Epigenetic Activation of BRCA1 by Genistein In Vivo and Triple Negative Breast Cancer Cells Linked to Antagonism toward Aryl Hydrocarbon Receptor

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Models
2.2. Cell Culture and Reagents
2.3. Western Blotting
2.4. mRNA Analyses
2.5. DNA Methylation
2.6. Chromatin Immunoprecipitation
2.7. Cell Viability Assay
2.8. Statistical Analysis
3. Results
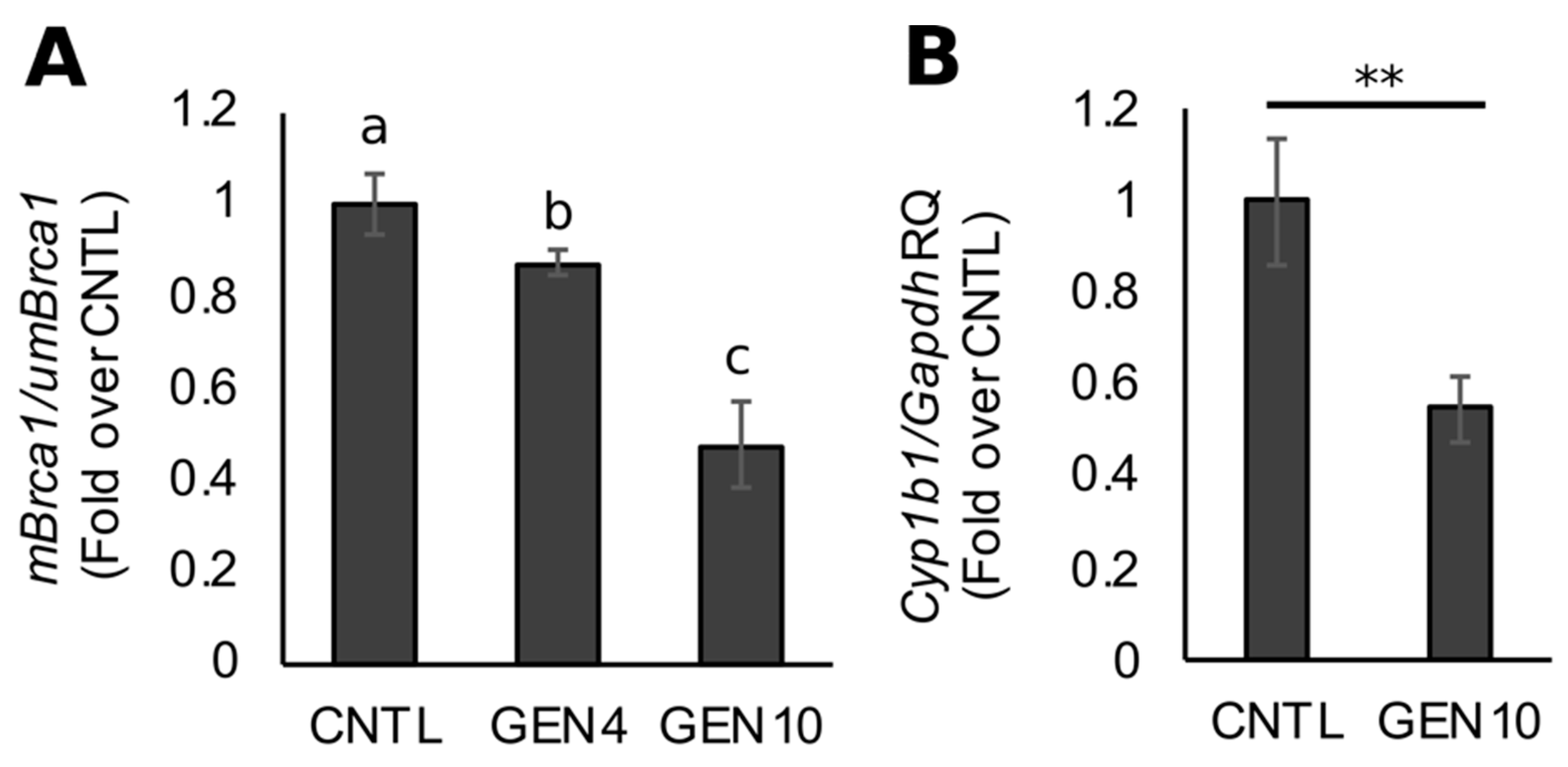
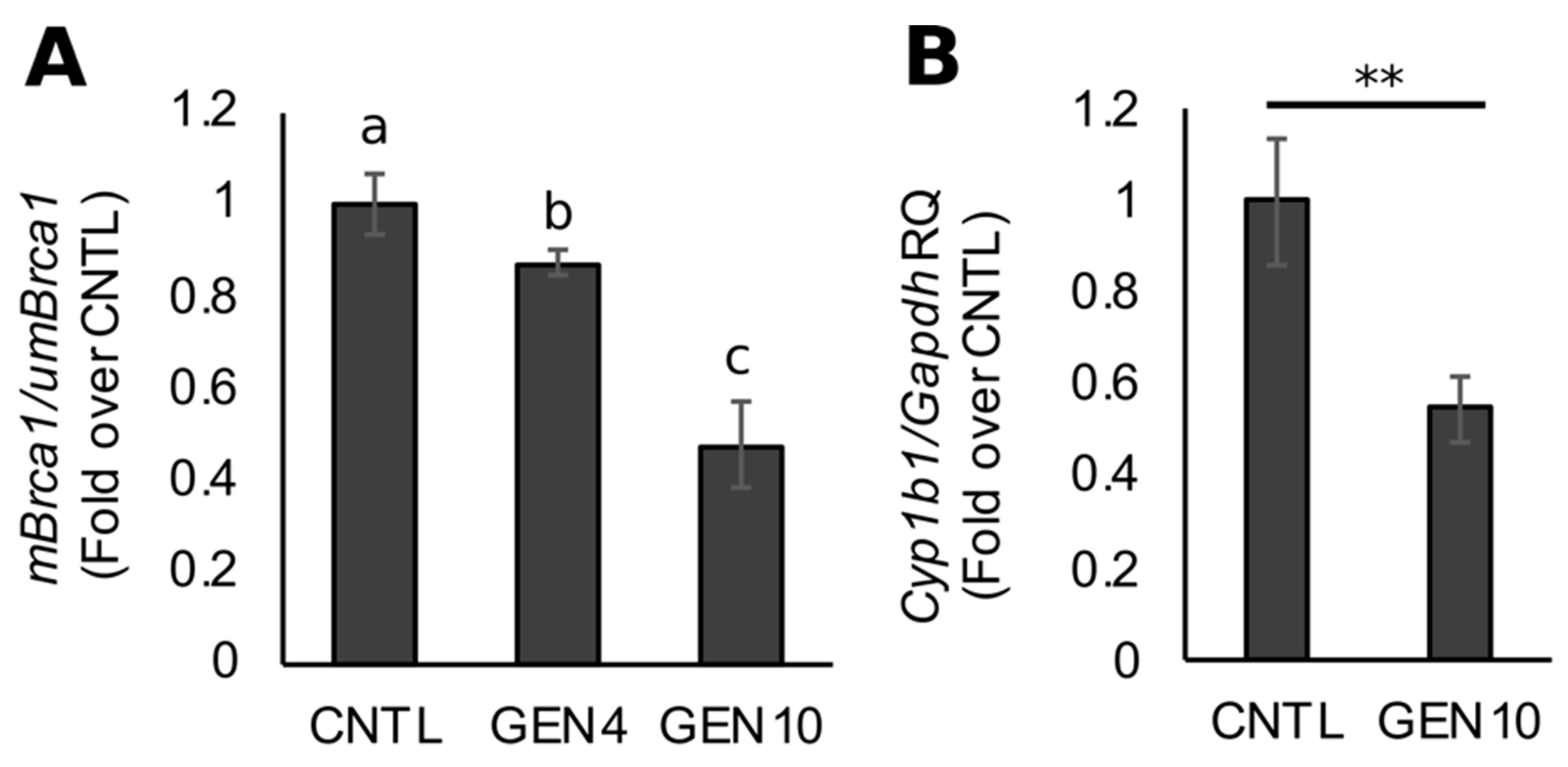
3.1. Lifetime Exposure to Dietary GEN Decreases Basal Brca1 Methylation in Mouse Mammary Tissue of Offspring
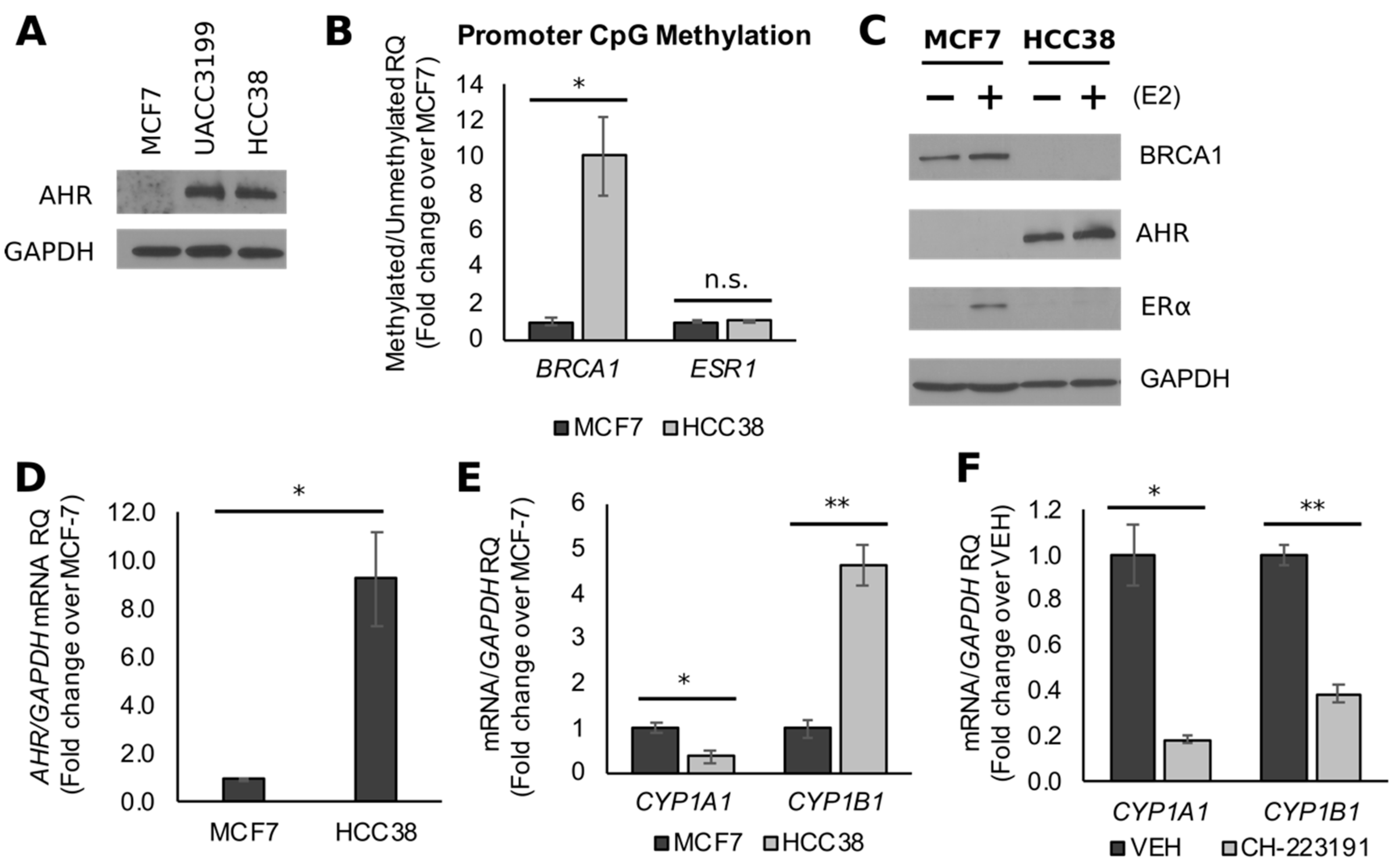
3.2. BRCA1 CpG Hypermethylation Associates with Constitutively Active AHR in TNBC Cells
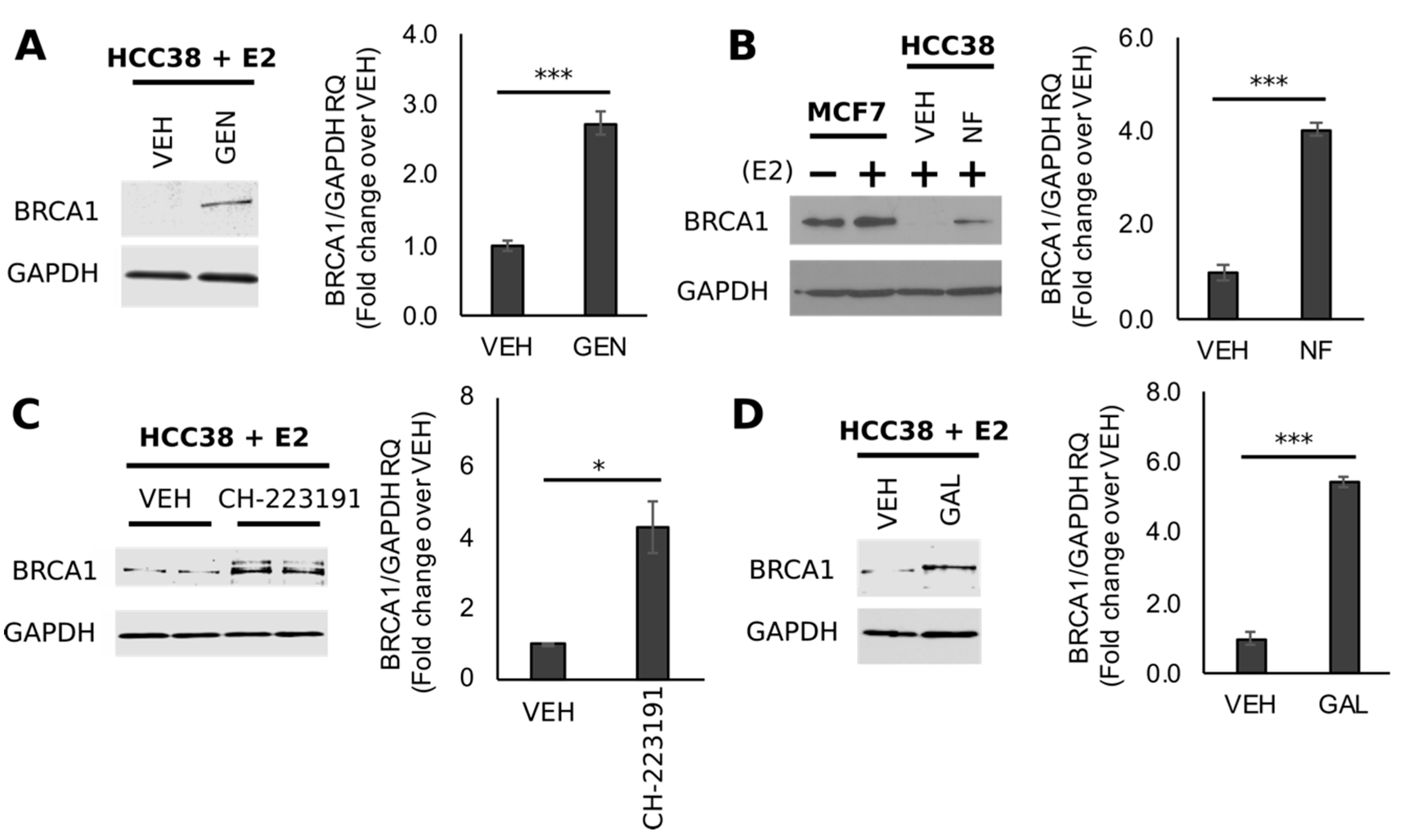
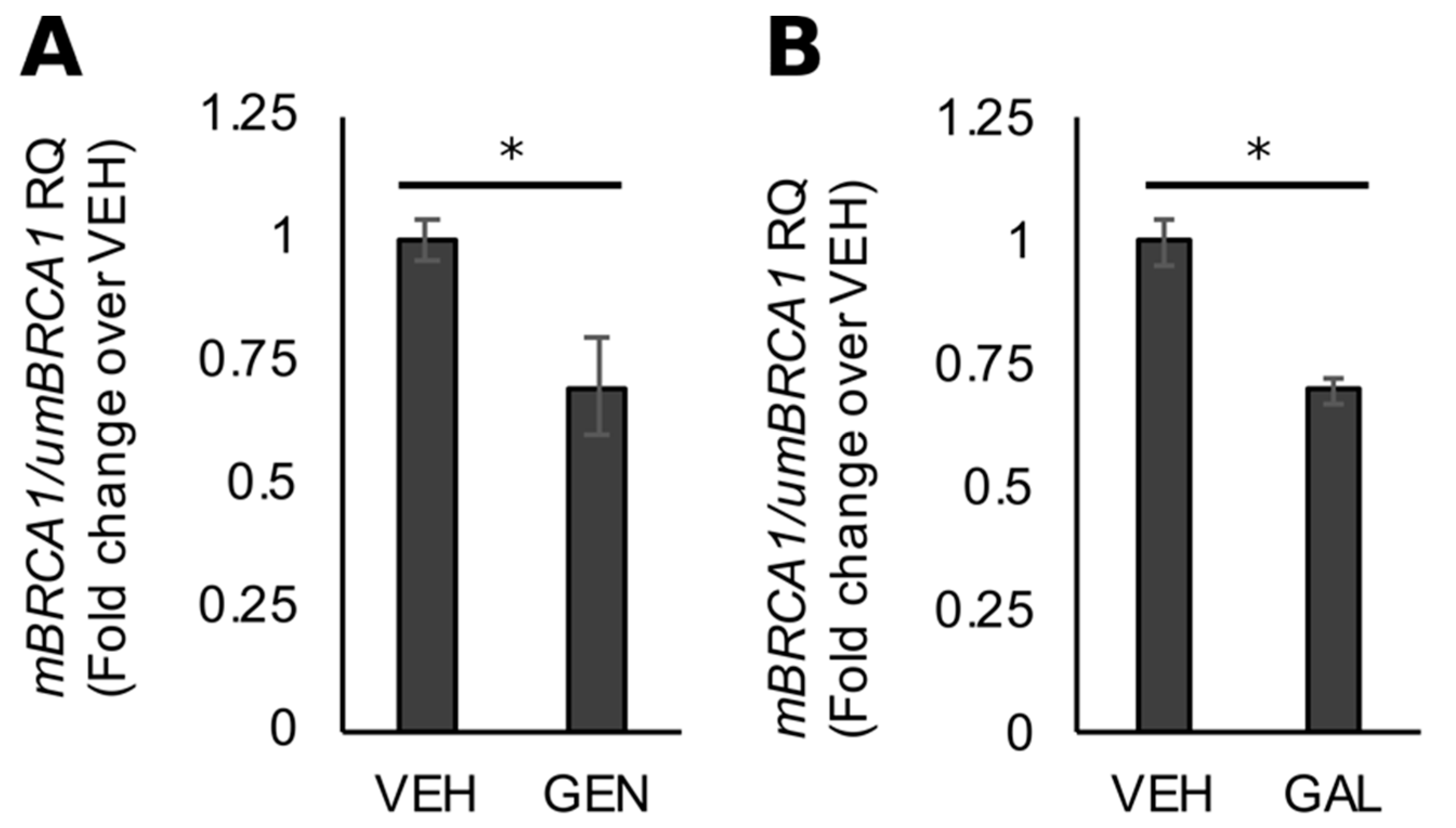
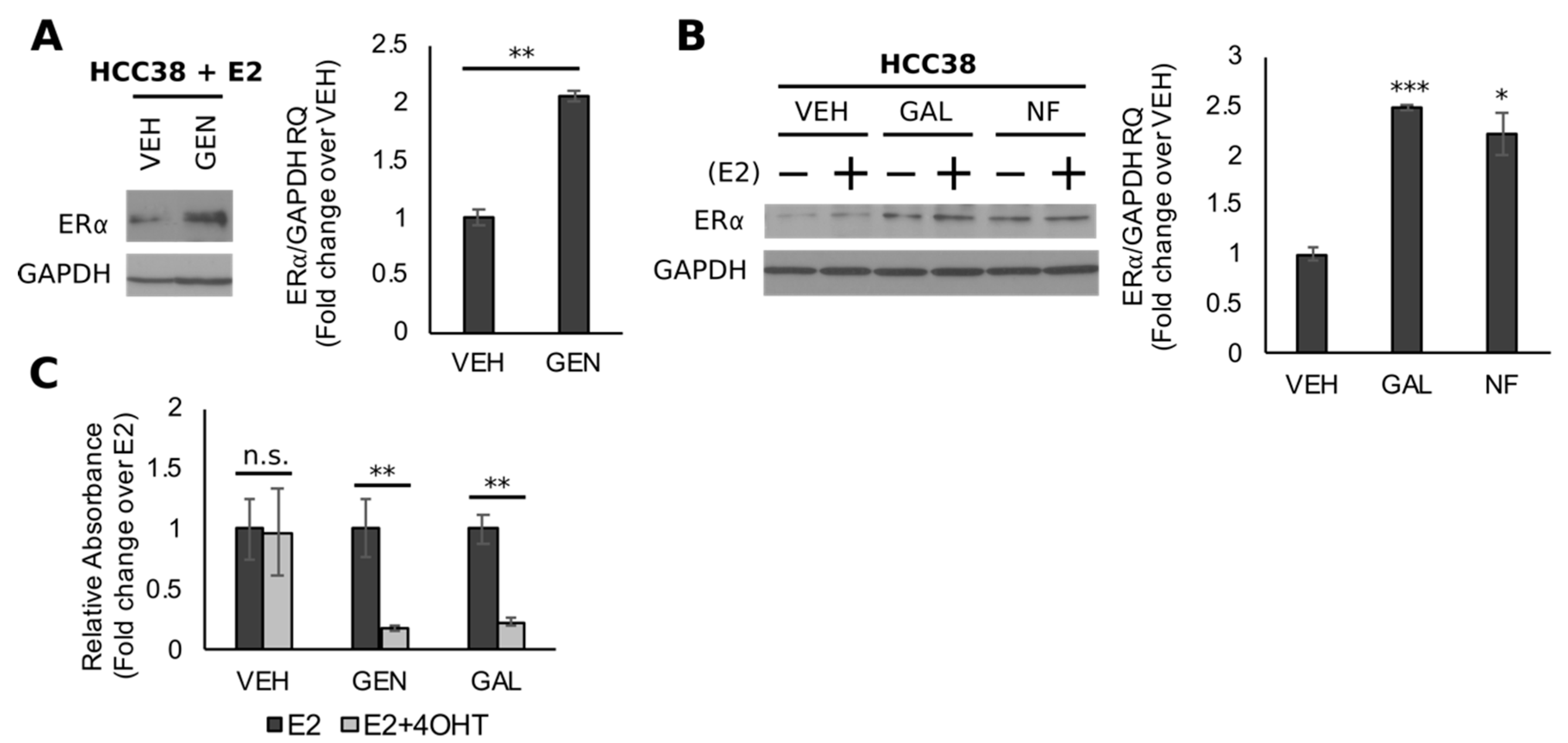
3.3. GEN Rescues BRCA1 Protein Levels in TNBC Cells with CpG Hypermethylated BRCA1
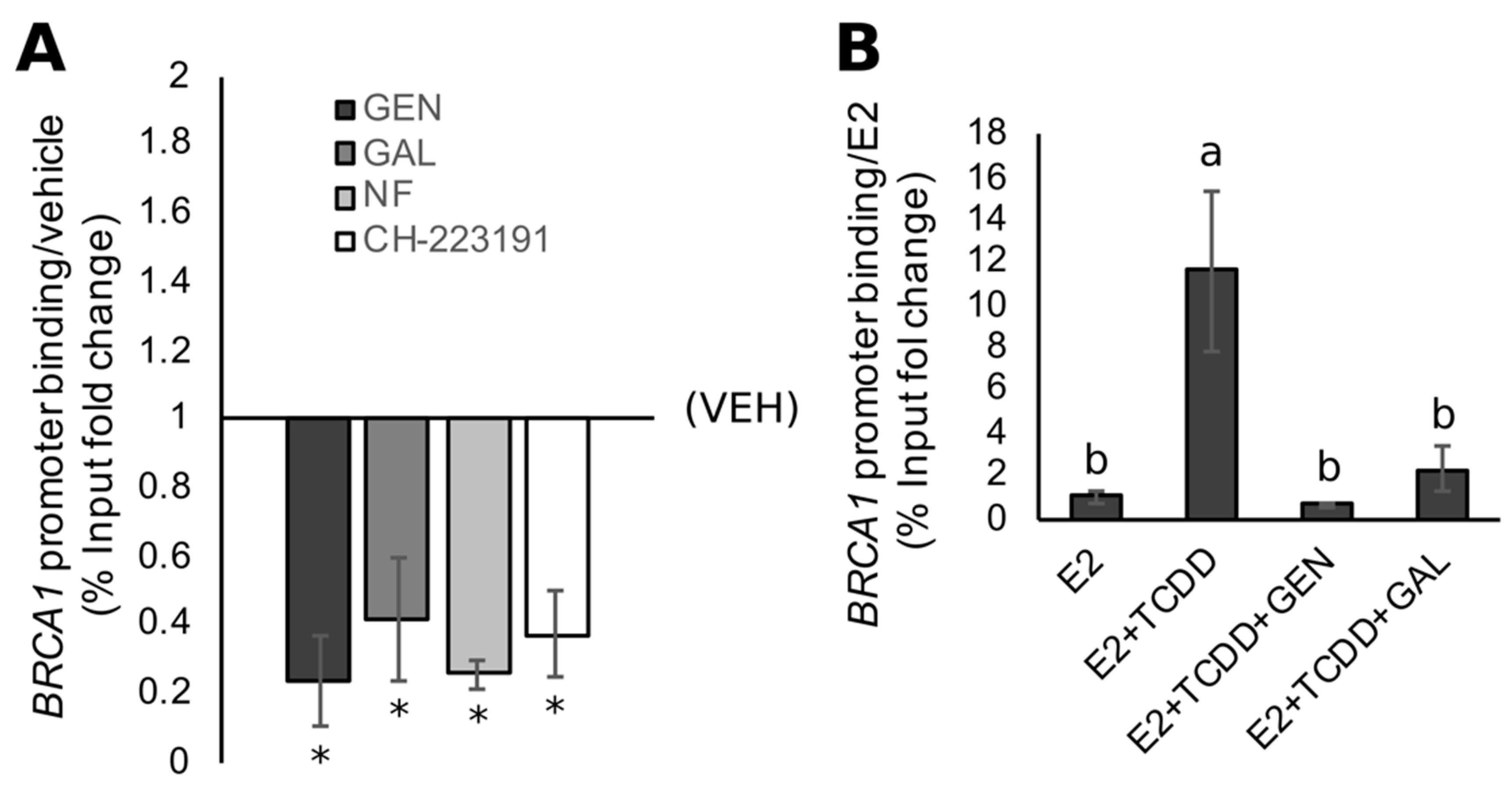
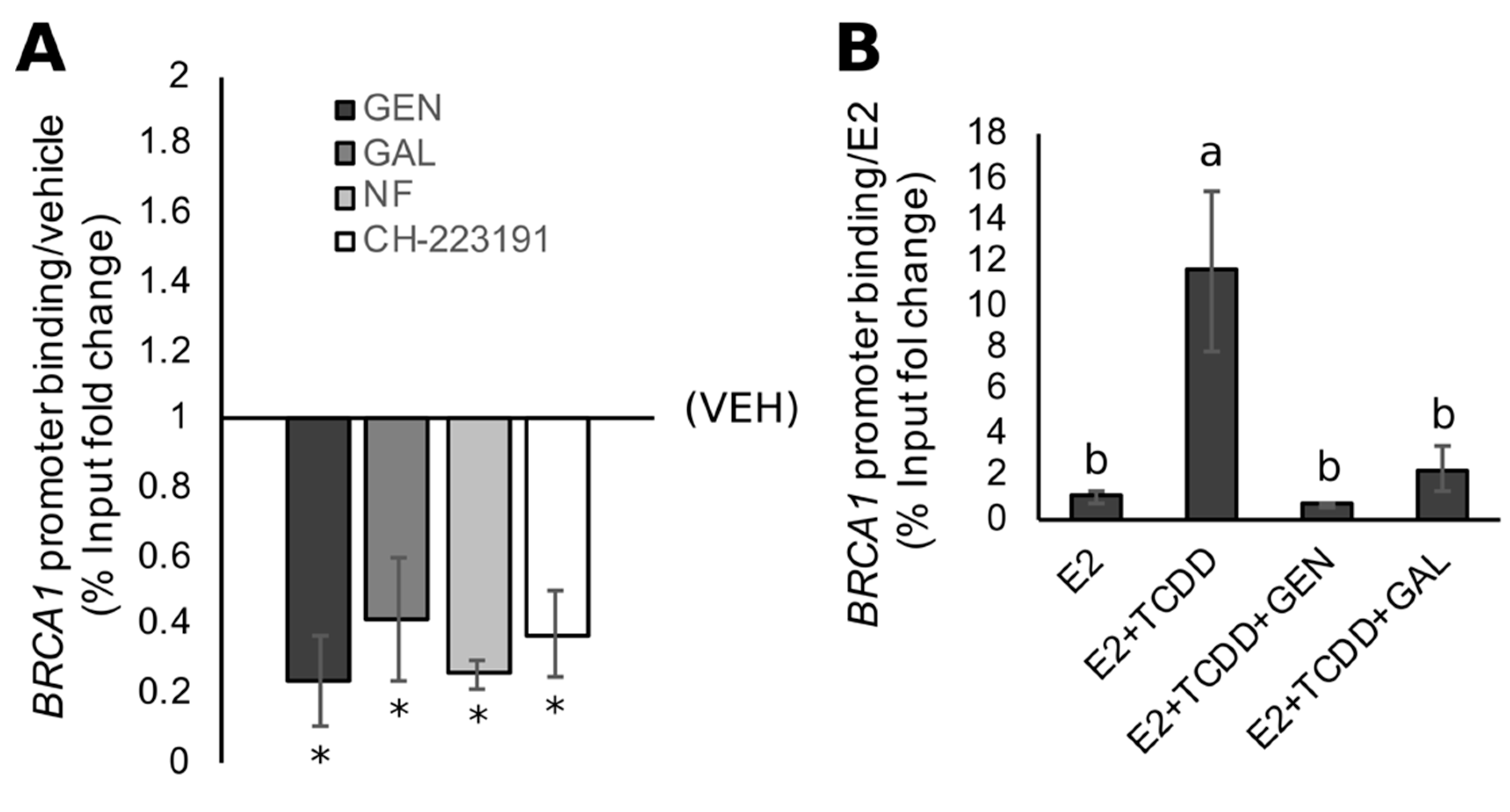
3.4. Rescue of BRCA1 Expression Is Linked to CpG Demethylation and AHR Inhibition in TNBC Cells
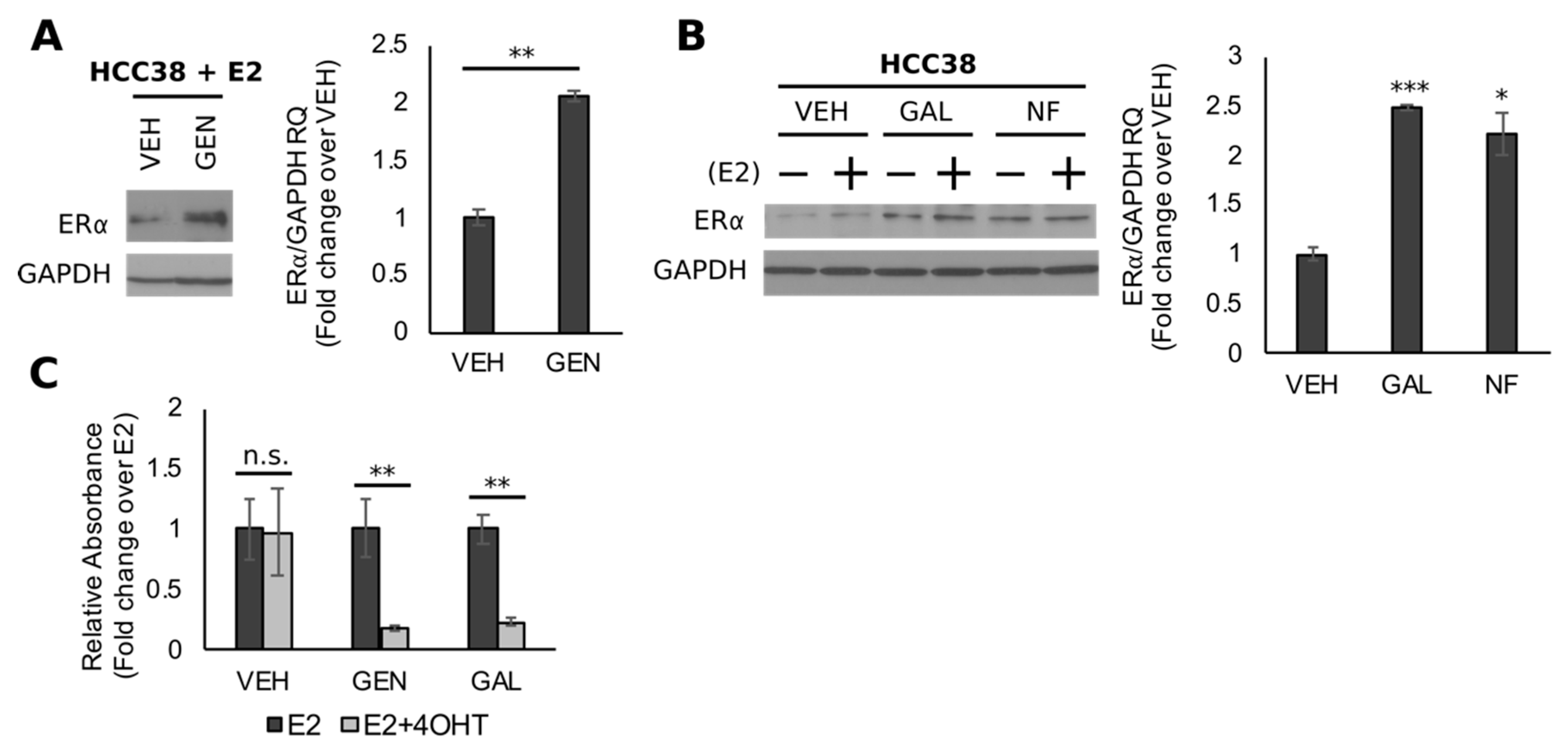
3.5. Genistein Upregulates ER and Sensitizes TNBC Cells to 4-OHT
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: Globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Guarneri, V.; Dieci, M.V.; Conte, P. Relapsed triple-negative breast cancer: Challenges and treatment strategies. Drugs 2013, 73, 1257–1265. [Google Scholar] [CrossRef] [PubMed]
- Nakashoji, A.; Matsui, A.; Nagayama, A.; Iwata, Y.; Sasahara, M.; Murata, Y. Clinical predictors of pathological complete response to neoadjuvant chemotherapy in triple-negative breast cancer. Oncol. Lett. 2017, 14, 4135–4141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savage, K.I.; Harkin, D.P. Brca1, a ‘complex’ protein involved in the maintenance of genomic stability. FEBS J. 2015, 282, 630–646. [Google Scholar] [CrossRef] [PubMed]
- Kuchenbaecker, K.B.; Hopper, J.L.; Barnes, D.R.; Phillips, K.A.; Mooij, T.M.; Roos-Blom, M.J.; Jervis, S.; van Leeuwen, F.E.; Milne, R.L.; Andrieu, N.; et al. Risks of breast, ovarian, and contralateral breast cancer for brca1 and brca2 mutation carriers. Jama 2017, 317, 2402–2416. [Google Scholar] [CrossRef] [PubMed]
- Silver, D.P.; Richardson, A.L.; Eklund, A.C.; Wang, Z.C.; Szallasi, Z.; Li, Q.; Juul, N.; Leong, C.O.; Calogrias, D.; Buraimoh, A.; et al. Efficacy of neoadjuvant cisplatin in triple-negative breast cancer. J. Clin. Oncol. 2010, 28, 1145–1153. [Google Scholar] [CrossRef]
- Tian, T.; Shan, L.; Yang, W.; Zhou, X.; Shui, R. Evaluation of the brcaness phenotype and its correlations with clinicopathological features in triple-negative breast cancers. Hum. Pathol. 2019, 84, 231–238. [Google Scholar] [CrossRef]
- Zhang, L.; Long, X. Association of brca1 promoter methylation with sporadic breast cancers: Evidence from 40 studies. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef]
