandrashekar, J.; Hoon, M.A.; Ryba, N.J.; Zuker, C.S. The receptors and cells for mammalian taste. Nature 2006, 444, 288. [Google Scholar] [CrossRef] [PubMed]
- Freunda, J.R.; Lee, R.J. Taste receptors in the upper airway. World J Otorhinolaryngol Head Neck Surg. 2018, 4, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Hoon, M.A.; Chandrashekar, J.; Mueller, K.L.; Cook, B.; Wu, D.; Zuker, C.S.; Ryba, N.J. Coding of sweet, bitter, and umami tastes: Different receptor cells sharing similar signaling pathways. Cell 2003, 112, 293–301. [Google Scholar] [CrossRef]
- Urwyler, S. Allosteric modulation of family C G-protein-coupled receptors: From molecular insights to therapeutic perspectives. Pharmacol. Rev. 2011, 63, 59–126. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, S.K.; McKinnon, P.J.; Margolskee, R.F. Gustducin is a taste-cell-specific G protein closely related to the transducins. Nature 1992, 357, 563–569. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, S.K.; McKinnon, P.J.; Spickofsky, N.; Danho, W.; Margolskee, R.F. Molecular cloning of G proteins and phosphodiesterases from rat taste cells. Physiol. Behav. 1994, 56, 1157–1164. [Google Scholar] [CrossRef]
- Kinnamon, S.C. Taste receptor signaling—From tongues to lungs. Acta Physiol. 2012, 204, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Finger, T.E.; Kinnamon, S.C. Taste isn’t just for taste buds anymore. F1000 Biol. Rep. 2011, 3, 20. [Google Scholar] [CrossRef] [PubMed]
- Margolskee, R.F.; Dyer, J.; Kokrashvili, Z.; Salmon, K.S.; Ilegems, E.; Daly, K.; Maillet, E.L.; Ninomiya, Y.; Mosinger, B.; Shirazi-Beechey, S.P. T1R3 and gustducin in gut sense sugars to regulate expression of Na+-glucose cotransporter 1. Proc. Natl. Acad. Sci. USA 2007, 104, 15075–15080. [Google Scholar] [CrossRef]
- Gerspach, A.C.; Steinert, R.E.; Schönenberger, L.; Graber-Maier, A.; Beglinger, C. The role of the gut sweet taste receptor in regulating GLP-1, PYY, and CCK release in humans. Am. J. Physiol. Endocrinol. Metab. 2011, 301, E317–E325. [Google Scholar] [CrossRef]
- Nakagawa, Y.; Nagasawa, M.; Yamada, S.; Hara, A.; Mogami, H.; Nikolaev, V.O.; Lohse, M.J.; Shigemura, N.; Ninomiya, Y.; Kojima, I. Sweet taste receptor expressed in pancreatic β-cells activates the calcium and cyclic AMP signaling systems and stimulates insulin secretion. PLoS ONE 2009, 4, e5106. [Google Scholar] [CrossRef] [PubMed]
- Negri, R.; Morini, G.; Greco, L. From the tongue to the gut. J. Pediatr. Gastroenterol. Nutr. 2011, 53, 601–605. [Google Scholar] [CrossRef]
- Han, P.; Bagenna, B.; Fu, M. The sweet taste signalling pathways in the oral cavity and the gastrointestinal tract affect human appetite and food intake: A review. Int. J. Food Sci. Nutr. 2019, 70, 125–135. [Google Scholar] [CrossRef]
- Cummings, D.E.; Overduin, J. Gastrointestinal regulation of food intake. J. Clin. Investig. 2007, 117, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Everard, A.; Duparc, T. Gut microbiota, enteroendocrine functions and metabolism. Curr. Opin. Pharmacol. 2013, 13, 935–940. [Google Scholar] [CrossRef] [PubMed]
- Steinert, R.E.; Feinle-Bisset, C.; Asarian, L.; Horowitz, M.; Beglinger, C.; Geary, N. Ghrelin, CCK, GLP-1, and PYY (3–36): Secretory controls and physiological roles in eating and glycemia in health, obesity, and after RYGB. Physiol. Rev. 2016, 97, 411–463. [Google Scholar] [CrossRef] [PubMed]
