Investigation of Antibacterial and Antiinflammatory Activities of Proanthocyanidins from Pelargonium sidoides DC Root Extract

 ,
,  , , , ,
, , , ,  , and
, and
Abstract
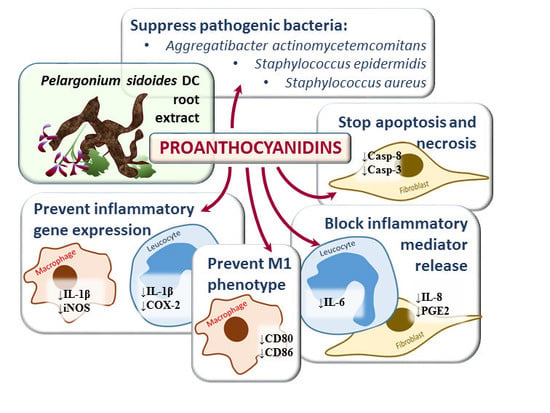
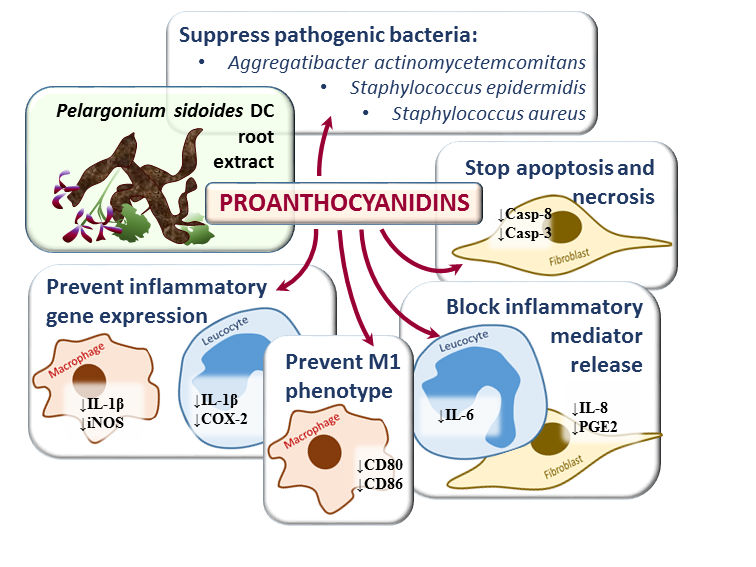
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Pelargonium sidoides Root Extract and Proanthocyanidin Fraction
2.2. Bacterial Strains and Growth Conditions
2.3. Antibacterial Efficiency Evaluation
2.4. Co-Cultures of Human Gingival Fibroblasts and Bacteria
2.5. Rat Gingival Fibroblast Cell Culture and Treatments
2.6. Bone Marrow-Derived Macrophages
2.7. Human Peripheral Blood Mononuclear Cells
2.8. Analysis of Cell Viability by Lactate Dehydrogenase Release, Alamarblue and MTT Assay
2.9. Necrosis Evaluation by Double Nuclear Staining
2.10. Apoptosis Evaluation by Annexin V staining
2.11. Caspase Activity Assessment
2.12. Detection of Secreted Inflammatory Mediators
2.13. Bone Marrow-Derived Macrophage Polarisation to M1 Phenotype and Analysis by Flow Cytometry
2.14. mRNA Isolation and Quantitative RT-PCR Analysis
2.15. Statistical Analysis
3. Results
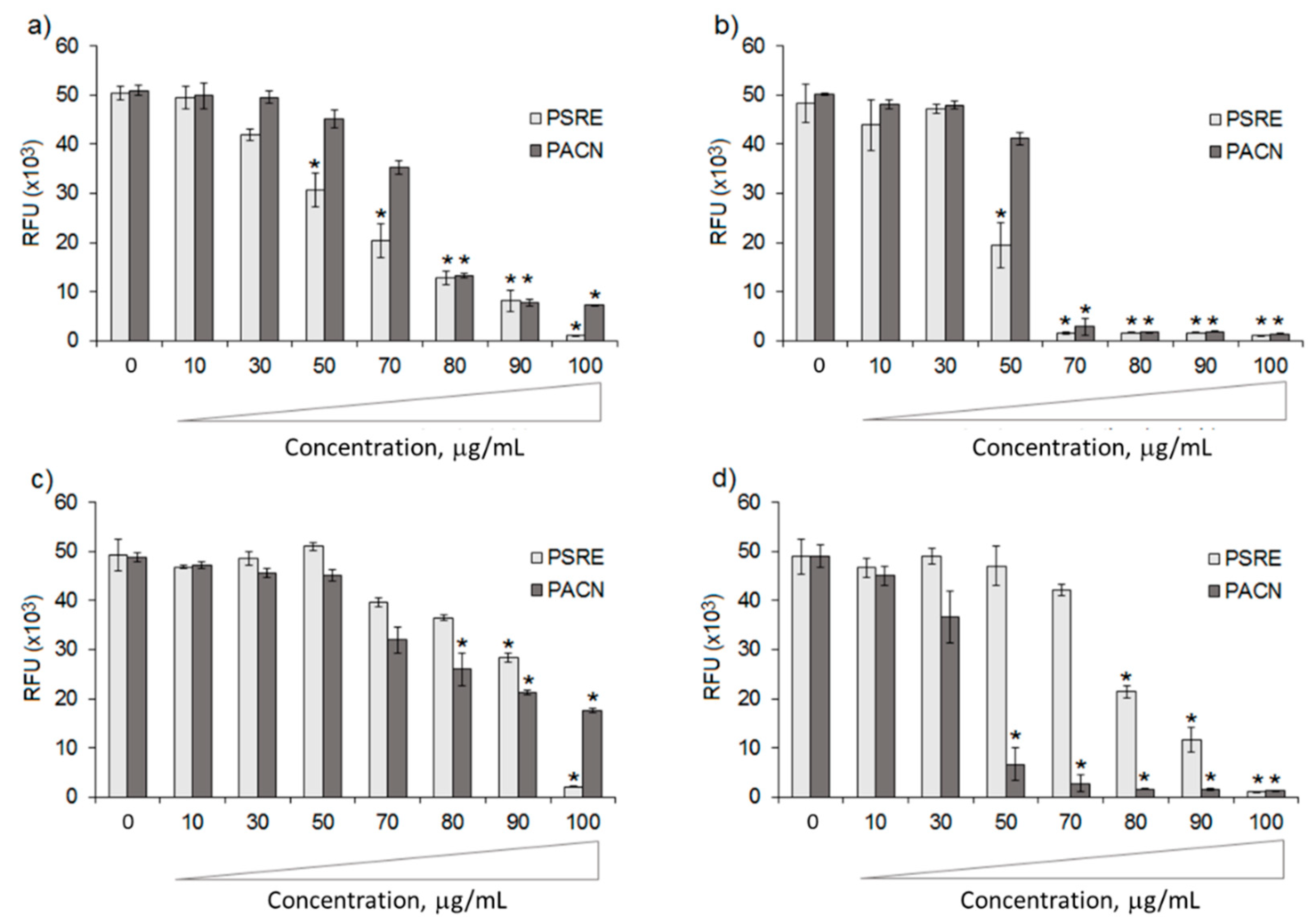
3.1. Antibacterial Activity of Proanthocyanidins from Pelargonium sidoides Root Extract
3.2. The Effect of Proanthocyanidins from Pelargonium sidoides Root Extract on Gingival Fibroblast Viability under Conditions of Bacterial Infection
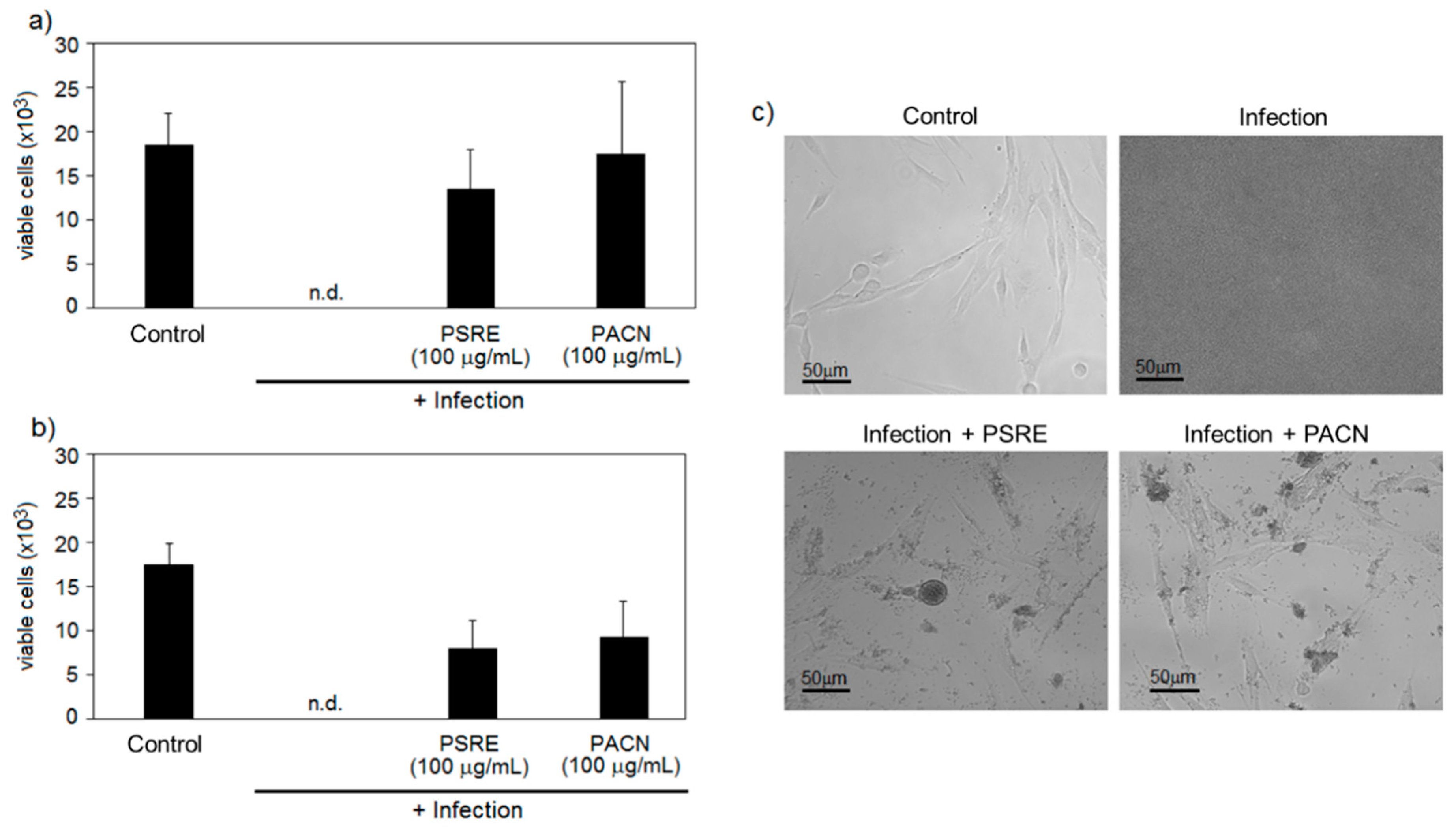
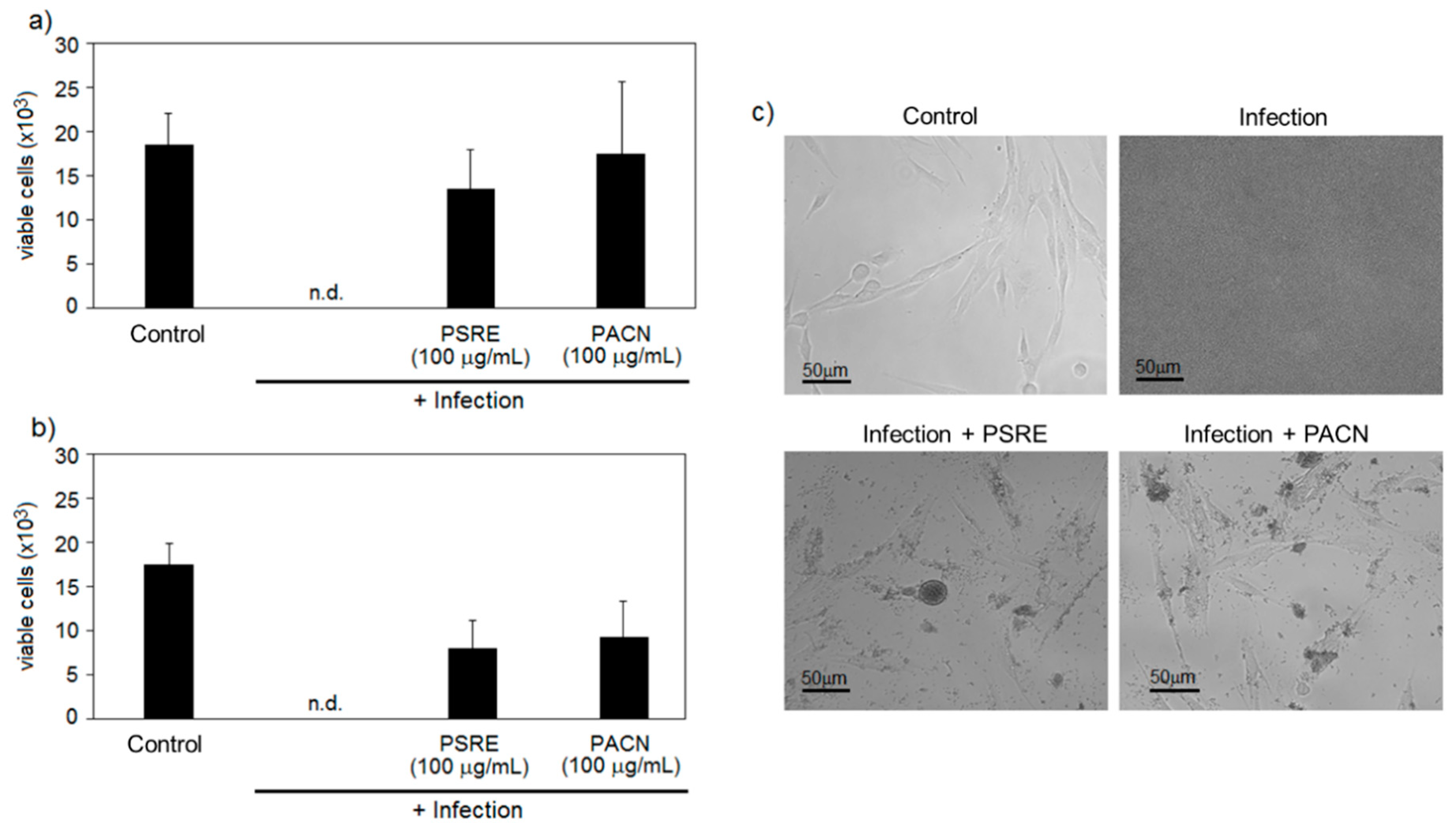
3.2.1. Pelargonium sidoides Root Extract and Proanthocyanidins Preserve Gingival Fibroblasts in the Presence of Bacteria
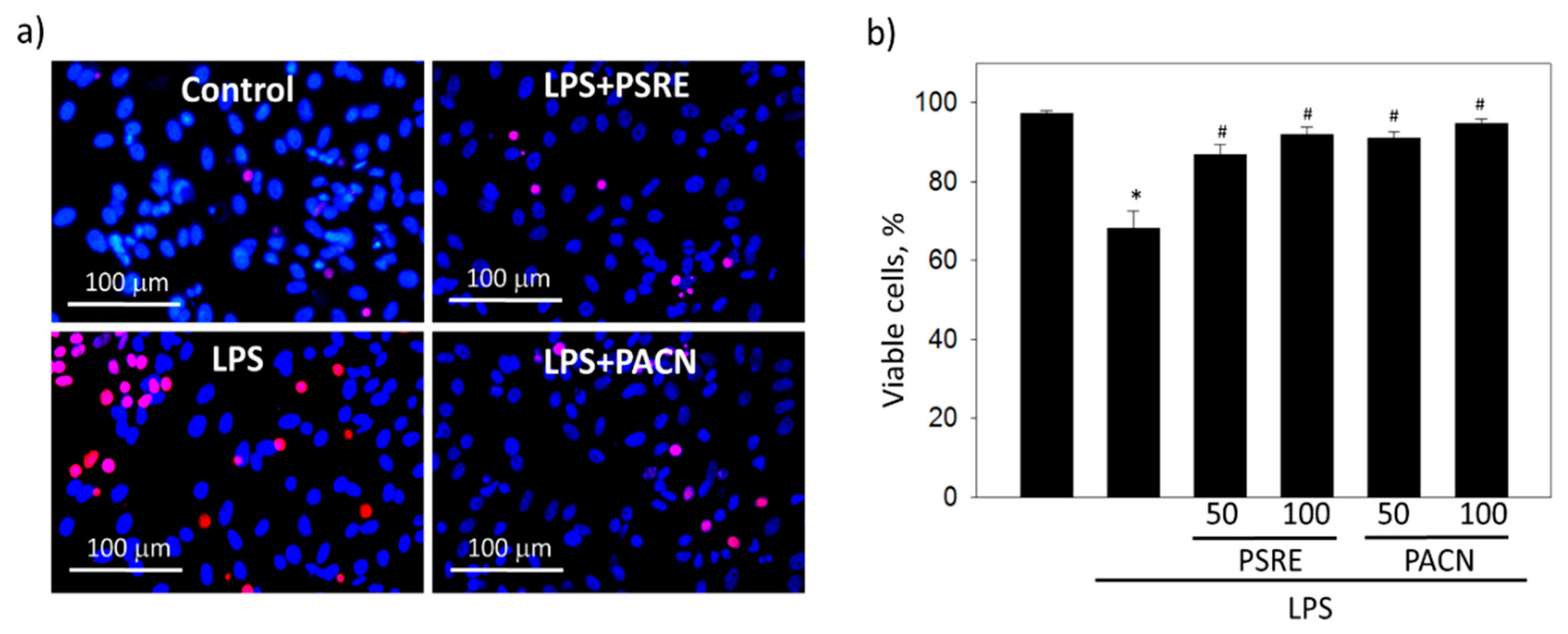
3.2.2. Pelargonium sidoides Root Extract and Proanthocyanidins Protect Gingival Fibroblasts from Necrotic Cell Death Induced by Bacterial Lipopolysaccharide
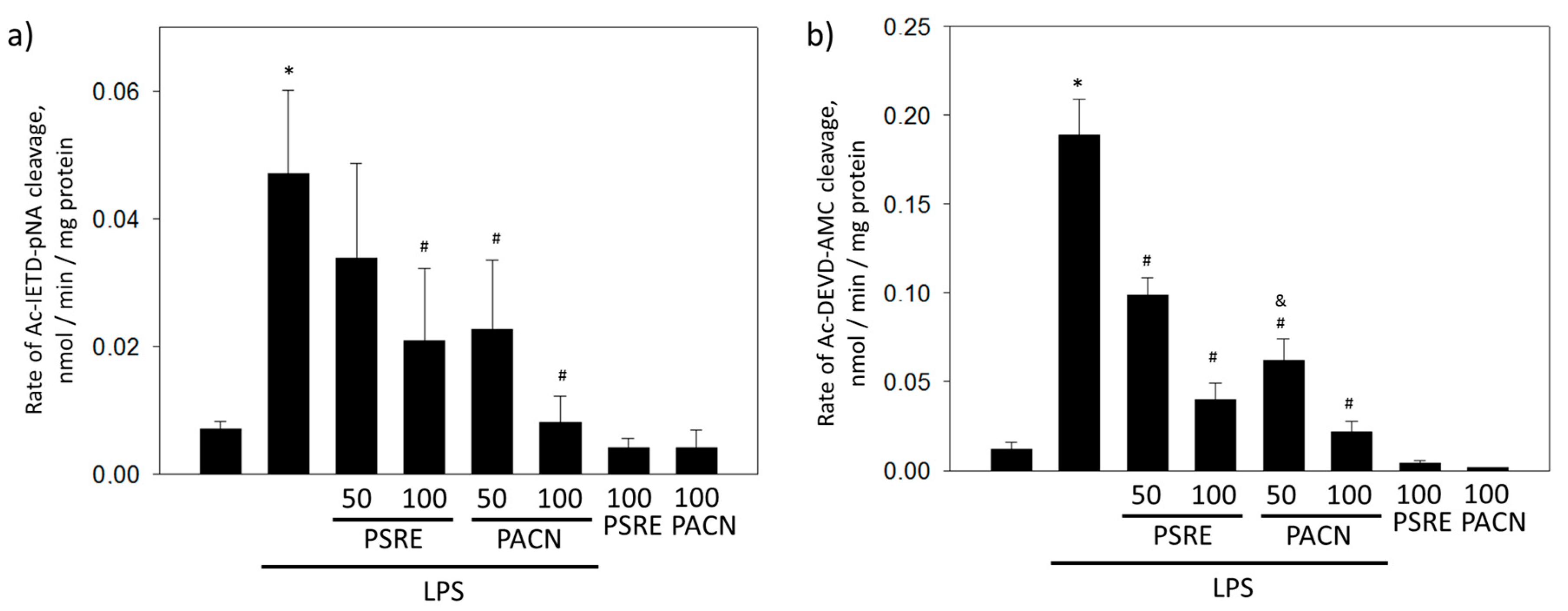
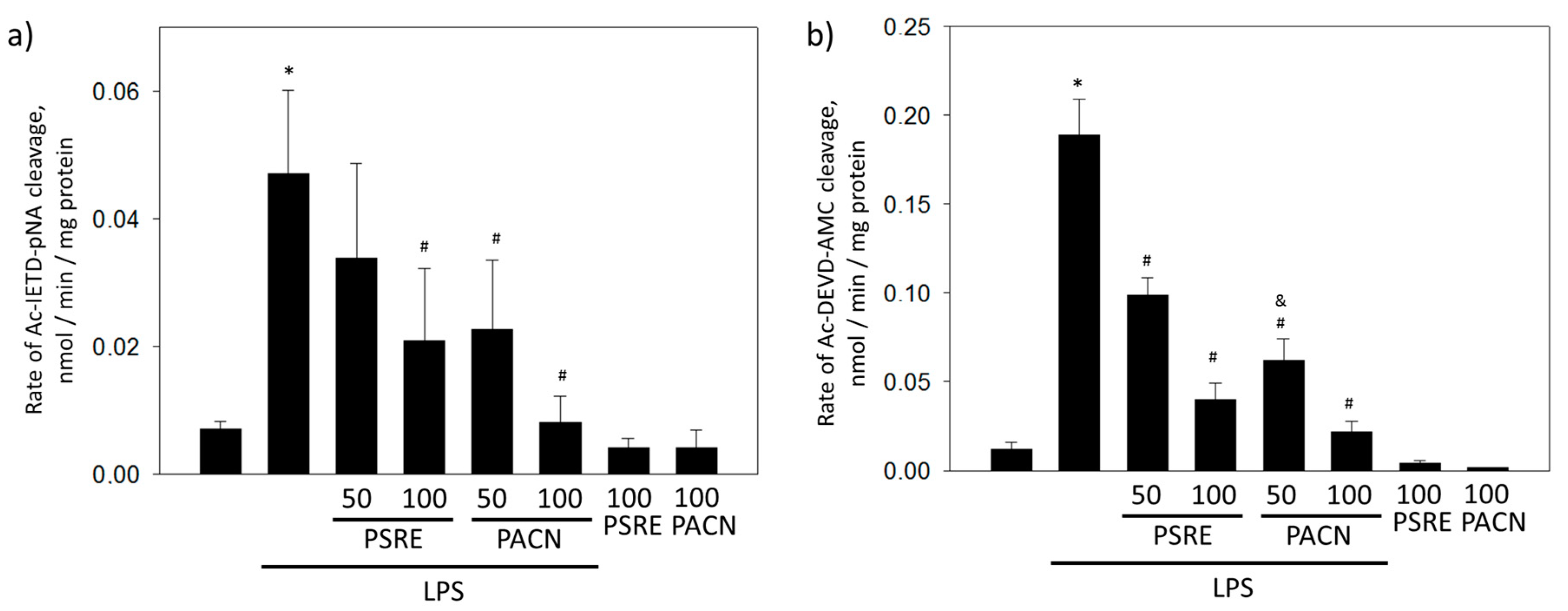
3.2.3. Pelargonium sidoides Root Extract and Proanthocyanidin Fraction Prevent Caspase Activity Induced by Bacterial Lipopolysaccharide
3.3. The Effect of Proanthocyanidins from Pelargonium sidoides Root Extract on Inflammatory Responses to Bacterial Lipopolysaccharide
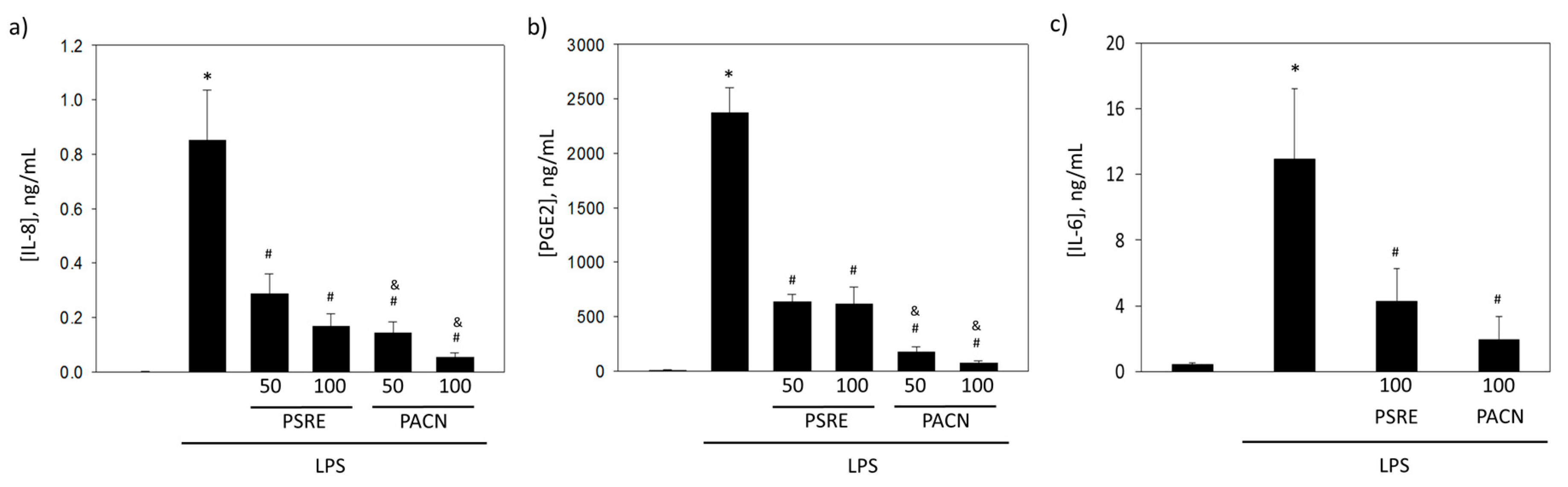
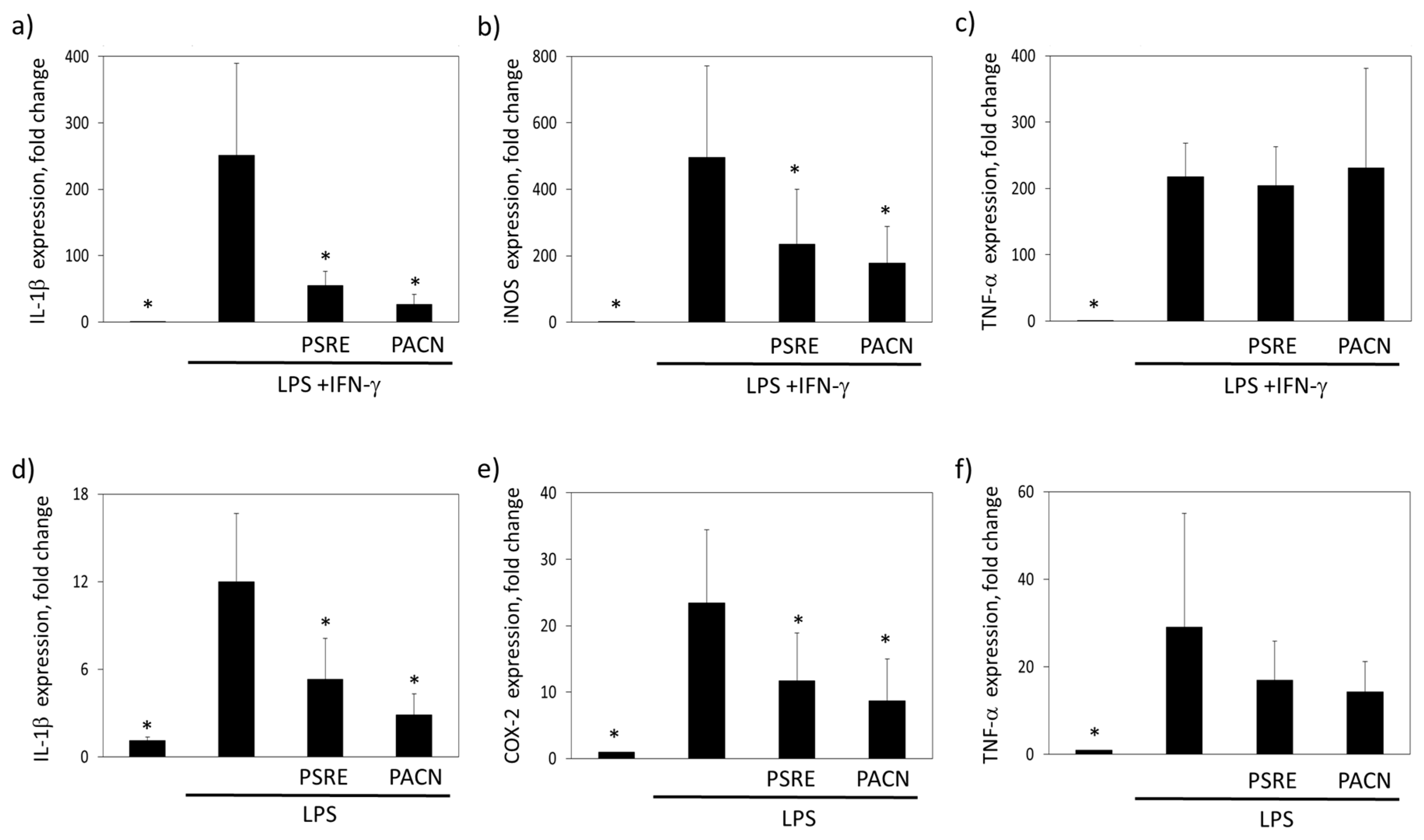