- Brianese, R.C.; Nakamura, K.D.M.; Almeida, F.; Ramalho, R.F.; Barros, B.D.F.; Ferreira, E.N.E.; Formiga, M.; de Andrade, V.P.; de Lima, V.C.C.; Carraro, D.M. Brca1 deficiency is a recurrent event in early-onset triple-negative breast cancer: A comprehensive analysis of germline mutations and somatic promoter methylation. Breast Cancer Res. Treat. 2018, 167, 803–814. [Google Scholar] [CrossRef]
- Lips, E.H.; Mulder, L.; Oonk, A.; van der Kolk, L.E.; Hogervorst, F.B.; Imholz, A.L.; Wesseling, J.; Rodenhuis, S.; Nederlof, P.M. Triple-negative breast cancer: Brcaness and concordance of clinical features with brca1-mutation carriers. Br. J. Cancer 2013, 108, 2172–2177. [Google Scholar] [CrossRef]
- Ignatov, T.; Poehlmann, A.; Ignatov, A.; Schinlauer, A.; Costa, S.D.; Roessner, A.; Kalinski, T.; Bischoff, J. Brca1 promoter methylation is a marker of better response to anthracycline-based therapy in sporadic tnbc. Breast Cancer Res. Treat. 2013, 141, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M.; Fraga, M.F.; Guo, M.; Garcia-Foncillas, J.; Hedenfalk, I.; Godwin, A.K.; Trojan, J.; Vaurs-Barriere, C.; Bignon, Y.J.; Ramus, S.; et al. DNA methylation patterns in hereditary human cancers mimic sporadic tumorigenesis. Hum. Mol. Genet. 2001, 10, 3001–3007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Heetvelde, M.; Van Bockstal, M.; Poppe, B.; Lambein, K.; Rosseel, T.; Atanesyan, L.; Deforce, D.; Van Den Berghe, I.; De Leeneer, K.; Van Dorpe, J.; et al. Accurate detection and quantification of epigenetic and genetic second hits in brca1 and brca2-associated hereditary breast and ovarian cancer reveals multiple co-acting second hits. Cancer Lett. 2018, 425, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Vos, S.; Moelans, C.B.; van Diest, P.J. Brca promoter methylation in sporadic versus brca germline mutation-related breast cancers. Breast Cancer Res. 2017, 19. [Google Scholar] [CrossRef]
- Rossi, R.E.; Pericleous, M.; Mandair, D.; Whyand, T.; Caplin, M.E. The role of dietary factors in prevention and progression of breast cancer. Anticancer. Res. 2014, 34, 6861–6875. [Google Scholar]
- Tham, D.G.C.; Haskell, W. Clinical review 97: Potential health benefits of dietary phytoestrogens: A review of the clinical, epidemiological, and mechanistic evidence. J. Clin. Endocrinol. Metab. 1998, 83, 2223–2235. [Google Scholar]
- Yang, Z.; Kulkarni, K.; Zhu, W.; Hu, M. Bioavailability and pharmacokinetics of genistein: Mechanistic studies on its adme. Anticancer. Agents Med. Chem. 2012, 12, 1264–1280. [Google Scholar] [CrossRef]
- Liu, X.O.; Huang, Y.B.; Gao, Y.; Chen, C.; Yan, Y.; Dai, H.J.; Song, F.J.; Wang, Y.G.; Wang, P.S.; Chen, K.X. Association between dietary factors and breast cancer risk among chinese females: Systematic review and meta-analysis. Asian Pac. J. Cancer Prev. 2014, 15, 1291–1298. [Google Scholar] [CrossRef]
- Qin, L.Q.; Xu, J.Y.; Wang, P.Y.; Hoshi, K. Soyfood intake in the prevention of breast cancer risk in women: A meta-analysis of observational epidemiological studies. J. Nutr. Sci. Vitaminol. 2006, 52, 428–436. [Google Scholar] [CrossRef]