- Fernstrom, J.D. Non-nutritive sweeteners and obesity. Annu. Rev. Food Sci. Technol. 2015, 6, 119–136. [Google Scholar] [CrossRef] [PubMed]
- Stearns, A.T.; Balakrishnan, A.; Rhoads, D.B.; Tavakkolizadeh, A. Rapid upregulation of sodium-glucose transporter SGLT1 in response to intestinal sweet taste stimulation. Ann. Surg. 2010, 251, 865–871. [Google Scholar] [CrossRef] [PubMed]
- Moran, A.W.; Al-Rammahi, M.A.; Arora, D.K.; Batchelor, D.J.; Coulter, E.A.; Daly, K.; Ionescu, C.; Bravo, D.; Shirazi-Beechey, S.P. Expression of Na+/glucose co-transporter 1 (SGLT1) is enhanced by supplementation of the diet of weaning piglets with artificial sweeteners. Br. J. Nutr. 2010, 104, 637–646. [Google Scholar] [CrossRef] [PubMed]
- Kreuch, D.; Keating, D.J.; Wu, T.; Horowitz, M.; Rayner, C.K.; Young, R.L. Gut mechanisms linking intestinal sweet sensing to glycemic control. Front. Endocrinol. 2018, 9, 741. [Google Scholar] [CrossRef] [PubMed]
- Decker, K.J. Potential Mechanisms for NNS-Induced Metabolic Deviances: Satiety Hormone Secretion and Alterations in the Gut Microbiota. DePaul Discov. 2018, 7, 5. [Google Scholar]
- Egan, J.M.; Margolskee, R.F. Taste cells of the gut and gastrointestinal chemosensation. Mol. Interv. 2008, 8, 78–81. [Google Scholar] [CrossRef]
- Bruzzese, E.; Volpicelli, M.; Squaglia, M.; Tartaglione, A.; Guarino, A. Impact of prebiotics on human health. Dig. Liver Dis. 2006, 38, S283–S287. [Google Scholar] [CrossRef]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Altamirano-Barrera, A.; Uribe, M.; Chávez-Tapia, N.C.; Nuño-Lámbarri, N. The role of the gut microbiota in the pathology and prevention of liver disease. J. Nutr. Biochem. 2018, 60, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Kallus, S.J.; Brandt, L.J. The intestinal microbiota and obesity. J. Clin. Gastroenterol. 2012, 46, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Larsen, N.; Vogensen, F.K.; Van Den Berg, F.W.; Nielsen, D.S.; Andreasen, A.S.; Pedersen, B.K.; Al-Soud, W.A.; Sørensen, S.J.; Hansen, L.H.; Jakobsen, M. Gut microbiota in human adults with type 2 diabetes differs from non-diabetic adults. PLoS ONE 2010, 5, e9085. [Google Scholar] [CrossRef] [PubMed]
- Eckburg, P.B.; Bik, E.M.; Bernstein, C.N.; Purdom, E.; Dethlefsen, L.; Sargent, M.; Gill, S.R.; Nelson, K.E.; Relman, D.A. Diversity of the human intestinal microbial flora. Science 2005, 308, 1635–1638. [Google Scholar] [CrossRef] [PubMed]
- Castaner, O.; Goday, A.; Park, Y.-M.; Lee, S.-H.; Magkos, F.; Shiow, S.-A.T.E.; Schröder, H. The gut microbiome profile in obesity: A systematic review. Int. J. Endocrinol. 2018, 2018, 9109451. [Google Scholar] [CrossRef] [PubMed]
- Ley, R.E.; Turnbaugh, P.J.; Klein, S.; Gordon, J.I. Microbial ecology: Human gut microbes associated with obesity. Nature 2006, 444, 1022–1023. [Google Scholar] [CrossRef]
- Kasai, C.; Sugimoto, K.; Moritani, I.; Tanaka, J.; Oya, Y.; Inoue, H.; Tameda, M.; Shiraki, K.; Ito, M.; Takei, Y.; et al. Comparison of the gut microbiota composition between obese and non-obese individuals in a Japanese population, as analyzed by terminal restriction fragment length polymorphism and next-generation sequencing. BMC Gastroenterol. 2015, 15, 100. [Google Scholar] [CrossRef] [PubMed]
