Mother or Father: Who Is in the Front Line? Mechanisms Underlying the Non-Genomic Transmission of Obesity/Diabetes via the Maternal or the Paternal Line



{kind=link}
{kind=link}
{kind=link}
Abstract
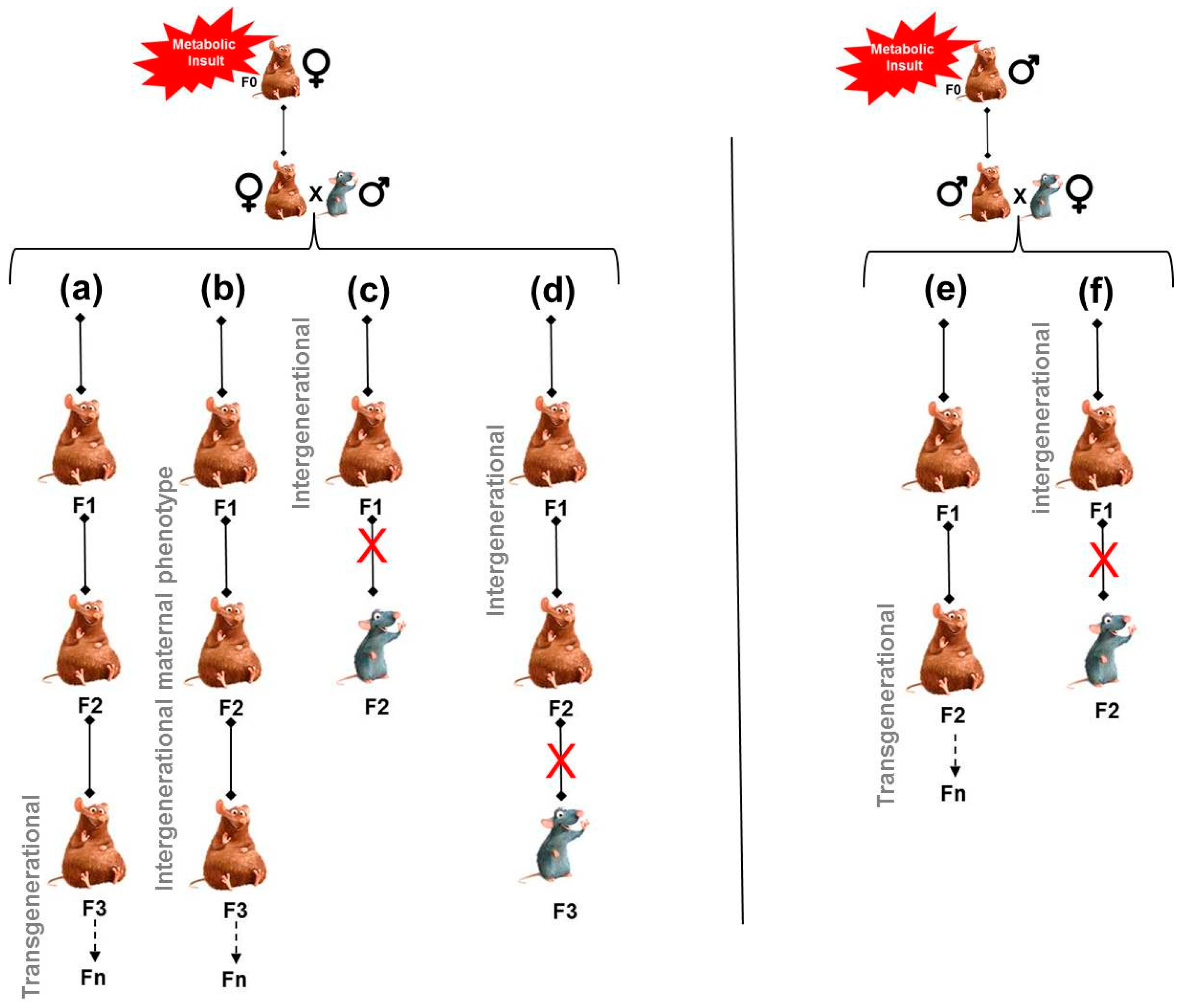
1. Obesity or Diabetes Risk Can Be Transmitted across Generations via the Maternal Line. Epidemiological and Experimental Evidence
2. Critical Windows of Exposure to Maternal Obesity/Diabetes Shape Metabolic Risk in the Offspring
3. Maternal Obesity or Diabetes Remodels Offspring Epigenome
4. How to Explain Risk Inheritance via the Maternal Line?
4.1. Persistence of Maternal Exposure to Adverse Environmental Conditions along Generations
4.2. Persistence of an Altered Maternal Phenotype along Generations
4.3. Involvement of Non-Genomic (Epigenomic) Modifications Induced by the Maternal Phenotype and Transmitted via the Oocyte
5. Obesity or Diabetes Risk Can Be Transmitted across Generations via the Paternal Line. Epidemiological and Experimental Evidence