- Trock, B.J.; Hilakivi-Clarke, L.; Clarke, R. Meta-analysis of soy intake and breast cancer risk. J. Natl. Cancer Inst. 2006, 98, 459–471. [Google Scholar] [CrossRef]
- Woo, H.D.; Park, S.; Oh, K.; Kim, H.J.; Shin, H.R.; Moon, H.K.; Kim, J. Diet and cancer risk in the korean population: A meta-analysis. Asian Pac. J. Cancer Prev. 2014, 15, 8509–8519. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.C.; Zheng, D.; Sun, J.J.; Zou, Z.K.; Ma, Z.L. Meta-analysis of studies on breast cancer risk and diet in chinese women. Int J. Clin. Exp. Med. 2015, 8, 73–85. [Google Scholar] [PubMed]
- Chen, M.; Rao, Y.; Zheng, Y.; Wei, S.; Li, Y.; Guo, T.; Yin, P. Association between soy isoflavone intake and breast cancer risk for pre- and post-menopausal women: A meta-analysis of epidemiological studies. PLoS ONE 2014, 9, e89288. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.Y.; Qin, L.Q. Soy isoflavones consumption and risk of breast cancer incidence or recurrence: A meta-analysis of prospective studies. Breast Cancer Res. Treat. 2011, 125, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Russo, M.; Russo, G.L.; Daglia, M.; Kasi, P.D.; Ravi, S.; Nabavi, S.F.; Nabavi, S.M. Understanding genistein in cancer: The “good” and the “bad” effects: A review. Food Chem. 2016, 196, 589–600. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Fan, J.; Cheng, L.; Hu, P.; Liu, R. The anticancer activity of genistein is increased in estrogen receptor beta 1-positive breast cancer cells. Onco Targets Ther. 2018, 11, 8153–8163. [Google Scholar] [CrossRef]
- Fang, Y.; Zhang, Q.; Wang, X.; Yang, X.; Wang, X.; Huang, Z.; Jiao, Y.; Wang, J. Quantitative phosphoproteomics reveals genistein as a modulator of cell cycle and DNA damage response pathways in triple-negative breast cancer cells. Int. J. Oncol. 2016, 48, 1016–1028. [Google Scholar] [CrossRef]
- Bosviel, R.; Dumollard, E.; Dechelotte, P.; Bignon, Y.J.; Bernard-Gallon, D. Can soy phytoestrogens decrease DNA methylation in brca1 and brca2 oncosuppressor genes in breast cancer? Omics 2012, 16, 235–244. [Google Scholar] [CrossRef]
- Lubecka, K.; Kaufman-Szymczyk, A.; Cebula-Obrzut, B.; Smolewski, P.; Szemraj, J.; Fabianowska-Majewska, K. Novel clofarabine-based combinations with polyphenols epigenetically reactivate retinoic acid receptor beta, inhibit cell growth, and induce apoptosis of breast cancer cells. Int. J. Mol. Sci 2018, 19, 3970. [Google Scholar] [CrossRef]
- King-Batoon, A.; Leszczynska, J.M.; Klein, C.B. Modulation of gene methylation by genistein or lycopene in breast cancer cells. Environ. Mol. Mutagen. 2008, 49, 36–45. [Google Scholar] [CrossRef]
- Xie, Q.; Bai, Q.; Zou, L.Y.; Zhang, Q.Y.; Zhou, Y.; Chang, H.; Yi, L.; Zhu, J.D.; Mi, M.T. Genistein inhibits DNA methylation and increases expression of tumor suppressor genes in human breast cancer cells. Genes Chromosomes Cancer 2014, 53, 422–431. [Google Scholar] [CrossRef] [PubMed]