- Suez, J.; Korem, T.; Zeevi, D.; Zilberman-Schapira, G.; Thaiss, C.A.; Maza, O.; Israeli, D.; Zmora, N.; Gilad, S.; Weinberger, A.; et al. Artificial sweeteners induce glucose intolerance by altering the gut microbiota. Nature 2014, 514, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.K.; Chin, K.-Y.; Suhaimi, F.H.; Fairus, A.; Ima-Nirwana, S. Animal models of metabolic syndrome: A review. Nutr. Metab. 2016, 13, 65. [Google Scholar] [CrossRef]
- Connor, S.C.; Hansen, M.K.; Corner, A.; Smith, R.F.; Ryan, T.E. Integration of metabolomics and transcriptomics data to aid biomarker discovery in type 2 diabetes. Mol. Biosyst. 2010, 6, 909–921. [Google Scholar] [CrossRef] [PubMed]
- Koropatkin, N.M.; Cameron, E.A.; Martens, E.C. How glycan metabolism shapes the human gut microbiota. Nat. Rev. Microbiol. 2012, 10, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Bibbò, S.; Dore, M.P.; Pes, G.M.; Delitala, G.; Delitala, A.P. Is there a role for gut microbiota in type 1 diabetes pathogenesis? Ann Med. 2017, 49, 11–22. [Google Scholar] [CrossRef]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic Endotoxemia Initiates Obesity and Insulin Resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef] [PubMed]
- Tanti, J.-F.; Ceppo, F.; Jager, J.; Berthou, F. Implication of inflammatory signaling pathways in obesity-induced insulin resistance. Front. Endocrinol. 2013, 3, 181. [Google Scholar] [CrossRef] [PubMed]
- Cotillard, A.; Kennedy, S.P.; Kong, L.C.; Prifti, E.; Pons, N.; Le Chatelier, E.; Almeida, M.; Quinquis, B.; Levenez, F.; Galleron, N.; et al. Dietary intervention impact on gut microbial gene richness. Nature 2013, 500, 585–588. [Google Scholar] [CrossRef]
- Muegge, B.D.; Kuczynski, J.; Knights, D.; Clemente, J.C.; Gonzalez, A.; Fontana, L.; Henrissat, B.; Knight, R.; Gordon, J.I. Diet drives convergence in gut microbiome functions across mammalian phylogeny and within humans. Science 2011, 332, 970–974. [Google Scholar] [CrossRef] [PubMed]
- Zinöcker, M.; Lindseth, I. The Western diet–microbiome-host interaction and its role in metabolic disease. Nutrients 2018, 10, 365. [Google Scholar] [CrossRef]
- Samuel, B.S.; Shaito, A.; Motoike, T.; Rey, F.E.; Backhed, F.; Manchester, J.K.; Hammer, R.E.; Williams, S.C.; Crowley, J.; Yanagisawa, M.; et al. Effects of the gut microbiota on host adiposity are modulated by the short-chain fatty-acid binding G protein-coupled receptor, Gpr41. Proc. Natl. Acad. Sci. USA 2008, 105, 16767–16772. [Google Scholar] [CrossRef]
- Sonnenburg, J.L.; Bäckhed, F. Diet–microbiota interactions as moderators of human metabolism. Nature 2016, 535, 56–64. [Google Scholar] [CrossRef]
- Brown, A.J.; Goldsworthy, S.M.; Barnes, A.A.; Eilert, M.M.; Tcheang, L.; Daniels, D.; Muir, A.I.; Wigglesworth, M.J.; Kinghorn, I.; Fraser, N.J.; et al. The orphan G protein-coupled receptors GPR41 and GPR43 are activated by propionate and other short chain carboxylic acids. J. Biol. Chem. 2003, 278, 11312–11319. [Google Scholar] [CrossRef] [PubMed]