6. Paternal Obesity or Diabetes Remodels Sperm Epigenome
6.1. Remodeling of DNA Methylation Profile
6.2. Remodeling of Chromatin
6.3. Remodeling of the Small Non-Coding RNA Expression Pattern
7. Mechanisms for the Transmission of Risk via the Paternal Line
7.1. Transferred by Sperm. Proof of Concept
7.2. Sperm-Borne RNAs Are Candidates That Could Transfer the Risk of Obesity or Diabetes. Proof of Concept
7.3. Transfer of Paternal Metabolic Traits by Sperm tRNAs
7.4. Transfer of Paternal Metabolic Traits by Sperm miRNAs
8. What Happens If Both Parents Are Obese or Diabetic?
9. Conclusions and Questions
Funding
Acknowledgments
Conflicts of Interest
References
- Voight, B.F.; Scott, L.J.; Steinthorsdottir, V.; Morris, A.P.; Dina, C.; Welch, R.P.; Zeggini, E.; Huth, C.; Aulchenko, Y.S.; Thorleifsson, G.; et al. Twelve type 2 diabetes susceptibility loci identified through large-scale association analysis. Nat. Genet. 2010, 42, 579–589. [Google Scholar] [CrossRef] [PubMed]
- Ling, C.; Groop, L. Epigenetics: A molecular link between environmental factors and type 2 diabetes. Diabetes 2009, 58, 2718–2725. [Google Scholar] [CrossRef] [PubMed]
- Groop, L.; Pociot, F. Genetics of diabetes—Are we missing the genes or the disease? Mol. Cell. Endocrinol. 2014, 382, 726–739. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Twinn, D.S.; Constância, M.; Ozanne, S.E. Intergenerational epigenetic inheritance in models of developmental programming of adult disease. Semin. Cell Dev. Biol. 2015, 43, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Rando, O.J.; Simmons, R.A. I’m eating for two: Parental dietary effects on offspring metabolism. Cell 2015, 161, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Drake, A.J.; Liu, L. Intergenerational transmission of programmed effects: Public health consequences. Trends Endocrinol. Metab. 2010, 21, 206–213. [Google Scholar] [CrossRef]
- Sales, V.M.; Ferguson-Smith, A.C.; Patti, M.E. Epigenetic mechanisms of transmission of metabolic disease across generations. Cell Metab. 2017, 25, 559–571. [Google Scholar] [CrossRef]
- Kahn, S.E.; Hull, R.L.; Utzschneide, KM. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature 2006, 444, 840–846. [Google Scholar] [CrossRef]
- Svensson, H.; Wetterling, L.; Bosaeus, M.; Oden, B.; Oden, A.; Jennische, E.; Edén, S.; Holmäng, A.; Lönn, M. Body fat mass and the proportion of very large adipocytes in pregnant women are associated with gestational insulin resistance. Int. J. Obes. 2016, 40, 646–653. [Google Scholar] [CrossRef]
- Micali, N.; Al Essimii, H.; Field, A.E.; Treasure, J. Pregnancy loss of control over eating: A longitudinal study of maternal and child outcomes. Am. J. Clin. Nutr. 2018, 108, 101–107. [Google Scholar] [CrossRef]
- Murrin, C.M.; Kelly, G.E.; Tremblay, R.E.; Kelleher, C.C. Body mass index and height over three generations: Evidence from the Lifeways cross-generational cohort study. BMC Public Health. 2012, 12, 81. [Google Scholar] [CrossRef] [PubMed]
- Sharp, G.C.; Salas, L.A.; Monnereau, C.; Allard, C.; Yousefi, P.; Everson, T.M.; Bohlin, J.; Xu, Z.; Huang, R.C.; Reese, S.E.; et al. Maternal BMI at the start of pregnancy and offspring epigenome-wide DNA methylation: Findings from the pregnancy and childhood epigenetics (PACE) consortium. Hum. Mol. Genet. 2017, 26, 4067–4085. [Google Scholar] [CrossRef] [PubMed]
- Guenard, F.; Deshaies, Y.; Cianflone, K.; Kral, J.G.; Marceau, P.; Vohl, M.C. Differential methylation in glucoregulatory genes of offspring born before vs. after maternal gastrointestinal bypass surgery. Proc. Natl. Acad. Sci. USA 2013, 110, 11439–11444. [Google Scholar] [CrossRef] [PubMed]
- Lumey, L.H.; Stein, A.D.; Kahn, H.S.; van der Pal-de Bruin, K.M.; Blauw, G.J.; Zybert, P.A.; Susser, E.S. Cohort profile: The Dutch Hunger Winter families study. Int. J. Epidemiol. 2007, 36, 1196–1204. [Google Scholar] [CrossRef] [PubMed]
- Ravelli, A.C.; van der Meulen, J.H.; Michels, R.P.; Osmond, C.; Barker, D.J.; Hales, C.N.; Bleker, O.P. Glucose tolerance in adults after prenatal exposure to famine. Lancet 1998, 351, 173–177. [Google Scholar] [CrossRef]
- Lumey, L.H.; Khalangot, M.D.; Vaiserman, A.M. Association between type 2 diabetes and prenatal exposure to the Ukraine famine of 1932-33: A retrospective cohort study. Lancet Diabetes Endocrinol. 2015, 3, 787–794. [Google Scholar] [CrossRef]
- Thurner, S.; Klimek, P.; Szell, M.; Duftschmid, G.; Endel, G.; Kautzky-Willer, A.; Kasper, D.C. Quantification of excess risk for diabetes for those born in times of hunger, in an entire population of a nation, across a century. Proc. Natl. Acad. Sci. USA 2013, 110, 4703–4707. [Google Scholar] [CrossRef]
- Li, Y.; He, Y.; Qi, L.; Jaddoe, V.W.; Feskens, E.J.; Yang, X.; Ma, G.; Hu, F.B. Exposure to the Chinese famine in early life and the risk of hyperglycemia and type 2 diabetes in adulthood. Diabetes 2010, 59, 2400–2406. [Google Scholar] [CrossRef]
- Hult, M.; Tornhammar, P.; Ueda, P.; Chima, C.; Bonamy, A.K.; Ozumba, B.; Norman, M. Hypertension, diabetes and overweight: Looming legacies of the Biafran famine. PLoS ONE 2010, 5, e13582. [Google Scholar] [CrossRef]
- Nicholas, L.M.; Morrison, L.; Rattanatray, L.; Zhang, S.; Ozanne, S.E.; McMillen, I.C. The early origins of obesity and insulin resistance: Timing, programming and mechanisms. Internat. J. Obesity 2016, 40, 229–238. [Google Scholar] [CrossRef]