- Luijten, M.; Thomsen, A.R.; van den Berg, J.A.; Wester, P.W.; Verhoef, A.; Nagelkerke, N.J.; Adlercreutz, H.; van Kranen, H.J.; Piersma, A.H.; Sorensen, I.K.; et al. Effects of soy-derived isoflavones and a high-fat diet on spontaneous mammary tumor development in tg.Nk (mmtv/c-neu) mice. Nutr. Cancer 2004, 50, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, A.R.; Mortensen, A.; Breinholt, V.M.; Lindecrona, R.H.; Penalvo, J.L.; Sorensen, I.K. Influence of prevastein, an isoflavone-rich soy product, on mammary gland development and tumorigenesis in tg.Nk (mmtv/c-neu) mice. Nutr. Cancer 2005, 52, 176–188. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Cook, K.L.; Warri, A.; Cruz, I.M.; Rosim, M.; Riskin, J.; Helferich, W.; Doerge, D.; Clarke, R.; Hilakivi-Clarke, L. Lifetime genistein intake increases the response of mammary tumors to tamoxifen in rats. Clin. Cancer Res. 2017, 23, 814–824. [Google Scholar] [CrossRef] [PubMed]
- De Assis, S.; Warri, A.; Benitez, C.; Helferich, W.; Hilakivi-Clarke, L. Protective effects of prepubertal genistein exposure on mammary tumorigenesis are dependent on brca1 expression. Cancer Prev. Res. (Phila) 2011, 4, 1436–1448. [Google Scholar] [CrossRef]
- Hockings, J.K.; Thorne, P.A.; Kemp, M.Q.; Morgan, S.S.; Selmin, O.; Romagnolo, D.F. The ligand status of the aromatic hydrocarbon receptor modulates transcriptional activation of brca-1 promoter by estrogen. Cancer Res. 2006, 66, 2224–2232. [Google Scholar] [CrossRef]
- Tian, J.; Feng, Y.; Fu, H.; Xie, H.Q.; Jiang, J.X.; Zhao, B. The aryl hydrocarbon receptor: A key bridging molecule of external and internal chemical signals. Environ. Sci. Technol. 2015, 49, 9518–9531. [Google Scholar] [CrossRef]
- Yueh, M.F.; Huang, Y.H.; Hiller, A.; Chen, S.; Nguyen, N.; Tukey, R.H. Involvement of the xenobiotic response element (xre) in ah receptor-mediated induction of human udp-glucuronosyltransferase 1a1. J. Biol. Chem. 2003, 278, 15001–15006. [Google Scholar] [CrossRef]
- Jeffy, B.D.; Hockings, J.K.; Kemp, M.Q.; Morgan, S.S.; Hager, J.A.; Beliakoff, J.; Whitesell, L.J.; Bowden, G.T.; Romagnolo, D.F. An estrogen receptor-alpha/p300 complex activates the brca-1 promoter at an ap-1 site that binds jun/fos transcription factors: Repressive effects of p53 on brca-1 transcription. Neoplasia 2005, 7, 873–882. [Google Scholar] [CrossRef]
- Papoutsis, A.J.; Borg, J.L.; Selmin, O.I.; Romagnolo, D.F. Brca-1 promoter hypermethylation and silencing induced by the aromatic hydrocarbon receptor-ligand tcdd are prevented by resveratrol in mcf-7 cells. J. Nutr. Biochem. 2012, 23, 1324–1332. [Google Scholar] [CrossRef]
- Papoutsis, A.J.; Lamore, S.D.; Wondrak, G.T.; Selmin, O.I.; Romagnolo, D.F. Resveratrol prevents epigenetic silencing of brca-1 by the aromatic hydrocarbon receptor in human breast cancer cells. J. Nutr. 2010, 140, 1607–1614. [Google Scholar] [CrossRef] [PubMed]
