Dietary Cows’ Milk Protein A1 Beta-Casein Increases the Incidence of T1D in NOD Mice

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Animal Experiments
2.2. Breeding Program
2.3. Blood Glucose Monitoring and Diabetes Incidence
2.4. Intraperitoneal Glucose Tolerance Test
2.5. Insulin Tolerance Test
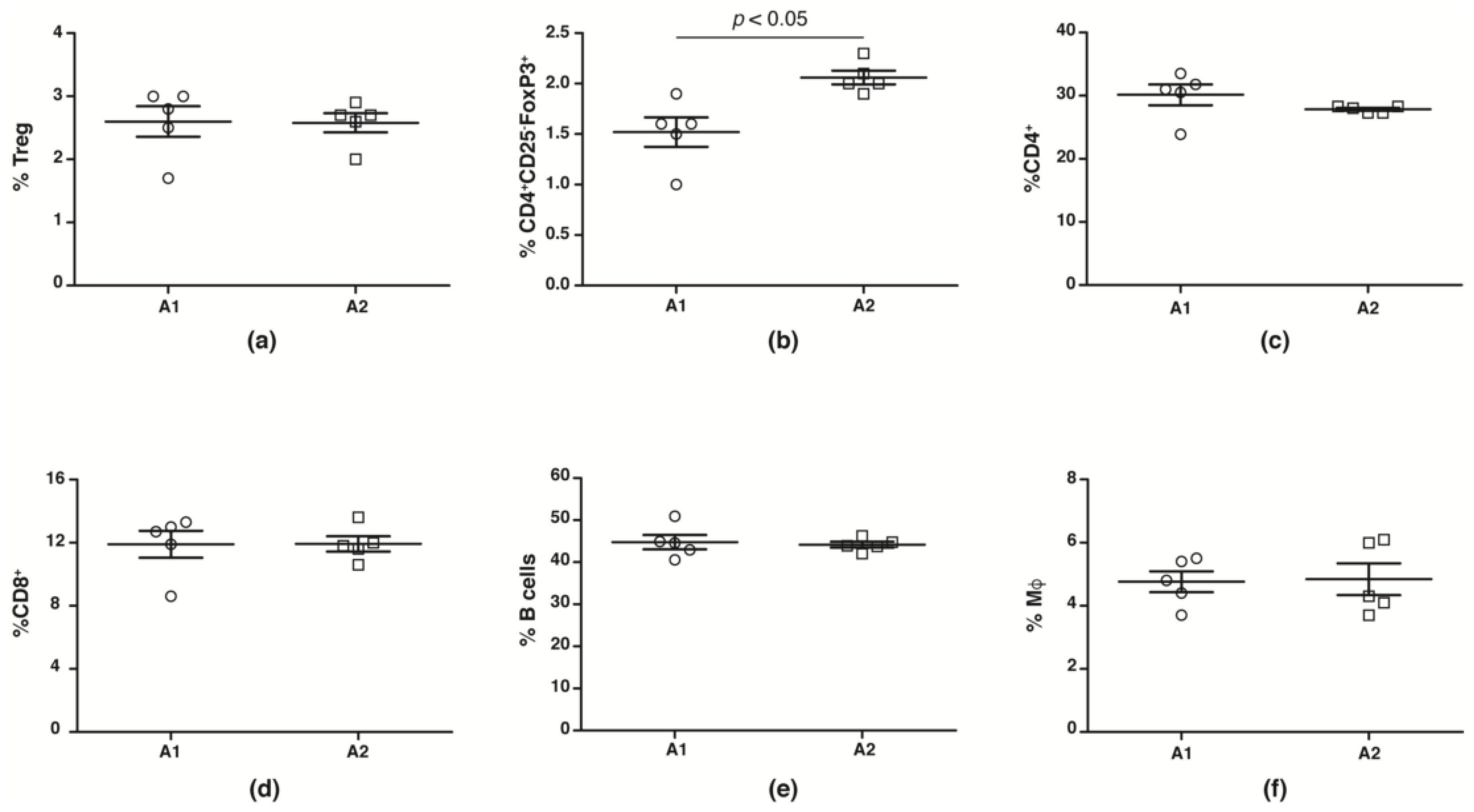
2.6. Immune Profiling
2.7. Treg Suppression Assays
2.8. Peptide Extraction from Whole Blood, Lymph Tissues and Mass Spectrometry
2.9. Bacterial DNA Preparation
2.10. High-Throughput Sequence Analysis
2.11. Histological Staining
2.12. Statistical Analysis
3. Results
3.1. Effect of A1 Beta-Casein Supplemented Diet on Incidence of T1D in NOD Mice
3.2. Isolation and Analysis of Peptides from Whole Blood and Lymph Tissues by Mass Spectrometry
3.3. The Effect of A1 Beta-Casein Supplemented Diet on Intestinal Microbial Communities of Female NOD Mice in the F0 Generation
3.4. A1 Beta-Casein Diet Consumption Did Not Alter the Gastrointestinal Integrity in Female NOD Mice in the F4 Generation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Jeker, L.T.; Bour-Jordan, H.; Bluestone, J.A. Breakdown in peripheral tolerance in type 1 diabetes in mice and humans. Cold Spring Harb. Perspect. Med. 2012, 2, a007807. [Google Scholar] [CrossRef] [PubMed]
- Mathis, D.; Vence, L.; Benoist, C. beta-Cell death during progression to diabetes. Nature 2001, 414, 792–798. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, M.A.; Eisenbarth, G.S.; Michels, A.W. Type 1 diabetes. Lancet 2014, 383, 69–82. [Google Scholar] [CrossRef]
- Polychronakos, C.; Li, Q. Understanding type 1 diabetes through genetics: Advances and prospects. Nat. Rev. Genet. 2011, 12, 781–792. [Google Scholar] [CrossRef] [PubMed]
- Burn, P. Type 1 diabetes. Nat. Rev. Drug Discov. 2010, 9, 187–188. [Google Scholar] [CrossRef] [PubMed]
- Patterson, C.C.; Dahlquist, G.G.; Gyurus, E.; Green, A.; Soltesz, G. Incidence trends for childhood type 1 diabetes in Europe during 1989–2003 and predicted new cases 2005-20: A multicentre prospective registration study. Lancet 2009, 373, 2027–2033. [Google Scholar] [CrossRef]
- Katsarou, A.; Gudbjornsdottir, S.; Rawshani, A.; Dabelea, D.; Bonifacio, E.; Anderson, B.J.; Jacobsen, L.M.; Schatz, D.A.; Lernmark, A. Type 1 diabetes mellitus. Nat. Rev. Dis. Primers 2017, 3, 17016. [Google Scholar] [CrossRef] [PubMed]