- Martin, A.O.; Simpson, J.L.; Ober, C.; Freinkel, N. Frequency of diabetes mellitus in probands with gestational diabetes: Possible maternal influence on the predisposition to gestational diabetes. Am. J. Obstet. Gynecol. 1985, 151, 471–475. [Google Scholar] [CrossRef]
- Alcolado, J.C.; Laji, K.; Gill-Randall, R. Maternal transmission of diabetes. Diabet. Med. 2002, 19, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Dabelea, D.; Hanson, R.L.; Lindsay, R.S.; Pettitt, D.J.; Imperatore, G.; Gabir, M.M.; Roumain, J.; Bennett, P.H.; Knowler, W.C. Intrauterine exposure to diabetes conveys risks for type 2 diabetes and obesity: A study of discordant sibships. Diabetes 2000, 49, 2208–2211. [Google Scholar] [CrossRef] [PubMed]
- Sobngwi, E.; Boudou, P.; Mauvais-Jarvis, F.; Leblanc, H.; Velho, G.; Vexiau, P.; Porcher, R.; Hadjadj, S.; Pratley, R.; Tataranni, P.A.; Calvo, F.; Gautier, J.F. Effect of a diabetic environment in utero on predisposition to type 2 diabetes. Lancet 2003, 361, 1861–1865. [Google Scholar] [CrossRef]
- Gautier, J.F.; Wilson, C.; Weyer, C.; Mott, D.; Knowler, W.C.; Cavaghan, M.; Polonsky, K.S.; Bogardus, C.; Pratley, R.E. Low acute insulin secretory responses in adult offspring of people with early onset type 2 diabetes. Diabetes 2001, 50, 1828–1833. [Google Scholar] [CrossRef] [PubMed]
- DeFronzo, R.A. Banting Lecture. From the triumvirate to the ominousoctet: A new paradigm for the treatment of type 2 diabetes mellitus. Diabetes 2009, 58, 773–795. [Google Scholar] [CrossRef] [PubMed]
- Hales, C.N.; Barker, D.J.; Clark, P.M.; Cox, L.J.; Fall, C.; Osmond, C.; Winter, P.D. Fetal and infant growth and impaired glucose tolerance at age 64. BMJ. 1991, 303, 1019–1022. [Google Scholar] [CrossRef]
- Hales, C.N.; Barker, D.J. Type 2 (non-insulin-dependent) diabetes mellitus: The thrifty phenotype hypothesis. Diabetologia 1992, 35, 595–601. [Google Scholar] [CrossRef]
- Economides, D.L.; Nicolaides, K.H.; Gahl, W.A.; Bernardini, I.; Evans, MI. Plasma insulin in appropriate and small for gestational age fetuses. Am. J. Obstet. Gynecol. 1989, 160, 1091–1094. [Google Scholar] [CrossRef]
- Brufani, C.; Grossi, A.; Fintini, D.; Tozzi, A.; Nocerino, V.; Patera, P.I.; Ubertini, G.; Porzio, O.; Barbetti, F.; Cappa, M. Obese children with low birth weight demonstrate impaired beta-cell function during oral glucose tolerance test. J. Clin. Endocrinol. Metab. 2009, 94, 4448–4452. [Google Scholar] [CrossRef]
- Van Assche, F.A.; De Prins, F.; Aerts, L.; Verjans, M. The endocrine pancreas in small-for dates infants. Br. J. Obstet. Gynaecol. 1977, 84, 751–753. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, P.; Morriseau, T.S.; Kereliuk, S.M.; Doucette, C.A.; Wicklow, B.A.; Dolinsky, V.W. Maternal obesity, diabetes during pregnancy and epigenetic mechanisms that influence the developmental origins of cardiometabolic disease in the offspring. Crit. Rev. Clin. Lab. Sci. 2018, 55, 71–101. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.A.; Bale, T.L. Maternal high-fat diet effects on third-generation female body size via the paternal lineage. Endocrinology 2011, 152, 2228–2236. [Google Scholar] [CrossRef] [PubMed]
- Gniuli, D.; Calcagno, A.; Caristo, M.E.; Mancuso, A.; Macchi, V.; Mingrone, G.; Vettor, R. Effects of high-fat diet exposure during fetal life on type 2 diabetes development in the progeny. J. Lipid Res. 2008, 49, 1936–1945. [Google Scholar] [CrossRef] [PubMed]
- Cerf, M.E.; Williams, K.; Nkomo, X.I.; Muller, C.J.; Du Toit, D.F.; Louw, J.; Wolfe-Coote, S.A. Islet cell response in the neonatal rat after exposure to a high-fat diet during pregnancy. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 288, R1122–R1128. [Google Scholar] [CrossRef] [PubMed]
- Nivoit, P.; Morens, C.; Van Assche, F.A.; Jansen, E.; Poston, L.; Remacle, C.; Reusens, B. Established diet induced obesity in female rats leads to offspring hyperphagia, adiposity and insulin resistance. Diabetologia 2009, 52, 1133–1142. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.D.; McConnell, J.; Khan, I.Y.; Holemans, K.; Lawrence, K.M.; Asare-Anane, H.; Persaud, S.J.; Jones, P.M.; Petrie, L.; Hanson, M.A.; et al. Impaired glucose homeostasis and mitochondrial abnormalities in offspring of rats fed a fat-rich diet in pregnancy. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 288, R134–R139. [Google Scholar] [CrossRef]
- Rooney, K.; Ozanne, S.E. Maternal over-nutrition and offspring obesity predisposition: Targets for preventative interventions. Internat. J. Obesity 2011, 35, 883–890. [Google Scholar] [CrossRef]
- Isganaitis, E.; Woo, M.; Ma, H.; Chen, M.; Kong, W.; Lytras, A.; Sales, V.; Decoste-Lopez, J.; Lee, K.J.; Leatherwood, C.; et al. Developmental programming by maternal insulin resistance: Hyperinsulinemia, glucose intolerance, and dysregulated lipid metabolism in male offspring of insulin-resistant mice. Diabetes 2014, 63, 688–700. [Google Scholar] [CrossRef]
- Jimenez-Chillaron, J.C.; Hernandez-Valencia, M.; Lightner, A.; Faucette, R.R.; Reamer, C.; Przybyla, R.; Ruest, S.; Barry, K.; Otis, J.P.; Patti, M.E. Reductions in caloric intake and early postnatal growth prevent glucose intolerance and obesity associated with low birthweight. Diabetologia 2006, 49, 1974–1984. [Google Scholar] [CrossRef]