- Romagnolo, D.F.; Donovan, M.G.; Papoutsis, A.J.; Doetschman, T.C.; Selmin, O.I. Genistein prevents brca1 cpg methylation and proliferation in human breast cancer cells with activated aromatic hydrocarbon receptor. Curr. Dev. Nutr. 2017, 1, e000562. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative pcr and the 2(-delta delta c(t)) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Rice, J.C.; Massey-Brown, K.S.; Futscher, B.W. Aberrant methylation of the brca1 cpg island promoter is associated with decreased brca1 mrna in sporadic breast cancer cells. Oncogene 1998, 17, 1807–1812. [Google Scholar] [CrossRef]
- Xu, J.; Huo, D.; Chen, Y.; Nwachukwu, C.; Collins, C.; Rowell, J.; Slamon, D.J.; Olopade, O.I. Cpg island methylation affects accessibility of the proximal brca1 promoter to transcription factors. Breast Cancer Res. Treat. 2010, 120, 593–601. [Google Scholar] [CrossRef]
- Romagnolo, D.F.; Papoutsis, A.J.; Laukaitis, C.; Selmin, O.I. Constitutive expression of ahr and brca-1 promoter cpg hypermethylation as biomarkers of erα-negative breast tumorigenesis. BMC Cancer 2015, 15. [Google Scholar] [CrossRef]
- Yang, X.; Solomon, S.; Fraser, L.R.; Trombino, A.F.; Liu, D.; Sonenshein, G.E.; Hestermann, E.V.; Sherr, D.H. Constitutive regulation of cyp1b1 by the aryl hydrocarbon receptor (ahr) in pre-malignant and malignant mammary tissue. J. Cell. Biochem. 2008, 104, 402–417. [Google Scholar] [CrossRef]
- Ashida, H.; Fukuda, I.; Yamashita, T.; Kanazawa, K. Flavones and flavonols at dietary levels inhibit a transformation of aryl hydrocarbon receptor induced by dioxin. FEBS Lett. 2000, 476, 213–217. [Google Scholar] [CrossRef]
- Tomblin, J.K.; Arthur, S.; Primerano, D.A.; Chaudhry, A.R.; Fan, J.; Denvir, J.; Salisbury, T.B. Aryl hydrocarbon receptor (ahr) regulation of l-type amino acid transporter 1 (lat-1) expression in mcf-7 and mda-mb-231 breast cancer cells. Biochem. Pharmacol. 2016, 106, 94–103. [Google Scholar] [CrossRef]
- Kasai, S.; Kikuchi, H. The inhibitory mechanisms of the tyrosine kinase inhibitors herbimycin a, genistein, and tyrphostin b48 with regard to the function of the aryl hydrocarbon receptor in caco-2 cells. Biosci. Biotechnol. Biochem. 2010, 74, 36–43. [Google Scholar] [CrossRef]
- Van der Heiden, E.; Bechoux, N.; Muller, M.; Sergent, T.; Schneider, Y.J.; Larondelle, Y.; Maghuin-Rogister, G.; Scippo, M.L. Food flavonoid aryl hydrocarbon receptor-mediated agonistic/antagonistic/synergic activities in human and rat reporter gene assays. Anal. Chim. Acta 2009, 637, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Hosey, A.M.; Gorski, J.J.; Murray, M.M.; Quinn, J.E.; Chung, W.Y.; Stewart, G.E.; James, C.R.; Farragher, S.M.; Mulligan, J.M.; Scott, A.N.; et al. Molecular basis for estrogen receptor alpha deficiency in brca1-linked breast cancer. J. Natl. Cancer Inst. 2007, 99, 1683–1694. [Google Scholar] [CrossRef] [PubMed]