- Delli, A.J.; Lernmark, Å. Chapter 39–Type 1 (Insulin-Dependent) Diabetes Mellitus: Etiology, Pathogenesis, Prediction, and Prevention. In Endocrinology: Adult and Pediatric, 7 ed.; Jameson, J.L., De Groot, L.J., de Kretser, D.M., Giudice, L.C., Grossman, A.B., Melmed, S., Potts, J.T., Weir, G.C., Eds.; W.B. Saunders: Philadelphia, PA, USA, 2016; pp. 672–690. [Google Scholar] [CrossRef]
- Bluestone, J.A.; Herold, K.; Eisenbarth, G. Genetics, pathogenesis and clinical interventions in type 1 diabetes. Nature 2010, 464, 1293–1300. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, D.S.; Krych, L.; Buschard, K.; Hansen, C.H.; Hansen, A.K. Beyond genetics. Influence of dietary factors and gut microbiota on type 1 diabetes. FEBS Lett. 2014, 588, 4234–4243. [Google Scholar] [CrossRef] [PubMed]
- Rewers, M.; Ludvigsson, J. Environmental risk factors for type 1 diabetes. Lancet 2016, 387, 2340–2348. [Google Scholar] [CrossRef]
- Mustonen, N.; Siljander, H.; Peet, A.; Tillmann, V.; Harkonen, T.; Ilonen, J.; Hyoty, H.; Knip, M. Early childhood infections precede development of beta-cell autoimmunity and type 1 diabetes in children with HLA-conferred disease risk. Pediatr. Diabetes 2017. [CrossRef] [PubMed]
- Zhao, G.; Vatanen, T.; Droit, L.; Park, A.; Kostic, A.D.; Poon, T.W.; Vlamakis, H.; Siljander, H.; Harkonen, T.; Hamalainen, A.M.; et al. Intestinal virome changes precede autoimmunity in type I diabetes-susceptible children. Proc. Natl. Acad. Sci. USA 2017, 114, E6166–E6175. [Google Scholar] [CrossRef] [PubMed]
- Kostic, A.D.; Gevers, D.; Siljander, H.; Vatanen, T.; Hyotylainen, T.; Hamalainen, A.M.; Peet, A.; Tillmann, V.; Poho, P.; Mattila, I.; et al. The dynamics of the human infant gut microbiome in development and in progression toward type 1 diabetes. Cell Host Microbe 2015, 17, 260–273. [Google Scholar] [CrossRef] [PubMed]
- Wen, L.; Ley, R.E.; Volchkov, P.Y.; Stranges, P.B.; Avanesyan, L.; Stonebraker, A.C.; Hu, C.; Wong, F.S.; Szot, G.L.; Bluestone, J.A.; et al. Innate immunity and intestinal microbiota in the development of Type 1 diabetes. Nature 2008, 455, 1109–1113. [Google Scholar] [CrossRef] [PubMed]