3.3.1. The effect of Pelargonium sidoides Root Extract and Proanthocyanidin Fraction on Lipopolysaccharide-Induced Secretion of Inflammatory Mediators
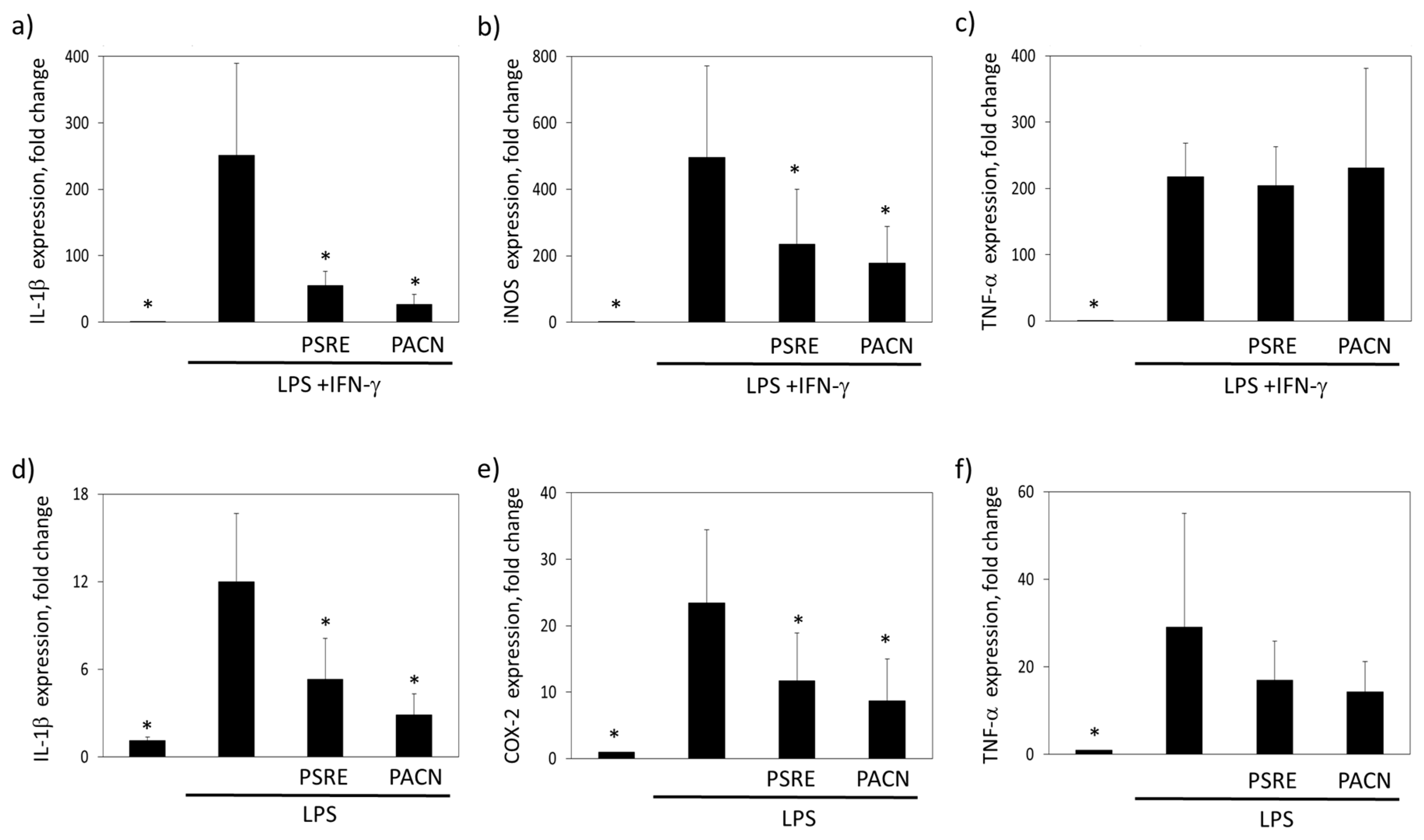
3.3.2. The Effect of Pelargonium sidoides Root Extract and Proanthocyanidin Fraction on Lipopolysaccharide-Induced Expression of Inflammation-Related Genes
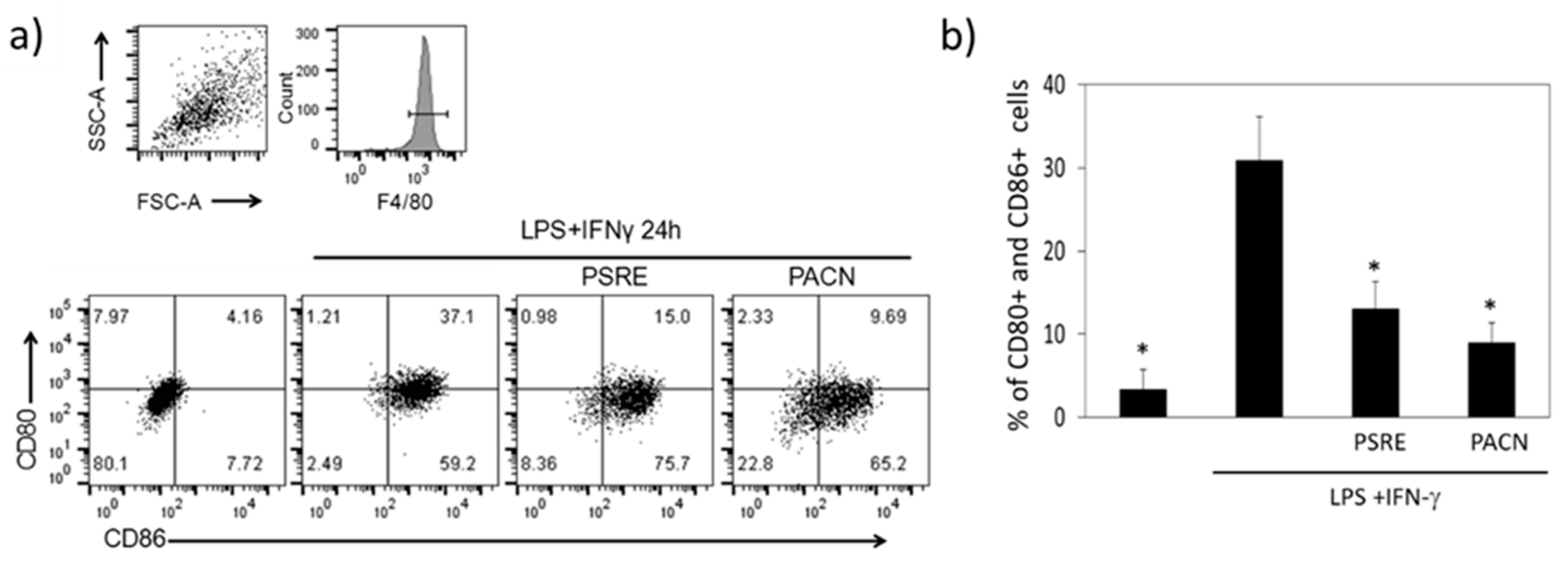
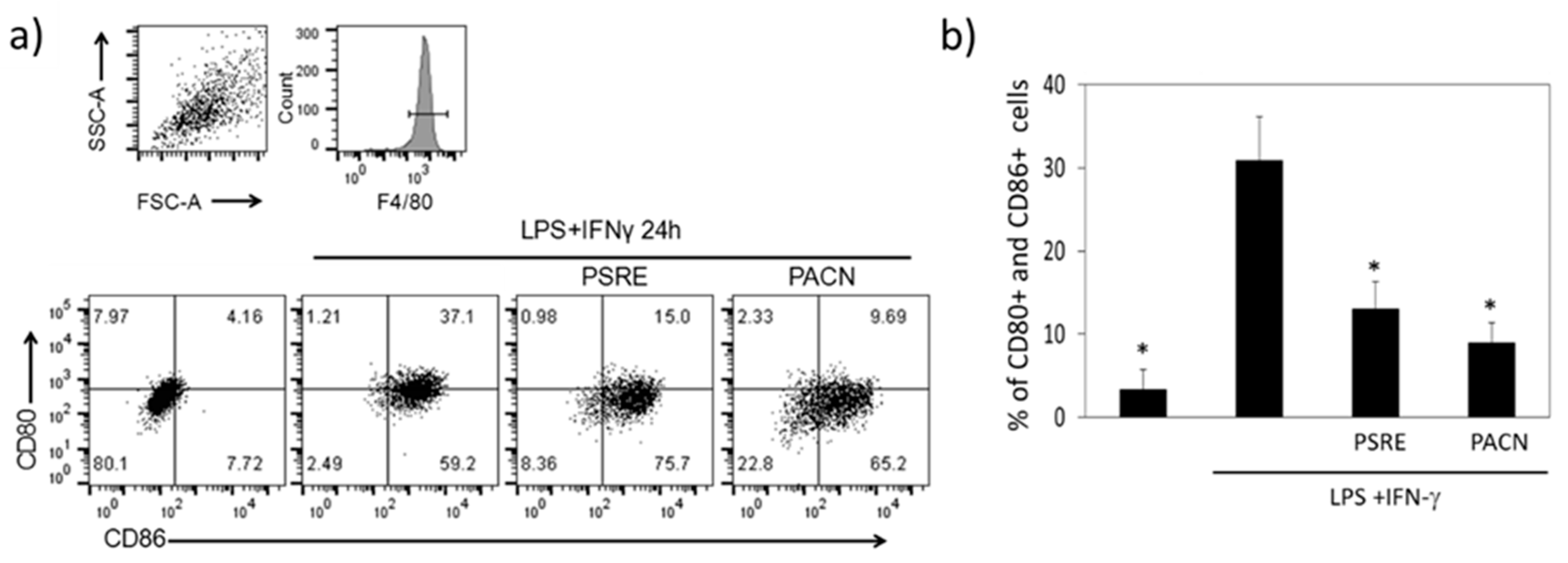
3.3.3. The Effect of Pelargonium sidoides Root Extract and Proanthocyanidin Fraction on Lipopolysaccharide-Induced Macrophage Conversion to M1 Phenotype
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Loesche, W.J.; Grossman, N.S. Periodontal Disease as a Specific, albeit Chronic, Infection: Diagnosis and Treatment. Clin. Microbiol. Rev. 2001, 14, 727. [Google Scholar] [CrossRef]
- Khan, S.A.; Kong, E.F.; Meiller, T.F.; Jabra-Rizk, M.A. Periodontal Diseases: Bug Induced, Host Promoted. PLoS Pathog. 2015, 11, e1004952. [Google Scholar] [CrossRef]
- Soares, G.M.S.; Figueiredo, L.C.; Faveri, M.; Cortelli, S.C.; Duarte, P.M.; Feres, M. Mechanisms of action of systemic antibiotics used in periodontal treatment and mechanisms of bacterial resistance to these drugs. J. Appl. Oral Sci. 2012, 20, 295–309. [Google Scholar] [CrossRef]
- Pareek, V.; Gupta, R.; Panwar, J. Do physico-chemical properties of silver nanoparticles decide their interaction with biological media and bactericidal action? A review. Mater. Sci. Eng. C 2018, 90, 739–749. [Google Scholar] [CrossRef]
- Nakatsuji, T.; Chen, T.H.; Narala, S.; Chun, K.A.; Two, A.M.; Yun, T.; Shafiq, F.; Kotol, P.F.; Bouslimani, A.; Melnik, A.V.; et al. Antimicrobials from human skin commensal bacteria protect against Staphylococcus aureus and are deficient in atopic dermatitis. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef]
- Avila, M.; Ojcius, D.M.; Yilmaz, O. The oral microbiota: Living with a permanent guest. DNA Cell Biol. 2009, 28, 405–411. [Google Scholar] [CrossRef]
- Brambilla, E.; Ionescu, A.; Gagliani, M.; Cochis, A.; Arciola, C.R.; Rimondini, L. Biofilm Formation on Composite Resins for Dental Restorations: An in Situ Study on the Effect of Chlorhexidine Mouthrinses. Int. J. Artif. Organs 2012, 35, 792–799. [Google Scholar] [CrossRef]