- Dent, R.; Hanna, W.M.; Trudeau, M.; Rawlinson, E.; Sun, P.; Narod, S.A. Pattern of metastatic spread in triple-negative breast cancer. Breast Cancer Res. Treat. 2009, 115, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Rakha, E.A.; Chan, S. Metastatic triple-negative breast cancer. Clin. Oncol (R Coll Radiol) 2011, 23, 587–600. [Google Scholar] [CrossRef]
- Schmadeka, R.; Harmon, B.E.; Singh, M. Triple-negative breast carcinoma: Current and emerging concepts. Am. J. Clin. Pathol. 2014, 141, 462–477. [Google Scholar] [CrossRef]
- Vacher, S.; Castagnet, P.; Chemlali, W.; Lallemand, F.; Meseure, D.; Pocard, M.; Bieche, I.; Perrot-Applanat, M. High ahr expression in breast tumors correlates with expression of genes from several signaling pathways namely inflammation and endogenous tryptophan metabolism. PLoS ONE 2018, 13, e0190619. [Google Scholar] [CrossRef]
- Trombino, A.F.; Near, R.I.; Matulka, R.A.; Yang, S.; Hafer, L.J.; Toselli, P.A.; Kim, D.W.; Rogers, A.E.; Sonenshein, G.E.; Sherr, D.H. Expression of the aryl hydrocarbon receptor/transcription factor (ahr) and ahr-regulated cyp1 gene transcripts in a rat model of mammary tumorigenesis. Breast Cancer Res. Treat. 2000, 63, 117–131. [Google Scholar] [CrossRef]
- Eltom, S.E.; Gasmelseed, A.A.; Saudoudi-Guentri, D. The aryl hydrocarbon receptor is over-expressed and constitutively activated in advanced breast carcinoma. Cancer Res. 2006, 66, 408. [Google Scholar]
- Li, Z.D.; Wang, K.; Yang, X.W.; Zhuang, Z.G.; Wang, J.J.; Tong, X.W. Expression of aryl hydrocarbon receptor in relation to p53 status and clinicopathological parameters in breast cancer. Int. J. Clin. Exp. Pathol. 2014, 7, 7931–7937. [Google Scholar]
- Saito, R.; Miki, Y.; Hata, S.; Takagi, K.; Iida, S.; Oba, Y.; Ono, K.; Ishida, T.; Suzuki, T.; Ohuchi, N.; et al. Aryl hydrocarbon receptor in breast cancer-a newly defined prognostic marker. Horm. Cancer 2014, 5, 11–21. [Google Scholar] [CrossRef]
- Brooks, J.; Eltom, S.E. Malignant transformation of mammary epithelial cells by ectopic overexpression of the aryl hydrocarbon receptor. Curr. Cancer Drug Targets 2011, 11, 654–669. [Google Scholar] [CrossRef] [PubMed]
- Goode, G.; Ballard, B.R.; Manning, H.C.; Freeman, M.L.; Kang, Y.; Eltom, S.E. Knockdown of aberrantly upregulated aryl hydrocarbon receptor reduces tumor growth and metastasis of mda-mb-231 human breast cancer cell line. Int. J. Cancer 2013, 133, 2769–2780. [Google Scholar] [CrossRef] [PubMed]
- Andlauer, W.; Kolb, J.; Stehle, P.; Furst, P. Absorption and metabolism of genistein in isolated rat small intestine. J. Nutr. 2000, 130, 843–846. [Google Scholar] [CrossRef] [PubMed]
- Andrade, J.E.; Twaddle, N.C.; Helferich, W.G.; Doerge, D.R. Absolute bioavailability of isoflavones from soy protein isolate-containing food in female balb/c mice. J. Agric. Food Chem. 2010, 58, 4529–4536. [Google Scholar] [CrossRef]
- Chen, J.; Wang, S.; Jia, X.; Bajimaya, S.; Lin, H.; Tam, V.H.; Hu, M. Disposition of flavonoids via recycling: Comparison of intestinal versus hepatic disposition. Drug Metab. Dispos. 2005, 33, 1777–1784. [Google Scholar] [CrossRef]