- Le Poul, E.; Loison, C.; Struyf, S.; Springael, J.Y.; Lannoy, V.; Decobecq, M.E.; Brezillon, S.; Dupriez, V.; Vassart, G.; Van Damme, J.; et al. Functional characterization of human receptors for short chain fatty acids and their role in polymorphonuclear cell activation. J. Biol. Chem. 2003, 278, 25481–25489. [Google Scholar] [CrossRef] [PubMed]
- Roberts, C.K.; Hevener, A.I.; Barnard, R.J. Metabolic Syndrome and Insulin Resistance: Underlying Causes and Modification by Exercise Training. Compr. Physiol. 2013, 3, 1–58. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Weiner, H.L. Control of the gut microbiome by fecal microRNA. Microb. Cell 2016, 3, 176–177. [Google Scholar] [CrossRef] [PubMed]
- Musso, G.; Gambino, R.; Cassader, M. Interactions between gut microbiota and host metabolism predisposing to obesity and diabetes. Annu. Rev. Med. 2011, 62, 361–380. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Miyamoto, N.; Shibata, K.; Valasek, M.A.; Motoike, T.; Kedzierski, R.M.; Yanagisawa, M. Short-chain fatty acids stimulate leptin production in adipocytes through the G protein-coupled receptor GPR41. Proc. Natl. Acad. Sci. USA. 2004, 101, 1045–1050. [Google Scholar] [CrossRef] [PubMed]
- Kimura, I.; Ozawa, K.; Inoue, D.; Imamura, T.; Kimura, K.; Maeda, T.; Terasawa, K.; Kashihara, D.; Hirano, K.; Tani, T.; et al. The gut microbiota suppresses insulin-mediated fat accumulation via the short-chain fatty acid receptor GPR43. Nat. Commun. 2013, 4, 1829. [Google Scholar] [CrossRef] [PubMed]
- Haxho, F.; Haq, S.; Szewczuk, M.R. Biased G protein-coupled receptor agonism mediates Neu1 sialidase and matrix metalloproteinase-9 crosstalk to induce transactivation of insulin receptor signaling. Cell. Signal. 2018, 43, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Liauchonak, I.; Dawoud, F.; Riat, Y.; Qorri, B.; Sambi, M.; Jain, J.; Kalaydina, R.-V.; Mendonza, N.; Bajwa, K.; Szewczuk, R.M. The Biased G-Protein-Coupled Receptor Agonism Bridges the Gap between the Insulin Receptor and the Metabolic Syndrome. Int. J. Mol. Sci. 2018, 19, 575. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Staszewski, L.; Xu, H.; Durick, K.; Zoller, M.; Adler, E. Human receptors for sweet and umami taste. Proc. Natl. Acad. Sci. USA 2002, 99, 4692–4696. [Google Scholar] [CrossRef] [PubMed]
- Thal, D.M.; Glukhova, A.; Sexton, P.M.; Christopoulos, A. Structural insights into G-protein-coupled receptor allostery. Nature 2018, 559, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Correll, C.C.; McKittrick, B.A. Biased Ligand Modulation of Seven Transmembrane Receptors (7TMRs): Functional Implications for Drug Discovery. J. Med. Chem. 2014, 57, 6887–6896. [Google Scholar] [CrossRef]
- Luttrell, L.M.; Maudsley, S.; Bohn, L.M. Fulfilling the Promise of “Biased” G Protein–Coupled Receptor Agonism. Mol. Pharmacol. 2015, 88, 579–588. [Google Scholar] [CrossRef] [PubMed]
- Lane, J.R.; May, L.T.; Parton, R.G.; Sexton, P.M.; Christopoulos, A. A kinetic view of GPCR allostery and biased agonism. Nat. Chem. Biol. 2017, 13, 929–937. [Google Scholar] [CrossRef] [PubMed]
- Edelstein, S.J.; Changeux, J.-P. Biased Allostery. Biophys. J. 2016, 111, 902–908. [Google Scholar] [CrossRef] [PubMed]
- Khoury, E.; Clément, S.; Laporte, S.A. Allosteric and biased G protein-coupled receptor signaling regulation: Potentials for new therapeutics. Front. Endocrinol. 2014, 5, 68. [Google Scholar] [CrossRef] [PubMed]