- Azzimonti, B.; Cochis, A.; Beyrouthy, M.; Iriti, M.; Uberti, F.; Sorrentino, R.; Landini, M.M.; Rimondini, L.; Varoni, E.M. Essential Oil from Berries of Lebanese Juniperus excelsa M. Bieb Displays Similar Antibacterial Activity to Chlorhexidine but Higher Cytocompatibility with Human Oral Primary Cells. Molecules 2015, 20, 9344–9357. [Google Scholar] [CrossRef]
- Singh, N.; Savita, S.; Rithesh, K.; Shivanand, S. Phytotherapy: A novel approach for treating periodontal disease. J. Pharm. Biomed. Sci. JPBMS. 2010, 6, 205–210. [Google Scholar]
- Santos-Buelga, C.; Scalbert, A. Proanthocyanidins and tannin-like compounds—Nature, occurrence, dietary intake and effects on nutrition and health. J. Sci. Food Agric. 2000, 80, 1094–1117. [Google Scholar] [CrossRef]
- Aron, P.M.; Kennedy, J.A. Flavan-3-ols: Nature, occurrence and biological activity. Mol. Nutr. Food Res. 2008, 52, 79–104. [Google Scholar] [CrossRef]
- Raudone, L.; Vilkickyte, G.; Pitkauskaite, L.; Raudonis, R.; Vainoriene, R.; Motiekaityte, V.; Raudone, L.; Vilkickyte, G.; Pitkauskaite, L.; Raudonis, R.; et al. Antioxidant Activities of Vaccinium vitis-idaea L. Leaves within Cultivars and Their Phenolic Compounds. Molecules 2019, 24, 844. [Google Scholar] [CrossRef]
- Balalaie, A.; Rezvani, M.B.; Basir, M.M. Dual function of proanthocyanidins as both MMP inhibitor and crosslinker in dentin biomodification: A literature review. Dent. Mater. J. 2018, 37, 173–182. [Google Scholar] [CrossRef]
- Shahzad, M.; Millhouse, E.; Culshaw, S.; Edwards, C.A.; Ramage, G.; Combet, E. Selected dietary (poly)phenols inhibit periodontal pathogen growth and biofilm formation. Food Funct. 2015, 6, 719–729. [Google Scholar] [CrossRef]
- Kolodziej, H. Aqueous ethanolic extract of the roots of Pelargonium sidoides—New scientific evidence for an old anti-infective phytopharmaceutical. Planta Med. 2008, 74, 661–666. [Google Scholar] [CrossRef]
- Kayser, O.; Kolodziej, H. Antibacterial Activity of Extracts and Constituents of Pelargonium sidoides and Pelargonium reniforme. Planta Med. 1997, 63, 508–510. [Google Scholar] [CrossRef]
- Kolodziej, H. Antimicrobial, antiviral and immunomodulatory activity studies of pelargonium sidoides (EPs® 7630) in the context of health promotion. Pharmaceuticals 2011, 4, 1295–1314. [Google Scholar] [CrossRef]