- Herold, K.C.; Vignali, D.A.A.; Cooke, A.; Bluestone, J.A. Type I Diabetes: Translating Mechanistic Observations into Effective Clinical Outcomes. Nat. Rev. Immunol. 2013, 13, 243–256. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, A.G.; Rewers, M.; Simell, O.; Simell, T.; Lempainen, J.; Steck, A.; Winkler, C.; Ilonen, J.; Veijola, R.; Knip, M.; et al. Seroconversion to multiple islet autoantibodies and risk of progression to diabetes in children. JAMA 2013, 309, 2473–2479. [Google Scholar] [CrossRef] [PubMed]
- Krischer, J.P.; Lynch, K.F.; Schatz, D.A.; Ilonen, J.; Lernmark, A.; Hagopian, W.A.; Rewers, M.J.; She, J.X.; Simell, O.G.; Toppari, J.; et al. The 6 year incidence of diabetes-associated autoantibodies in genetically at-risk children: The TEDDY study. Diabetologia 2015, 58, 980–987. [Google Scholar] [CrossRef] [PubMed]
- Knip, M.; Simell, O. Environmental Triggers of Type 1 Diabetes. Cold Spring Harb. Perspect. Med. 2012, 2, a007690. [Google Scholar] [CrossRef] [PubMed]
- Chia, J.S.J.; McRae, J.L.; Kukuljan, S.; Woodford, K.; Elliott, R.B.; Swinburn, B.; Dwyer, K.M. A1 beta-casein milk protein and other environmental pre-disposing factors for type 1 diabetes. Nutr. Diabetes 2017, 7, e274. [Google Scholar] [CrossRef] [PubMed]
- Lamb, M.M.; Miller, M.; Seifert, J.A.; Frederiksen, B.; Kroehl, M.; Rewers, M.; Norris, J.M. The effect of childhood cow’s milk intake and HLA-DR genotype on risk of islet autoimmunity and type 1 diabetes: The Diabetes Autoimmunity Study in the Young. Pediatr. Diabetes 2015, 16, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Knip, M.; Virtanen, S.M.; Seppa, K.; Ilonen, J.; Savilahti, E.; Vaarala, O.; Reunanen, A.; Teramo, K.; Hamalainen, A.M.; Paronen, J.; et al. Dietary intervention in infancy and later signs of beta-cell autoimmunity. N. Engl. J. Med. 2010, 363, 1900–1908. [Google Scholar] [CrossRef] [PubMed]
- Kramer, M.S.; Kakuma, R. The optimal duration of exclusive breastfeeding: A systematic review. Adv. Exp. Med. Biol. 2004, 554, 63–77. [Google Scholar] [PubMed]
- Chowdhury, R.; Sinha, B.; Sankar, M.J.; Taneja, S.; Bhandari, N.; Rollins, N.; Bahl, R.; Martines, J. Breastfeeding and maternal health outcomes: A systematic review and meta-analysis. Acta Paediatr. 2015, 104, 96–113. [Google Scholar] [CrossRef] [PubMed]
- Haug, A.; Hostmark, A.T.; Harstad, O.M. Bovine milk in human nutrition—A review. Lipids Health Dis. 2007, 6, 25. [Google Scholar] [CrossRef] [PubMed]
- Huppertz, T.; Fox, P.F.; Kelly, A.L. 3–The caseins: Structure, stability, and functionality A2–Yada, Rickey Y. In Proteins in Food Processing, 7th ed.; Woodhead Publishing: Cambridge, UK, 2018; pp. 49–92. [Google Scholar]