- Onfroy, L.; Galandrin, S.; Pontier, S.M.; Seguelas, M.-H.; N’Guyen, D.; Sénard, J.-M.; Galés, C. G protein stoichiometry dictates biased agonism through distinct receptor-G protein partitioning. Sci. Rep. 2017, 7, 7885. [Google Scholar] [CrossRef] [PubMed]
- Galandrin, S.; Bouvier, M. Distinct Signaling Profiles of β1and β2 Adrenergic Receptor Ligands toward Adenylyl Cyclase and Mitogen-Activated Protein Kinase Reveals the Pluridimensionality of Efficacy. Mol. Pharmacol. 2006, 70, 1575–1584. [Google Scholar] [CrossRef] [PubMed]
- Kala, R.; Peek, G.W.; Hardy, T.M.; Tollefsbol, T.O. MicroRNAs: An emerging science in cancer epigenetics. J. Clin. Bioinform. 2013, 3, 6. [Google Scholar] [CrossRef] [PubMed]
- Armand-Labit, V.; Pradines, A. Circulating cell-free microRNAs as clinical cancer biomarkers. Biomol. Concepts 2017, 8, 61–81. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Kolfschoten, I.G.M.; Roggli, E.; Nesca, V.; Regazzi, R. Role and therapeutic potential of microRNAs in diabetes. Diabetes Obes. Metab. 2009, 11, 118–129. [Google Scholar] [CrossRef] [PubMed]
- Zacharewicz, E.; Lamon, S.; Russell, A.P. MicroRNAs in skeletal muscle and their regulation with exercise, ageing, and disease. Front. Physiol. 2013, 4, 266. [Google Scholar] [CrossRef] [PubMed]
- Miao, C.; Zhang, G.; Xie, Z.; Chang, J. MicroRNAs in the pathogenesis of type 2 diabetes: New research progress and future direction. Can. J. Physiol. Pharmacol. 2017, 96, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Latreille, M.; Hausser, J.; Stützer, I.; Zhang, Q.; Hastoy, B.; Gargani, S.; Kerr-Conte, J.; Pattou, F.; Zavolan, M.; Esguerra, J.L.S.; et al. MicroRNA-7a regulates pancreatic β cell function. J. Clin. Investig. 2014, 124, 2722–2735. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, C.; Nakatsuka, A.; Eguchi, J.; Teshigawara, S.; Kanzaki, M.; Katayama, A.; Yamaguchi, S.; Takahashi, N.; Murakami, K.; Ogawa, D.; et al. Identification of Circulating miR-101, miR-375 and miR-802 as Biomarkers for Type 2 Diabetes. Metabolism 2015, 64, 489–497. [Google Scholar] [CrossRef]
- Kilic, I.D.; Dodurga, Y.; Uludag, B.; Alihanoglu, Y.I.; Yildiz, B.S.; Enli, Y.; Secme, M.; Bostancı, H.E. microRNA-143 and -223 in obesity. Gene 2015, 560, 140–142. [Google Scholar] [CrossRef]
- Liu, S.; da Cunha, A.P.; Rezende, R.M.; Cialic, R.; Wei, Z.; Bry, L.; Comstock, L.E.; Gandhi, R.; Weiner, H.L. The Host Shapes the Gut Microbiota via Fecal MicroRNA. Cell Host Microbe 2016, 19, 32–43. [Google Scholar] [CrossRef]
- Valdes, A.M.; Walter, J.; Segal, E.; Spector, T.D. Role of the gut microbiota in nutrition and health. BMJ 2018, 361, k2179. [Google Scholar] [CrossRef]
- Rowland, I.; Gibson, G.; Heinken, A.; Scott, K.; Swann, J.; Thiele, I.; Tuohy, K. Gut microbiota functions: Metabolism of nutrients and other food components. J. Eur. J. Nutr. 2018, 57, 1–24. [Google Scholar] [CrossRef]
- Bhatia, P.; Raina, S.; Chugh, J.; Sharma, S. miRNAs: Early prognostic biomarkers for Type 2 diabetes mellitus? Biomark. Med. 2015, 9, 1025–1040. [Google Scholar] [CrossRef] [PubMed]