- Janecki, A.; Conrad, A.; Engels, I.; Frank, U.; Kolodziej, H. Evaluation of an aqueous-ethanolic extract from Pelargonium sidoides (EPs® 7630) for its activity against group A-streptococci adhesion to human HEp-2 epithelial cells. J. Ethnopharmacol. 2011, 133, 147–152. [Google Scholar] [CrossRef]
- Savickiene, N.; Jekabsone, A.; Raudone, L.; Abdelgeliel, A.; Cochis, A.; Rimondini, L.; Makarova, E.; Grinberga, S.; Pugovics, O.; Dambrova, M.; et al. Efficacy of Proanthocyanidins from Pelargonium sidoides Root Extract in Reducing P. gingivalis Viability While Preserving Oral Commensal S. salivarius. Materials 2018, 11, 1499. [Google Scholar] [CrossRef]
- Gristina, A.G.; Naylor, P.T.; Myrvik, Q. The Race for the Surface: Microbes, Tissue Cells, and Biomaterials. In Molecular Mechanisms of Microbial Adhesion; Springer: New York, NY, USA, 1989; pp. 177–211. [Google Scholar]
- Hellström, J.; Sinkkonen, J.; Maarit Karonen, A.; Mattila, P. Isolation and Structure Elucidation of Procyanidin Oligomers from Saskatoon Berries (Amelanchier alnifolia). J. Agric. Food Chem. 2006. [Google Scholar] [CrossRef]
- Buttery, J.E.; Lim, H.H.; De Witt, G.F. The use of NADH as a standard in a modified PMS-INT colorimetric assay of lactate dehydrogenase. Clin. Chim. Acta 1976, 73, 109–115. [Google Scholar] [CrossRef]
- Chang, H.Y.; Yang, X. Proteases for cell suicide: Functions and regulation of caspases. Microbiol. Mol. Biol. Rev. 2000, 64, 821–846. [Google Scholar] [CrossRef] [PubMed]
- Rashmi, S.; Alka, D.; Ramakant, S. Neutrophils in health and disease: An overview. J. Oral Maxillofac. Pathol. 2006, 10, 3. [Google Scholar] [CrossRef]
- Dongari-Bagtzoglou, A.I.; Ebersole, J.L. Increased Presence of Interleukin-6 (IL-6) and IL-8 Secreting Fibroblast Subpopulations in Adult Periodontitis. J. Periodontol. 1998, 69, 899–910. [Google Scholar] [CrossRef]
- Arango Duque, G.; Descoteaux, A. Macrophage cytokines: Involvement in immunity and infectious diseases. Front. Immunol. 2014, 5, 491. [Google Scholar] [CrossRef]
- Xue, Q.; Yan, Y.; Zhang, R.; Xiong, H. Regulation of iNOS on Immune Cells and Its Role in Diseases. Int. J. Mol. Sci. 2018, 19, 3805. [Google Scholar] [CrossRef]