- Coldham, N.G.; Sauer, M.J. Pharmacokinetics of [(14)c]genistein in the rat: Gender-related differences, potential mechanisms of biological action, and implications for human health. Toxicol. Appl. Pharmacol. 2000, 164, 206–215. [Google Scholar] [CrossRef]
- Coldham, N.G.; Zhang, A.Q.; Key, P.; Sauer, M.J. Absolute bioavailability of [14c] genistein in the rat; plasma pharmacokinetics of parent compound, genistein glucuronide and total radioactivity. Eur J. Drug Metab. Pharmacokinet. 2002, 27, 249–258. [Google Scholar] [CrossRef]
- Hosoda, K.; Furuta, T.; Yokokawa, A.; Ogura, K.; Hiratsuka, A.; Ishii, K. Plasma profiling of intact isoflavone metabolites by high-performance liquid chromatography and mass spectrometric identification of flavone glycosides daidzin and genistin in human plasma after administration of kinako. Drug Metab. Dispos. 2008, 36, 1485–1495. [Google Scholar] [CrossRef]
- Yang, Z.; Zhu, W.; Gao, S.; Xu, H.; Wu, B.; Kulkarni, K.; Singh, R.; Tang, L.; Hu, M. Simultaneous determination of genistein and its four phase ii metabolites in blood by a sensitive and robust uplc-ms/ms method: Application to an oral bioavailability study of genistein in mice. J. Pharm. Biomed. Anal. 2010, 53, 81–89. [Google Scholar] [CrossRef]
- Hosoda, K.; Furuta, T.; Yokokawa, A.; Ishii, K. Identification and quantification of daidzein-7-glucuronide-4′-sulfate, genistein-7-glucuronide-4′-sulfate and genistein-4′,7-diglucuronide as major metabolites in human plasma after administration of kinako. Anal. Bioanal. Chem. 2010, 397, 1563–1572. [Google Scholar] [CrossRef]
- Chang, H.C.; Churchwell, M.I.; Delclos, K.B.; Newbold, R.R.; Doerge, D.R. Mass spectrometric determination of genistein tissue distribution in diet-exposed sprague-dawley rats. J. Nutr. 2000, 130, 1963–1970. [Google Scholar] [CrossRef] [PubMed]
- Warri, A.; Saarinen, N.M.; Makela, S.; Hilakivi-Clarke, L. The role of early life genistein exposures in modifying breast cancer risk. Br. J. Cancer 2008, 98, 1485–1493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novikov, O.; Wang, Z.; Stanford, E.A.; Parks, A.J.; Ramirez-Cardenas, A.; Landesman, E.; Laklouk, I.; Sarita-Reyes, C.; Gusenleitner, D.; Li, A.; et al. An aryl hydrocarbon receptor-mediated amplification loop that enforces cell migration in er(−)/pr(−)/her2(−) human breast cancer cells. Mol. Pharmacol. 2016, 90, 674–688. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.F.A.; Haarmann-Stemmann, T. The aryl hydrocarbon receptor repressor - more than a simple feedback inhibitor of ahr signaling: Clues for its role in inflammation and cancer. Curr. Opin. Toxicol. 2017, 2, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Holder, C.L.; Churchwell, M.I.; Doerge, D.R. Quantification of soy isoflavones, genistein and daidzein, and conjugates in rat blood using lc/es-ms. J. Agric. Food Chem. 1999, 47, 3764–3770. [Google Scholar] [CrossRef] [PubMed]