- Guay, C.; Regazzi, R. Circulating microRNAs as novel biomarkers for diabetes mellitus. Nat. Rev. Endocrinol. 2013, 9, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Mirra, P.; Raciti, G.A.; Nigro, C.; Fiory, F.; D’Esposito, V.; Formisano, P.; Beguinot, F.; Miele, C. Circulating miRNAs as intercellular messengers, potential biomarkers and therapeutic targets for Type 2 diabetes. Epigenomics 2015, 7, 653–667. [Google Scholar] [CrossRef] [PubMed]
- Jansen, F.; Yang, X.; Hoelscher, M.; Cattelan, A.; Schmitz, T.; Proebsting, S.; Wenzel, D.; Vosen, S.; Franklin, B.S.; Fleischmann, B.K.; et al. Endothelial Microparticle–Mediated Transfer of MicroRNA-126 Promotes Vascular Endothelial Cell Repair via SPRED1 and Is Abrogated in Glucose-Damaged Endothelial Microparticles. Circulation 2013, 128, 2026–2038. [Google Scholar] [CrossRef] [PubMed]
- Aller, E.E.J.G.; Abete, I.; Astrup, A.; Martinez, J.A.; van Baak, M.A. Starches, sugars and obesity. Nutrients 2011, 3, 341–369. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q. Gain weight by “going diet?” Artificial sweeteners and the neurobiology of sugar cravings: Neuroscience 2010. Yale J. Biol. Med. 2010, 83, 101–108. [Google Scholar] [PubMed]
- Ritze, Y.; Bárdos, G.; D’Haese, J.G.; Ernst, B.; Thurnheer, M.; Schultes, B.; Bischoff, S.C. Effect of high sugar intake on glucose transporter and weight regulating hormones in mice and humans. PLoS ONE 2014, 9, e101702. [Google Scholar] [CrossRef]
- Renwick, A.G.; Sims, J.; Snodin, D.J. Sucralose metabolism and pharmacokinetics in man. Food Chem. Toxicol. 2000, 38, 31–41. [Google Scholar] [CrossRef]
- Pepino, M.Y.; Bourne, C. Nonnutritive sweeteners, energy balance and glucose homeostasis. Curr. Opin. Clin. Nutr. Metab. Care 2011, 14, 391–395. [Google Scholar] [CrossRef]
- Moran, A.W.; Al-Rammahi, M.; Zhang, C.; Bravo, D.; Calsamiglia, S.; Shirazi-Beechey, S.P. Sweet taste receptor expression in ruminant intestine and its activation by artificial sweeteners to regulate glucose absorption. J. Dairy Sci. 2014, 97, 4955–4972. [Google Scholar] [CrossRef]
- Booth, D. Conditioned satiety in the rat. J. Comp. Physiol. Psychol. 1972, 81, 457–471. [Google Scholar] [CrossRef] [PubMed]
- Treesukosol, Y.; Moran, T.H. Cross-Generalization Profile to Orosensory Stimuli of Rats Conditioned to Avoid a High Fat/High Sugar Diet. Chem. Senses 2018, 43, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Swithers, S.E.; Martin, A.A.; Davidson, T.L. High-intensity sweeteners and energy balance. Physiol. Behav. 2010, 100, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Green, E.; Murphy, C. Altered processing of sweet taste in the brain of diet soda drinkers. Physiol. Behav. 2012, 107, 560–567. [Google Scholar] [CrossRef]
- Davidson, T.L.; Sample, C.H.; Swithers, S.E. An application of Pavlovian principles to the problems of obesity and cognitive decline. Neurobiol. Learn. Mem. 2014, 108, 172–184. [Google Scholar] [CrossRef] [PubMed]
- Swithers, S.E.; Laboy, A.F.; Clark, K.; Cooper, S.; Davidson, T.L. Experience with the high-intensity sweetener saccharin impairs glucose homeostasis and GLP-1 release in rats. Behav. Brain Res. 2012, 233, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Teff, K.L.; Engelman, K. Oral sensory stimulation improves glucose tolerance in humans: Effects on insulin, C-peptide, and glucagon. Am. J. Physiol. Regul. Integr. Comp. Physiol. 1996, 270, R1371–R1379. [Google Scholar] [CrossRef]