- Jimenez-Chillaron, J.C.; Hernandez-Valencia, M.; Reamer, C.; Fisher, S.; Joszi, A.; Hirshman, M.; Oge, A.; Walrond, S.; Przybyla, R.; Boozer, C.; et al. Beta-cell secretory dysfunction in the pathogenesis of low birth weight-associated diabetes: A murine model. Diabetes 2005, 54, 702–711. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Chillaron, J.C.; Isganaitis, E.; Charalambous, M.; Gesta, S.; Pentinat-Pelegrin, T.; Faucette, R.R.; Otis, J.P.; Chow, A.; Diaz, R.; Ferguson-Smith, A.; et al. Intergenerational transmission of glucose intolerance and obesity by in utero undernutrition in mice. Diabetes 2009, 58, 460–468. [Google Scholar] [CrossRef] [PubMed]
- Hanafi, M.Y.; Saleh, M.M.; Saad, M.I.; Abdelkhalek, T.M.; Kamel, M.A. Transgenerational effects of obesity and malnourishment on diabetes risk in F2 generation. Mol. Cell. Biochem. 2016, 412, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Hardikar, A.A.; Satoor, S.N.; Karandikar, M.S.; Joglekar, M.V.; Puranik, A.S.; Wong, W.; Kumar, S.; Limaye, A.; Bhat, D.S.; Januszewski, A.S.; et al. Multigenerational undernutrition increases susceptibility to obesity and diabetes that is not reversed after dietary recuperation. Cell Metab. 2015, 22, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Verhaeghe, J.; Peeters, T.L.; Vandeputte, M.M.; Rombauts, W.; Bouillon, R.; VanAssche, F.A. Maternal and foetal endocrine pancreas in the spontaneously diabetic BB rat. Biol. Neonate 1989, 55, 298–308. [Google Scholar] [CrossRef]
- Serradas, P.; Gangnerau, M.N.; Giroix, M.H.; Saulnier, C.; Portha, B. Impaired pancreatic beta-cell function in the foetal GK rat. Impact of diabetic inheritance. J. Clin. Invest. 1998, 101, 899–904. [Google Scholar] [CrossRef] [PubMed]
- Aerts, A.; Van Assche, F.A. Animal evidence for the transgenerational development of diabetes mellitus. Int. J. Biochem. Cell Biol. 2006, 38, 894–903. [Google Scholar] [CrossRef]
- Han, J.; Xu, J.; Long, Y.S.; Epstein, P.N.; Liu, Q.Y. Rat maternal diabetes impairs pancreatic beta-cell function in the offspring. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E228–E236. [Google Scholar] [CrossRef]
- Grill, V.; Johansson, B.; Jalkanen, P.; Eriksson, U.J. Influence of severe diabetes mellitus early in pregnancy in the rat: Effects on insulin sensitivity and insulin secretion in the offspring. Diabetologia 1991, 34, 373–378. [Google Scholar] [CrossRef]
- Ktorza, A.; Gauguier, D.; Bihoreau, M.T.; Berthault, M.F.; Picon, L. Adult off-spring from mildly hyperglycaemic rats show impairment of glucoseregulation and insulin secretion which is transmissible to the next gener-ation. In Frontiers in Diabetic Research. Lessons from Animal Diabetes; Shafrir, E., Ed.; Smith-Gordon and Company Ltd.: London, UK, 1990; pp. 555–560. [Google Scholar]
- Chavey, A.; Ah Kioon, M.D.; Bailbé, D.; Movassat, J.; Portha, B. Maternal diabetes, programming of beta-cell disorders and intergenerational risk of type 2 diabetes. Diabetes Metab. 2014, 40, 323–330. [Google Scholar] [CrossRef]
- Chavey, A.; Bailbé, D.; Maulny, L.; Renard, J.P.; Movassat, J.; Portha, B. A euglycaemic/non-diabetic perinatal environment does not alleviate early beta cell maldevelopment and type 2 diabetes risk in the GK/Par rat model. Diabetologia 2013, 56, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Portha, B.; Giroix, M.H.; Tourrel-Cuzin, C.; Le Stunff, H.; Movassat, J. The GK rat: A prototype for the study of non-overweight type 2 diabetes. Methods Mol. Biol. 2012, 933, 125–159. [Google Scholar] [PubMed]
- Goto, Y.; Kakizaki, M.; Masaki, N. Spontaneous diabetes produced by selective breeding of normal Wistar rats. Proc. Jpn. Acad. 1975, 51, 80–85. [Google Scholar] [CrossRef]
- Srinivasan, M.; Patel, M.S. Metabolic programming in the immediate postnatal period. Trends Endocrinol. Metab. 2008, 19, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Jungheim, E.S.; Moley, K.H. Current knowledge of obesity’s effects in the pre- and periconceptional periods and avenues for future research. Am. J. Obstet. Gynecol. 2010, 203, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Sasson, I.E.; Vitins, A.P.; Mainigi, M.A.; Moley, K.H.; Simmons, R.A. Pre-gestational vs. gestational exposure to maternal obesity differentially programs the offspring in mice. Diabetologia 2015, 58, 615–624. [Google Scholar] [CrossRef] [PubMed]
- Kaelin, W.G.; McKnight, S.L. Influence of metabolism on epigenetics and disease. Cell 2013, 153, 56–69. [Google Scholar] [CrossRef]
- Heijmans, B.T.; Tobi, E.W.; Stein, A.D.; Putter, H.; Blauw, G.J.; Susser, E.S.; Slagboom, P.E.; Lumey, L.H. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc. Natl. Acad. Sci. USA 2008, 105, 17046–17049. [Google Scholar] [CrossRef]
- Tobi, E.W.; Lumey, L.H.; Talens, R.P.; Kremer, D.; Putter, H.; Stein, A.D.; Slagboom, P.E.; Heijmans, B.T. DNA methylation differences after exposure prenatal famine are common and timing—and sex-specific. Hum. Mol. Genet. 2009, 18, 4046–4053. [Google Scholar] [CrossRef]
- Petropoulos, S.; Guillemin, C.; Ergaz, Z.; Dimov, S.; Suderman, M.; Weinstein-Fudim, L.; Ornoy, A.; Szyf, M. Gestational diabetes alters DNA methylation profiles in human and rat: Identification of key pathways involved in endocrine system disorders, insulin signaling, diabetes signaling, and ILK signaling. Endocrinology 2015, 156, 2222–2238. [Google Scholar] [CrossRef]
- Carreras-Badosa, G.; Bonmatí, A.; Ortega, F.J.; Mercader, J.M.; Guindo-Martínez, M.; Torrents, D.; Prats-Puig, A.; Martinez-Calcerrada, J.M.; Platero-Gutierrez, E.; De Zegher, F.; et al. Altered circulating miRNA expression profile in pregestational and gestational obesity. J. Clin. Endocrinol. Metab. 2015, 100, E1446–E1456. [Google Scholar] [CrossRef] [PubMed]