- Adlercreutz, H.; Fotsis, T.; Watanabe, S.; Lampe, J.; Wahala, K.; Makela, T.; Hase, T. Determination of lignans and isoflavonoids in plasma by isotope dilution gas chromatography-mass spectrometry. Cancer Detect. Prev. 1994, 18, 259–271. [Google Scholar]
- Pop, E.A.; Fischer, L.M.; Coan, A.D.; Gitzinger, M.; Nakamura, J.; Zeisel, S.H. Effects of a high daily dose of soy isoflavones on DNA damage, apoptosis and estrogenic outcomes in healthy, postmenopausal women—A phase i clinical trial. Menopause 2008, 15, 684–692. [Google Scholar] [CrossRef]
- Kasai, A.; Hiramatsu, N.; Hayakawa, K.; Yao, J.; Kitamura, M. Blockade of the dioxin pathway by herbal medicine formula bupleuri minor: Identification of active entities for suppression of ahr activation. Biol. Pharm. Bull. 2008, 31, 838–846. [Google Scholar] [CrossRef]
- Dunlap, T.L.; Howell, C.E.; Mukand, N.; Chen, S.N.; Pauli, G.F.; Dietz, B.M.; Bolton, J.L. Red clover aryl hydrocarbon receptor (ahr) and estrogen receptor (er) agonists enhance genotoxic estrogen metabolism. Chem. Res. Toxicol. 2017, 30, 2084–2092. [Google Scholar] [CrossRef]
- Zhang, S.; Qin, C.; Safe, S.H. Flavonoids as aryl hydrocarbon receptor agonists/antagonists: Effects of structure and cell context. Environ. Health Perspect. 2003, 111, 1877–1882. [Google Scholar] [CrossRef]
- Ciolino, H.P.; Yeh, G.C. The flavonoid galangin is an inhibitor of cyp1a1 activity and an agonist/antagonist of the aryl hydrocarbon receptor. Br. J. Cancer 1999, 79, 1340–1346. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Meeran, S.M.; Patel, S.N.; Chen, H.; Hardy, T.M.; Tollefsbol, T.O. Epigenetic reactivation of estrogen receptor-alpha (eralpha) by genistein enhances hormonal therapy sensitivity in eralpha-negative breast cancer. Mol. Cancer 2013, 12, 9. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Donovan, M.G.; Selmin, O.I.; Doetschman, T.C.; Romagnolo, D.F. Epigenetic Activation of BRCA1 by Genistein In Vivo and Triple Negative Breast Cancer Cells Linked to Antagonism toward Aryl Hydrocarbon Receptor. Nutrients 2019, 11, 2559. https://doi.org/10.3390/nu11112559
Donovan MG, Selmin OI, Doetschman TC, Romagnolo DF. Epigenetic Activation of BRCA1 by Genistein In Vivo and Triple Negative Breast Cancer Cells Linked to Antagonism toward Aryl Hydrocarbon Receptor. Nutrients. 2019; 11(11):2559. https://doi.org/10.3390/nu11112559
Chicago/Turabian StyleDonovan, Micah G., Ornella I. Selmin, Thomas C. Doetschman, and Donato F. Romagnolo. 2019. "Epigenetic Activation of BRCA1 by Genistein In Vivo and Triple Negative Breast Cancer Cells Linked to Antagonism toward Aryl Hydrocarbon Receptor" Nutrients 11, no. 11: 2559. https://doi.org/10.3390/nu11112559
APA StyleDonovan, M. G., Selmin, O. I., Doetschman, T. C., & Romagnolo, D. F. (2019). Epigenetic Activation of BRCA1 by Genistein In Vivo and Triple Negative Breast Cancer Cells Linked to Antagonism toward Aryl Hydrocarbon Receptor. Nutrients, 11(11), 2559. https://doi.org/10.3390/nu11112559