- Held, T.K.; Weihua, X.; Yuan, L.; Kalvakolanu, D.V.; Cross, A.S. Gamma interferon augments macrophage activation by lipopolysaccharide by two distinct mechanisms, at the signal transduction level and via an autocrine mechanism involving tumor necrosis factor alpha and interleukin-1. Infect. Immun. 1999, 67, 206–212. [Google Scholar]
- Barrett, J.P.; Costello, D.A.; O’Sullivan, J.; Cowley, T.R.; Lynch, M.A. Bone marrow-derived macrophages from aged rats are more responsive to inflammatory stimuli. J. Neuroinflammation 2015, 12, 67. [Google Scholar] [CrossRef]
- Zhou, L.; Bi, C.; Gao, L.; An, Y.; Chen, F.; Chen, F. Macrophage polarization in human gingival tissue in response to periodontal disease. Oral Dis. 2019, 25, 265–273. [Google Scholar] [CrossRef]
- Orecchioni, M.; Ghosheh, Y.; Pramod, A.B.; Ley, K. Macrophage Polarization: Different Gene Signatures in M1(LPS+) vs. Classically and M2(LPS–) vs. Alternatively Activated Macrophages. Front. Immunol. 2019, 10, 1084. [Google Scholar] [CrossRef]
- Lambert, C.; Preijers, F.W.M.B.; Yanikkaya Demirel, G.; Sack, U. Monocytes and macrophages in flow: An ESCCA initiative on advanced analyses of monocyte lineage using flow cytometry. Cytom. Part B Clin. Cytom. 2017, 92, 180–188. [Google Scholar] [CrossRef]
- Ventola, C.L. The antibiotic resistance crisis: Part 1: Causes and threats. Pharm. Ther. 2015, 40, 277–283. [Google Scholar]
- Shaddox, L.M.; Walker, C.B. Treating chronic periodontitis: Current status, challenges, and future directions. Clin. Cosmet. Investig. Dent. 2010, 2, 79–91. [Google Scholar] [CrossRef]
- Seymour, R.A. Effects of medications on the periodontal tissues in health and disease. Periodontol. 2000 2006, 40, 120–129. [Google Scholar] [CrossRef]
- Irie, K.; Novince, C.M.; Darveau, R.P. Impact of the Oral Commensal Flora on Alveolar Bone Homeostasis. J. Dent. Res. 2014, 93, 801–806. [Google Scholar] [CrossRef] [Green Version]
- Patini, R.; Cattani, P.; Marchetti, S.; Isola, G.; Quaranta, G.; Gallenzi, P. Evaluation of Predation Capability of Periodontopathogens Bacteria by Bdellovibrio Bacteriovorus HD100. An in Vitro Study. Materials 2019, 12, 2008. [Google Scholar] [CrossRef] [Green Version]