- Fowden, A.L.; Forhead, A.J. Endocrine mechanisms of intrauterine programming. Reproduction 2004, 127, 515–526. [Google Scholar] [CrossRef] [PubMed]
- McMillen, I.C.; Robinson, J.S. Developmental origins of the metabolic syndrome: Prediction, plasticity, and programming. Physiol. Rev. 2005, 85, 571–633. [Google Scholar] [CrossRef] [PubMed]
- Bansal, A.; Simmons, R.A. Epigenetics and developmental origins of diabetes: Correlation or causation? Am. J. Physiol. Endocrinol. Metab. 2018, 315, E15–E28. [Google Scholar] [CrossRef] [PubMed]
- Simmons, R.A.; Templeton, L.J.; Gertz, S.J. Intrauterine growth retardation leads to the development of type 2 diabetes in the rat. Diabetes 2001, 50, 2279–2286. [Google Scholar] [CrossRef]
- Park, J.H.; Stoffers, D.A.; Nicholls, R.D.; Simmons, RA. Development of type 2 diabetes following intrauterine growth retardation in rats is associated with progressive epigenetic silencing of Pdx1. J. Clin. Invest. 2008, 118, 2316–2324. [Google Scholar] [CrossRef]
- Thompson, R.F.; Fazzari, M.J.; Niu, H.; Barzilai, N.; Simmons, R.A.; Greally, J.M. Experimental intrauterine growth restriction induces alterations in DNA methylation and gene expression in pancreatic islets of rats. J. Biol. Chem. 2010, 285, 15111–15118. [Google Scholar] [CrossRef]
- Bernardo, A.S.; Hay, C.W.; Docherty, K. Pancreatic transcription factors and their role in the birth, life and survival of the pancreatic beta cell. Mol. Cell. Endocrinol. 2008, 294, 1–9. [Google Scholar] [CrossRef]
- Yang, B.T.; Dayeh, T.A.; Volkov, P.A.; Kirkpatrick, C.L.; Malmgren, S.; Jing, X.; Renström, E.; Wollheim, C.B.; Nitert, M.D.; Ling, C. Increased DNA methylation and decreased expression of PDX-1 in pancreatic islets from patients with type 2 diabetes. Mol. Endocrinol. 2012, 26, 1203–1212. [Google Scholar] [CrossRef]
- MacLennan, N.K.; James, S.J.; Melnyk, S.; Piroozi, A.; Jernigan, S.; Hsu, J.L.; Janke, S.M.; Pham, T.D.; Lane, R.H. Uteroplacental insufficiency alters DNA methylation, one-carbon metabolism, and histone acetylation in IUGR rats. Physiol. Genomics 2004, 18, 43–50. [Google Scholar] [CrossRef]
- Fu, Q.; McKnight, R.A.; Yu, X.; Wang, L.; Callaway, C.W.; Lane, R.H. Uteroplacental insufficiency induces site-specific changes in histone H3 covalent modifications and affects DNA-histone H3 positioning in day 0 IUGR rat liver. Physiol. Genomics 2004, 20, 108–116. [Google Scholar] [CrossRef]
- Lane, R.H.; MacLennan, N.K.; Hsu, J.L.; Janke, S.M.; Pham, T.D. Increased hepatic peroxisome proliferator-activated receptor coactivator-1 gene expression in a rat model of intrauterine growth retardation and subsequent insulin resistance. Endocrinology 2002, 143, 2486–2490. [Google Scholar] [CrossRef]
- Pinney, S.E.; Jaeckle Santos, L.J.; Han, Y.; Stoffers, D.A.; Simmons, R.A. Exendin-4 increases histone acetylase activity and reverses epigenetic modifications that silence Pdx1 in the intrauterine growth retarded rat. Diabetologia 2011, 54, 2606–2614. [Google Scholar] [CrossRef] [PubMed]
- Benyshek, D.C.; Johnston, C.S.; Martin, J.F. Glucose metabolism is altered in the adequately-nourished grand-offspring (F3 generation) of rats malnourished during gestation and perinatal life. Diabetologia 2006, 49, 1117–1119. [Google Scholar] [CrossRef]
- Zambrano, E.; Martínez-Samayoa, P.M.; Bautista, C.J.; Deás, M.; Guillén, L.; Rodríguez-González, G.L.; Guzmán, C.; Larrea, F.; Nathanielsz, P.W. Sex differences in transgenerational alterations of growth and metabolism in progeny (F2) of female offspring (F1) of rats fed a low protein diet during pregnancy and lactation. J. Physiol. 2005, 566, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Drake, A.J.; Walker, B.R.; Seckl, J.R. Intergenerational consequences of foetal programming by in utero exposure to glucocorticoids in rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 288, R34–R38. [Google Scholar] [CrossRef] [PubMed]
- Gluckman, P.D.; Hanson, M.A.; Beedle, A.S. Non-genomic transgenerational inheritance of disease risk. BioEssays 2007, 29, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Ozanne, S.E.; Constancia, M. Mechanisms of disease: The developmental origins of disease and the role of the epigenotype. Nat. Clin. Pract. Endocrinol. Metab. 2007, 3, 539–546. [Google Scholar] [CrossRef] [PubMed]
- Pinney, S.E.; Simmons, R.A. Epigenetic mechanisms in the development of type 2 diabetes. Trends Endocrinol. Metab. 2009, 21, 223–229. [Google Scholar] [CrossRef]
- Waterland, R.A.; Michels, K.B. Epigenetic epidemiology of the developmental origins hypothesis. Annu. Rev. Nutr. 2007, 27, 363–388. [Google Scholar] [CrossRef] [PubMed]
- Benyshek, D.C. The “early life” origins of obesity-related health disorders: New discoveries regarding the intergenerational transmission of developmentally programmed traits in the global cardiometabolic health crisis. Am. J. Phys. Anthropol. 2013, 152 (Suppl. 57), 79–93. [Google Scholar] [CrossRef] [PubMed]
- Burdge, G.C.; Slater-Jefferies, J.; Torrens, C.; Phillips, E.S.; Hanson, M.A.; Lillycrop, K.A. Dietary protein restriction of pregnant rats in the F0 generation induces altered methylation of hepatic gene promoters in the adult male offspring in the F1 and F2 generations. Br. J. Nutr. 2007, 97, 435–439. [Google Scholar] [CrossRef] [PubMed]
- Skinner, M.K. What is an epigenetic transgenerational phenotype? F3 or F2. Reprod. Toxicol. 2008, 1, 2–6. [Google Scholar] [CrossRef] [PubMed]