- Welcome, M.O. Emerging role of the neuronal sweet taste receptor heterodimer, T1R2+T1R3, in cognitive functioning. J. Res. Med. Dent. Sci. 2018, 6, 264–272. [Google Scholar]
- Zhou, L.; Huang, W.; Xu, Y.; Gao, C.; Zhang, T.; Guo, M.; Liu, Y.; Ding, J.; Qin, L.; Xu, Z.; et al. Sweet Taste Receptors Mediated ROS-NLRP3 Inflammasome Signaling Activation: Implications for Diabetic Nephropathy. J. Diabetes Res. 2018, 2018, 7078214. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Name | Brand Names | Applications in Food Industry | Relative Sweetness (Measured to Sucrose) |
|---|---|---|---|
| High-intensity Sweeteners | |||
| Saccharin | Sweet and Low®, Sweet Twin®, Sweet’N Low®, Necta Sweet® | Beverages, bases, and mixes for many food products, table sugar substitute | 200–700× |
| Aspartame * | Nutrasweet®, Equal®, Sugar Twin® | Soft drinks, chewing gum, pudding, cereals, instant coffee Also distributed as a “General Purpose Sweetener” | 200× |
| Acesulfame-potassium (Ace-K) | Sunett®, Sweet One® | Beverages, candy, frozen desserts, baked goods Heat stable so it can be used in baking | 200× |
| Sucralose | Splenda® | 600× | |
| Neotame | Newtame® | Beverages, candy gum | 7000–13,000× |
| Advantame | N/A | Baked goods, beverages, frozen desserts, frosting, chewing gum, candy, pudding, jelly and jam, gelatin | 20,000× |
| Sugar Alcohols | |||
| Erythritol | Fondant, ice cream, gum, tabletop sweeteners, chocolate, dairy products, jelly, beverages | 0.60×–0.70× | |
| Glycerol | Dairy products, processed fruits, energy bars, jam, fondant Often used as a thickening agent and to provide texture to food | ||
| Mannitol | Infant formula, frozen fish, precooked pasta, butter, chocolate flavored coatings | 0.50×–0.70× | |
| Sorbitol (Glucitol) * | Used as emulsifier | 0.66× | |
| Xylitol | Hard candy, chewing gum, mints, ice cream, chocolate, cookies, beverages, table sugar substitute | 1× | |
| Natural Sweeteners | |||
| Steviol glycosides | Natural constituents of leaves of Stevia rebaudiana (Bertoni) plant, commonly known as Stevia | Beverages, chewing gum, candy | 200–400× |
| Luo Han Guo Monk fruit extracts | Siraitia grosvenorii Swingle fruit extract (SGFE) | Tea | 100–250× |
| Lucuma powder | Beverages, pudding, granola, pastry, baked goods | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liauchonak, I.; Qorri, B.; Dawoud, F.; Riat, Y.; Szewczuk, M.R. Non-Nutritive Sweeteners and Their Implications on the Development of Metabolic Syndrome. Nutrients 2019, 11, 644. https://doi.org/10.3390/nu11030644
Liauchonak I, Qorri B, Dawoud F, Riat Y, Szewczuk MR. Non-Nutritive Sweeteners and Their Implications on the Development of Metabolic Syndrome. Nutrients. 2019; 11(3):644. https://doi.org/10.3390/nu11030644
Chicago/Turabian StyleLiauchonak, Iryna, Bessi Qorri, Fady Dawoud, Yatin Riat, and Myron R. Szewczuk. 2019. "Non-Nutritive Sweeteners and Their Implications on the Development of Metabolic Syndrome" Nutrients 11, no. 3: 644. https://doi.org/10.3390/nu11030644
APA StyleLiauchonak, I., Qorri, B., Dawoud, F., Riat, Y., & Szewczuk, M. R. (2019). Non-Nutritive Sweeteners and Their Implications on the Development of Metabolic Syndrome. Nutrients, 11(3), 644. https://doi.org/10.3390/nu11030644