- Moyo, M.; Van Staden, J. Medicinal properties and conservation of Pelargonium sidoides DC. J. Ethnopharmacol. 2014, 152, 243–255. [Google Scholar] [CrossRef]
- Kolodziej, H.; Kayser, O.; Radtke, O.A.; Kiderlen, A.F.; Koch, E. Pharmacological profile of extracts of Pelargonium sidoides and their constituents. Phytomedicine 2003, 10, 18–24. [Google Scholar] [CrossRef]
- Benso, B. Virulence factors associated with Aggregatibacter actinomycetemcomitans and their role in promoting periodontal diseases. Virulence 2017, 8, 111–114. [Google Scholar] [CrossRef] [Green Version]
- Lacombe, A.; Wu, V.C.H. The potential of berries to serve as selective inhibitors of pathogens and promoters of beneficial microorganisms. Food Qual. Saf. 2017, 1, 3–12. [Google Scholar] [CrossRef]
- Thapa, D.; Losa, R.; Zweifel, B.; Wallace, R.J. Sensitivity of pathogenic and commensal bacteria from the human colon to essential oils. Microbiology 2012, 158, 2870–2877. [Google Scholar] [CrossRef] [Green Version]
- Maisuria, V.B.; Los Santos, Y.L.; Tufenkji, N.; Déziel, E. Cranberry-derived proanthocyanidins impair virulence and inhibit quorum sensing of Pseudomonas aeruginosa. Sci. Rep. 2016, 6, 30169. [Google Scholar] [CrossRef] [Green Version]
- Krachler, A.M.; Orth, K. Targeting the bacteria-host interface: Strategies in anti-adhesion therapy. Virulence 2013, 4, 284–294. [Google Scholar] [CrossRef] [Green Version]
- Di Pasqua, R.; Betts, G.; Hoskins, N.; Edwards, M.; Ercolini, D.; Mauriello, G. Membrane Toxicity of Antimicrobial Compounds from Essential Oils. J. Agric. Food Chem. 2007, 55, 4863–4870. [Google Scholar] [CrossRef]
- Maisuria, V.B.; Okshevsky, M.; Déziel, E.; Tufenkji, N. Proanthocyanidin Interferes with Intrinsic Antibiotic Resistance Mechanisms of Gram-Negative Bacteria. Adv. Sci. 2019, 6, 1802333. [Google Scholar] [CrossRef]
- Trentin, D.S.; Silva, D.B.; Frasson, A.P.; Rzhepishevska, O.; da Silva, M.V.; Pulcini, E.D.L.; James, G.; Soares, G.V.; Tasca, T.; Ramstedt, M.; et al. Natural Green coating inhibits adhesion of clinically important bacteria. Sci. Rep. 2015, 5, 8287. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Gao, S.S.; Yang, H.J.; Wang, M.; Cheng, B.F.; Feng, Z.W.; Wang, L. Neuroprotective Effects of Proanthocyanidins, Natural Flavonoids Derived From Plants, on Rotenone-Induced Oxidative Stress and Apoptotic Cell Death in Human Neuroblastoma SH-SY5Y Cells. Front. Neurosci. 2018, 12, 369. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Chen, W.; Zhang, X.; Zheng, Y.; Yu, F.; Liu, Y.; Wang, Y. Grape seed proanthocyanidins induce mitochondrial pathway-mediated apoptosis in human colorectal carcinoma cells. Oncol. Lett. 2017, 14, 5853–5860. [Google Scholar] [CrossRef] [Green Version]
- Ji, B.C.; Hsu, W.H.; Yang, J.S.; Hsia, T.C.; Lu, C.C.; Chiang, J.H.; Yang, J.L.; Lin, C.H.; Lin, J.J.; Suen, L.J.W.; et al. Gallic Acid Induces Apoptosis via Caspase-3 and Mitochondrion-Dependent Pathways in Vitro and Suppresses Lung Xenograft Tumor Growth in Vivo. J. Agric. Food Chem. 2009, 57, 7596–7604. [Google Scholar] [CrossRef]
- Hikiji, H.; Takato, T.; Shimizu, T.; Ishii, S. The roles of prostanoids, leukotrienes, and platelet-activating factor in bone metabolism and disease. Prog. Lipid Res. 2008, 47, 107–126. [Google Scholar] [CrossRef]
- Noguchi, K.; Ishikawa, I. The roles of cyclooxygenase-2 and prostaglandin E2 in periodontal disease. Periodontol. 2000 2007, 43, 85–101. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, H.; Mehrotra, M.; Alander, C.; Lerner, U.; Pilbeam, C.; Raisz, L. Effects of prostaglandin E2 and lipopolysaccharide on osteoclastogenesis in RAW 264.7 cells. Prostaglandins Leukot. Essent. Fat. Acids 2007, 77, 181–186. [Google Scholar] [CrossRef] [Green Version]
- Hienz, S.A.; Paliwal, S.; Ivanovski, S. Mechanisms of Bone Resorption in Periodontitis. J. Immunol. Res. 2015, 2015, 615486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, A.; Park, J.Y.; Fenno, C.; Kapila, Y.L. Porphyromonas gingivalis, gamma interferon, and a proapoptotic fibronectin matrix form a synergistic trio that induces c-Jun N-terminal kinase 1-mediated nitric oxide generation and cell death. Infect. Immun. 2008, 76, 5514–5523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.M.; Min, B.G.; Jung, J.Y.; Jegal, K.H.; Lee, C.W.; Kim, K.Y.; Kim, Y.W.; Choi, Y.W.; Cho, I.J.; Ku, S.K.; et al. Combination of Pelargonium sidoides and Coptis chinensis root inhibits nuclear factor kappa B-mediated inflammatory response in vitro and in vivo. BMC Complement. Altern. Med. 2018, 18. [Google Scholar] [CrossRef] [Green Version]