- Hoile, S.P.; Lillycrop, K.A.; Thomas, N.A.; Hanson, M.A.; Burdge, G.C. Dietary protein restriction during F0 pregnancy in rats induces transgenerational changes in the hepatic transcriptome in female offspring. PLoS ONE 2011, 6, e21668. [Google Scholar] [CrossRef] [PubMed]
- Huypens, P.; Sass, S.; Wu, M.; Dyckhoff, D.; Tschöp, M.; Theis, F.; Marschall, S.; Hrabě de Angelis, M.; Beckers, J. Epigenetic germline inheritance of diet-induced obesity and insulin resistance. Nat. Genet. 2016, 48, 497–499. [Google Scholar] [CrossRef] [PubMed]
- Lane, M.; Zander-Fox, D.L.; Robker, R.L.; McPherson, N.O. Peri-conception parental obesity, reproductive health, and transgenerational impacts. Trends Endocrinol. Metab. 2015, 26, 84–90. [Google Scholar] [CrossRef]
- Minge, C.E.; Bennett, B.D.; Norman, R.J.; Robker, R.L. Peroxisome proliferator-activated receptor-gamma agonist rosiglitazone reverses the adverse effects of diet-induced obesity on oocyte quality. Endocrinology 2008, 149, 2646–2656. [Google Scholar] [CrossRef]
- Wyman, A.; Pinto, A.B.; Sheridan, R.; Moley, K.H. One-cell zygote transfer from diabetic to non diabetic mouse results in congenital malformations and growth retardation in offspring. Endocrinology 2008, 149, 466–469. [Google Scholar] [CrossRef]
- Igosheva, N.; Abramov, A.Y.; Poston, L.; Eckert, J.J.; Fleming, T.P.; Duchen, M.R.; McConnell, J. Maternal diet-induced obesity alters mitochondrial activity and redox status in mouse oocytes and zygotes. PLoS ONE 2010, 5, e10074. [Google Scholar] [CrossRef]
- Luzzo, K.M.; Wang, Q.; Purcell, S.H.; Chi, M.; Jimenez, P.T.; Grindler, N.; Schedl, T.; Moley, K.H. High fat diet induced developmental defects in the mouse: Oocyte meiotic aneuploidy and fetal growth retardation/brain defects. PLoS ONE 2012, 7, e49217. [Google Scholar] [CrossRef]
- Wu, L.L.; Dunning, K.R.; Yang, X.; Russell, D.L.; Lane, M.; Norman, R.J.; Robker, R.L. High-fat diet causes lipotoxicity responses in cumulus-oocyte complexes and decreased fertilization rates. Endocrinology 2010, 151, 5438–5445. [Google Scholar] [CrossRef] [PubMed]
- Bermejo-Alvarez, P.; Rosenfeld, C.S.; Roberts, R.M. Effect of maternal obesity on estrous cyclicity, embryo development and blastocyst gene expression in a mouse model. Hum Reprod. 2012, 27, 3513–3522. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.L.; Russell, D.L.; Wong, S.L.; Chen, M.; Tsai, T.S.; St John, J.C.; Norman, R.J.; Febbraio, M.A.; Carroll, J.; Robker, R.L. Mitochondrial dysfunction in oocytes of obese mothers: Transmission to offspring and reversal by pharmacological endoplasmic reticulum stress inhibitors. Development 2015, 142, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.J.; Zhu, C.C.; Duan, X.; Liu, H.L.; Wang, Q.; Sun, S.C. Both diet and gene mutation induced obesity affect oocyte quality in mice. Sci. Rep. 2016, 6, 18858. [Google Scholar] [CrossRef] [PubMed]
- Branco, M.R.; King, M.; Perez-Garcia, V.; Bogutz, A.B.; Caley, M.; Fineberg, E.; Lefebvre, L.; Cook, S.J.; Dean, W.; Hemberger, M.; et al. Maternal DNA methylation regulates early trophoblast development. Dev. Cell. 2016, 36, 152–163. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Yan, M.; Cao, Z.; Li, X.; Zhang, Y.; Shi, J.; Feng, G.H.; Peng, H.; Zhang, X.; Zhang, Y.; et al. Sperm tsRNAs contribute to intergenerational inheritance of an acquired metabolic disorder. Science 2016, 351, 397–400. [Google Scholar] [CrossRef] [PubMed]
- Carone, B.R.; Fauquier, L.; Habib, N.; Shea, J.M.; Hart, C.E.; Li, R.; Bock, C.; Li, C.; Gu, H.; Zamore, P.D.; et al. Paternally induced transgenerational environmental reprogramming of metabolic gene expression in mammals. Cell 2010, 143, 1084–1096. [Google Scholar] [CrossRef] [PubMed]
- Grandjean, V.; Fourré, S.; De Abreu, D.A.; Derieppe, M.A.; Remy, J.J.; Rassoulzadegan, M. RNA-mediated paternal heredity of diet-induced obesity and metabolic disorders. Sci. Rep. 2015, 5, 18193. [Google Scholar] [CrossRef]
- Chen, L.; Magliano, D.J.; Zimmet, P.Z. The worldwide epidemiology of type 2 diabetes mellitus-present and future perspectives. Nat. Rev. Endocrinol. 2012, 8, 228–236. [Google Scholar] [CrossRef]
- Soubry, A. POHaD: Why we should study future fathers. Environ. Epigenetics 2018, 4. [Google Scholar] [CrossRef]
- Painter, R.C.; Osmond, C.; Gluckman, P.; Hanson, M.; Phillips, D.I.; Roseboom, T.J. Transgenerational effects of prenatal exposure to the Dutch famine on neonatal adiposity and health in later life. BJOG 2008, 115, 1243–1249. [Google Scholar] [CrossRef] [PubMed]
- Veenendaal, M.V.; Painter, R.C.; de Rooij, S.R.; Bossuyt, P.M.; van der Post, J.A.; Gluckman, P.D.; Hanson, M.A.; Roseboom, T.J. Transgenerational effects of prenatal exposure to the 1944-45 Dutch famine. BJOG 2013, 120, 548–553. [Google Scholar] [CrossRef] [PubMed]
- Kaati, G.; Bygren, L.O.; Pembrey, M.; Sjostrom, M. Transgenerational response to nutrition, early life circumstances and longevity. Eur. J. Hum. Genet. 2007, 15, 784–790. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.F.; Lin, R.C.; Laybutt, D.R.; Barres, R.; Owens, J.A.; Morris, M.J. Chronic high-fat diet in fathers programs β-cell dysfunction in female rat offspring. Nature 2010, 467, 963–966. [Google Scholar] [CrossRef] [PubMed]
- Fullston, T.; Ohlsson Teague, E.M.; Palmer, N.O.; De Blasio, M.J.; Mitchell, M.; Corbett, M.; Print, C.G.; Owens, J.A.; Lane, M. Paternal obesity initiates metabolic disturbances in two generations of mice with incomplete penetrance to the F2 generation and alters the transcriptional profile of testis and sperm microRNA content. FASEB J. 2013, 27, 4226–4243. [Google Scholar] [CrossRef] [PubMed]
- Linn, T.; Loewk, E.; Schneider, K.; Federlin, K. Spontaneous glucose intolerance in the progeny of low dose streptozotocin-induced diabetic mice. Diabetologia 1993, 36, 1245–1251. [Google Scholar] [CrossRef]
- Calderari, S.; Gangnerau, M.N.; Meile, M.J.; Portha, B.; Serradas, P. Is defective pancreatic beta-cell mass environmentally programmed in Goto-Kakizaki rat model of type 2 diabetes? Insights from crossbreeding studies during suckling period. Pancreas 2006, 33, 412–417. [Google Scholar] [CrossRef]
- Wei, Y.; Yang, C.R.; Wei, Y.P.; Zhao, Z.A.; Hou, Y.; Schatten, H.; Sun, Q.Y. Paternally induced transgenerational inheritance of susceptibility to diabetes in mammals. Proc. Natl. Acad. Sci. USA 2014, 111, 1873–1878. [Google Scholar] [CrossRef]
- de Castro Barbosa, T.; Ingerslev, L.R.; Alm, P.S.; Versteyhe, S.; Massart, J.; Rasmussen, M.; Donkin, I.; Sjögren, R.; Mudry, J.M.; Vetterli, L.; et al. High-fat diet reprograms the epigenome of rat spermatozoa and transgenerationally affects metabolism of the offspring. Mol. Metab. 2015, 5, 184–197. [Google Scholar] [CrossRef]
- Radford, E.J.; Ito, M.; Shi, H.; Corish, J.A.; Yamazawa, K.; Isganaitis, E.; Seisenberger, S.; Hore, T.A.; Reik, W.; Erkek, S.; et al. In utero effects. In utero undernourishment perturbs the adult sperm methylome and intergenerational metabolism. Science 2014, 345, 1255903. [Google Scholar] [CrossRef]
- Shea, J.M.; Serra, R.W.; Carone, B.R.; Shulha, H.P.; Kucukural, A.; Ziller, M.J.; Vallaster, M.P.; Gu, H.; Tapper, A.R.; Gardner, P.D.; et al. Genetic and epigenetic variation, but not diet, shape the sperm methylome. Dev. Cell 2015, 35, 750–758. [Google Scholar] [CrossRef] [PubMed]
- Donkin, I.; Versteyhe, S.; Ingerslev, L.R.; Qian, K.; Mechta, M.; Nordkap, L.; Mortensen, B.; Appel, E.V.; Jørgensen, N.; Kristiansen, V.B.; et al. Obesity and Bariatric Surgery Drive Epigenetic Variation of Spermatozoa in Humans. Cell Metab. 2016, 23, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Bao, J.; Bedford, M.T. Epigenetic regulation of the histone-to-protamine transition during spermiogenesis. Reproduction 2016, 151, R55–R70. [Google Scholar] [CrossRef] [PubMed]
- Terashima, M.; Barbour, S.; Ren, J.; Yu, W.; Han, Y.; Muegge, K. Effect of high fat diet on paternal sperm histone distribution and male offspring liver gene expression. Epigenetics 2015, 10, 861–871. [Google Scholar] [CrossRef] [PubMed]
- Palmer, N.O.; Fullston, T.; Mitchell, M.; Setchell, B.P.; Lane, M. SIRT6 in mouse spermatogenesis is modulated by diet-induced obesity. Reprod. Fertil. Dev. 2011, 23, 929–939. [Google Scholar] [CrossRef] [PubMed]
- Öst, A.; Lempradl, A.; Casas, E.; Weigert, M.; Tiko, T.; Deniz, M.; Pantano, L.; Boenisch, U.; Itskov, P.M.; Stoeckius, M.; et al. Paternal diet defines offspring chromatin state and intergenerational obesity. Cell 2014, 159, 1352–1364. [Google Scholar] [CrossRef]
- Holoch, D.; Moazed, D. RNA-mediated epigenetic regulation of gene expression. Nat. Rev. Genet. 2015, 16, 71–84. [Google Scholar] [CrossRef]
- Grandjean, V.; Gounon, P.; Wagner, N.; Martin, L.; Wagner, K.D.; Bernex, F.; Cuzin, F.; Rassoulzadegan, M. The miR-124-Sox9 paramutation: RNA-mediated epigenetic control of embryonic and adult growth. Development 2009, 136, 3647–3655. [Google Scholar] [CrossRef]
- Rassoulzadegan, M.; Grandjean, V.; Gounon, P.; Vincent, S.; Gillot, I.; Cuzin, F. RNA-mediated non-mendelian inheritance of an epigenetic change in the mouse. Nature 2006, 441, 469–474. [Google Scholar] [CrossRef]
- Yuan, S.; Oliver, D.; Schuster, A.; Zheng, H.; Yan, W. Breeding scheme and maternal small RNAs affect the efficiency of transgenerational inheritance of a paramutation in mice. Sci. Rep. 2015, 5, 9266. [Google Scholar] [CrossRef]
- Rodgers, A.B.; Morgan, C.P.; Leu, N.A.; Bale, T.L. Transgenerational epigenetic programming via sperm microRNA recapitulates effects of paternal stress. Proc. Natl. Acad. Sci. USA 2015, 112, 13699–13704. [Google Scholar] [CrossRef] [PubMed]
- Gapp, K.; Jawaid, A.; Sarkies, P.; Bohacek, J.; Pelczar, P.; Prados, J.; Farinelli, L.; Miska, E.; Mansuy, I.M. Implication of sperm RNAs in transgenerational inheritance of the effects of early trauma in mice. Nat. Neurosci. 2014, 17, 667–669. [Google Scholar] [CrossRef] [PubMed]
- Krawetz, S.A.; Kruger, A.; Lalancette, C.; Tagett, R.; Anton, E.; Draghici, S.; Diamond, M.P. A survey of small RNAs in human sperm. Hum. Reprod. 2011, 26, 3401–3412. [Google Scholar] [CrossRef] [PubMed]
- Herrera, B.M.; Lockstone, H.E.; Taylor, J.M.; Ria, M.; Barrett, A.; Collins, S.; Kaisaki, P.; Argoud, K.; Fernandez, C.; Travers, M.E.; et al. Global microRNA expression profiles in insulin target tissues in a spontaneous rat model of type 2 diabetes. Diabetologia 2010, 53, 1099–1109. [Google Scholar] [CrossRef] [PubMed]
- Hulsmans, M.; De Keyzer, D.; Holvoet, P. MicroRNAs regulating oxidative stress and inflammation in relation to obesity and atherosclerosis. FASEB J. 2011, 25, 2515–2527. [Google Scholar] [CrossRef] [PubMed]
- Robertson, S.A. Seminal plasma and male factor signalling in the female reproductive tract. Cell Tissue Res. 2005, 322, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, R.M. Environmental/lifestyle effects on spermatogenesis. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2010, 365, 1697–1712. [Google Scholar] [CrossRef] [PubMed]