- Dinarello, C.A. Proinflammatory Cytokines. Chest 2000, 118, 503–508. [Google Scholar] [CrossRef]
- Garlet, G.P.; Cardoso, C.R.B.; Campanelli, A.P.; Ferreira, B.R.; Avila-Campos, M.J.; Cunha, F.Q.; Silva, J.S. The dual role of p55 tumour necrosis factor-alpha receptor in Actinobacillus actinomycetemcomitans-induced experimental periodontitis: Host protection and tissue destruction. Clin. Exp. Immunol. 2007, 147, 128–138. [Google Scholar] [CrossRef]
- Graves, D.T.; Cochran, D. The Contribution of Interleukin-1 and Tumor Necrosis Factor to Periodontal Tissue Destruction. J. Periodontol. 2003, 74, 391–401. [Google Scholar] [CrossRef]
- Yucel-Lindberg, T.; Båge, T. Inflammatory mediators in the pathogenesis of periodontitis. Expert Rev. Mol. Med. 2013, 15, e7. [Google Scholar] [CrossRef] [Green Version]
- Ambriz-Ambriz-Pérez, D.L.; Leyva-Lopez, N.; Gutierrez-Grijalva, E.P.; Heredia, J.B. Phenolic compounds: Natural alternative in inflammation treatment. A Review. Cogent Food Agric. 2016, 2. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jekabsone, A.; Sile, I.; Cochis, A.; Makrecka-Kuka, M.; Laucaityte, G.; Makarova, E.; Rimondini, L.; Bernotiene, R.; Raudone, L.; Vedlugaite, E.; et al. Investigation of Antibacterial and Antiinflammatory Activities of Proanthocyanidins from Pelargonium sidoides DC Root Extract. Nutrients 2019, 11, 2829. https://doi.org/10.3390/nu11112829
Jekabsone A, Sile I, Cochis A, Makrecka-Kuka M, Laucaityte G, Makarova E, Rimondini L, Bernotiene R, Raudone L, Vedlugaite E, et al. Investigation of Antibacterial and Antiinflammatory Activities of Proanthocyanidins from Pelargonium sidoides DC Root Extract. Nutrients. 2019; 11(11):2829. https://doi.org/10.3390/nu11112829
Chicago/Turabian StyleJekabsone, Aiste, Inga Sile, Andrea Cochis, Marina Makrecka-Kuka, Goda Laucaityte, Elina Makarova, Lia Rimondini, Rasa Bernotiene, Lina Raudone, Evelina Vedlugaite, and et al. 2019. "Investigation of Antibacterial and Antiinflammatory Activities of Proanthocyanidins from Pelargonium sidoides DC Root Extract" Nutrients 11, no. 11: 2829. https://doi.org/10.3390/nu11112829