- Hammoud, A.O.; Gibson, M.; Stanford, J.; White, G.; Carrell, D.T.; Peterson, M. In vitro fertilization availability and utilization in the United States: A study of demographic, social, and economic factors. Fertil. Steril. 2009, 9, 1630–1635. [Google Scholar] [CrossRef]
- Kort, H.I.; Massey, J.B.; Elsner, C.W.; Mitchell-Leef, D.; Shapiro, D.B.; Witt, M.A.; Roudebush, W.E. Impact of body mass index values on sperm quantity and quality. J. Androl. 2006, 27, 450–452. [Google Scholar] [CrossRef]
- Chavarro, J.E.; Furtado, J.; Toth, T.L.; Ford, J.; Keller, M.; Campos, H.; Hauser, R. Trans-fatty acid levels in sperm are associated with sperm concentration among men from an infertility clinic. Fertil. Steril. 2011, 95, 1794–1797. [Google Scholar] [CrossRef]
- Ghanayem, B.I.; Bai, P.; Kissling, G.E.; Travlos, G.; Hoffler, U. Diet-induced obesity in male mice is associated with reduced fertility and potentiation of acrylamide-induced reproductive toxicity. Biol. Reprod. 2010, 82, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, M.; Bakos, H.W.; Lane, M. Paternal diet-induced obesity impairs embryo development and implantation in the mouse. Fertil. Steril. 2011, 95, 1349–1353. [Google Scholar] [CrossRef]
- Rato, L.; Alves, M.G.; Dias, T.R.; Lopes, G.; Cavaco, J.E.; Socorro, S.; Oliveira, P.F. High-energy diets may induce a pre-diabetic state altering testicular glycolytic metabolic profile and male reproductive parameters. Andrology 2013, 1, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Bakos, H.W.; Henshaw, R.C.; Mitchell, M.; Lane, M. Paternal body mass index is associated with decreased blastocyst development and reduced live birth rates following assisted reproductive technology. Fertil. Steril. 2011, 95, 1700–1704. [Google Scholar] [CrossRef]
- Lim, J.P.; Brune, A. Bridging the transgenerational gap with epigenetic memory. Trends in Genetics TIG 2013, 29, 176–186. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Shi, J.; Zhang, Y.; Zhang, H.; Liao, S.; Li, W.; Lei, L.; Han, C.; Ning, L.; Cao, Y.; et al. A novel class of tRNA-derived small RNAs extremely enriched in mature mouse sperm. Cell Res. 2012, 22, 1609–1612. [Google Scholar] [CrossRef] [PubMed]
- Sharma, U.; Conine, C.C.; Shea, J.M.; Boskovic, A.; Derr, A.G.; Bing, X.Y.; Belleannee, C.; Kucukural, A.; Serra, R.W.; Sun, F.; et al. Biogenesis and function of tRNA fragments during sperm maturation and fertilization in mammals. Science 2016, 351, 391–396. [Google Scholar] [CrossRef]
- Elbarbary, R.A.; Takaku, H.; Uchiumi, N.; Tamiya, H.; Abe, M.; Takahashi, M.; Nishida, H.; Nashimoto, M. Modulation of gene expression by human cytosolic tRNase Z(L) through 5’-half-tRNA. PLoS ONE 2009, 4, e5908. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Medvid, R.; Melton, C.; Jaenisch, R.; Blelloch, R. DGCR8 is essential for microRNA biogenesis and silencing of embryonic stem cell self-renewal. Nat. Genet. 2007, 39, 380–385. [Google Scholar] [CrossRef]
- Wagner, K.D.; Wagner, N.; Ghanbarian, H.; Grandjean, V.; Gounon, P.; Cuzin, F.; Rassoulzadegan, M. RNA induction and inheritance of epigenetic cardiac hypertrophy in the mouse. Dev. Cell 2008, 14, 962–969. [Google Scholar] [CrossRef]
- Kupka, M.S.; Gnoth, C.; Buehler, K.; Dahncke, W.; Kruessel, J.S. Impact of female and male obesity on IVF/ICSI: Results of 700,000 ART-cycles in Germany. Gynecol. Endocrinol. 2011, 27, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Schliep, K.C.; Mumford, S.L.; Ahrens, K.A.; Hotaling, J.M.; Carrell, D.T.; Link, M.; Hinkle, S.N.; Kissell, K.; Porucznik, C.A.; Hammoud, A.O. Effect of male and female body mass index on pregnancy and live birth success after in vitro fertilization. Fertil. Steril. 2014, 103, 388–395. [Google Scholar] [CrossRef] [PubMed]
- Ornellas, F.; Souza-Mello, V.; Mandarim-de-Lacerda, C.A.; Aguila, M.B. Programming of obesity and comorbidities in the progeny: Lessons from a model of diet-induced obese parents. PLoS ONE 2015, 10, e0124737. [Google Scholar] [CrossRef] [PubMed]
- McPherson, N.O.; Bell, V.G.; Zander-Fox, D.L.; Fullston, T.; Wu, L.L.; Robker, R.L.; Lane, M. When two obese parents are worse than one! Impacts on embryo and fetal development. Am. J. Physiol. Endocrinol. Metab. 2015, 309, E568–E581. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Portha, B.; Grandjean, V.; Movassat, J. Mother or Father: Who Is in the Front Line? Mechanisms Underlying the Non-Genomic Transmission of Obesity/Diabetes via the Maternal or the Paternal Line. Nutrients 2019, 11, 233. https://doi.org/10.3390/nu11020233
Portha B, Grandjean V, Movassat J. Mother or Father: Who Is in the Front Line? Mechanisms Underlying the Non-Genomic Transmission of Obesity/Diabetes via the Maternal or the Paternal Line. Nutrients. 2019; 11(2):233. https://doi.org/10.3390/nu11020233
Chicago/Turabian StylePortha, Bernard, Valérie Grandjean, and Jamileh Movassat. 2019. "Mother or Father: Who Is in the Front Line? Mechanisms Underlying the Non-Genomic Transmission of Obesity/Diabetes via the Maternal or the Paternal Line" Nutrients 11, no. 2: 233. https://doi.org/10.3390/nu11020233
APA StylePortha, B., Grandjean, V., & Movassat, J. (2019). Mother or Father: Who Is in the Front Line? Mechanisms Underlying the Non-Genomic Transmission of Obesity/Diabetes via the Maternal or the Paternal Line. Nutrients, 11(2), 233. https://doi.org/10.3390/nu11020233