- Kaminski, S.; Cieslinska, A.; Kostyra, E. Polymorphism of bovine beta-casein and its potential effect on human health. J. Appl. Genet. 2007, 48, 189–198. [Google Scholar] [CrossRef] [PubMed]
- De Noni, R.J.; FitzGerald, H.J.T.; Korhonen, Y.; Le Roux, C.T.; Livesey, I.; Thorsdottir, D.; Tomé, R.W. Scientific Report of EFSA prepared by a DATEX Working Group on the potential health impact of β-casomorphins and related peptides. Eur. Food Saf. Auth. 2009, 231, 1–107. [Google Scholar]
- Brantl, V.; Teschemacher, H.; Henschen, A.; Lottspeich, F. Novel opioid peptides derived from casein (beta-casomorphins). I. Isolation from bovine casein peptone. Hoppe-Seyler Z. Physiol. Chem. 1979, 360, 1211–1216. [Google Scholar] [CrossRef] [PubMed]
- Henschen, A.; Lottspeich, F.; Brantl, V.; Teschemacher, H. Novel opioid peptides derived from casein (beta-casomorphins). II. Structure of active components from bovine casein peptone. Hoppe-Seyler Z. Physiol. Chem. 1979, 360, 1217–1224. [Google Scholar] [PubMed]
- Boutrou, R.; Gaudichon, C.; Dupont, D.; Jardin, J.; Airinei, G.; Marsset-Baglieri, A.; Benamouzig, R.; Tome, D.; Leonil, J. Sequential release of milk protein-derived bioactive peptides in the jejunum in healthy humans. Am. J. Clin. Nutr. 2013, 97, 1314–1323. [Google Scholar] [CrossRef] [PubMed]
- Clemens, R.A. Milk A1 and A2 peptides and diabetes. Nestle Nutr. Workshop Ser. Paediatr. Prog. 2011, 67, 187–195. [Google Scholar] [CrossRef]
- Elliott, R.B.; Harris, D.P.; Hill, J.P.; Bibby, N.J.; Wasmuth, H.E. Type I (insulin-dependent) diabetes mellitus and cow milk: Casein variant consumption. Diabetologia 1999, 42, 292–296. [Google Scholar] [CrossRef] [PubMed]
- Knip, M. Environmental triggers and determinants of beta-cell autoimmunity and type 1 diabetes. Rev. Endocr. Metab. Disord. 2003, 4, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Harrison, L.C.; Honeyman, M.C. Cow’s milk and type 1 diabetes: The real debate is about mucosal immune function. Diabetes 1999, 48, 1501–1507. [Google Scholar] [CrossRef] [PubMed]
- Laugesen, M.; Elliott, R. Ischaemic heart disease, Type 1 diabetes, and cow milk A1 beta-casein. N. Zeal. Med. J. 2003, 116, U295. [Google Scholar]
- Elliott, R.B.; Reddy, S.N.; Bibby, N.J.; Kida, K. Dietary prevention of diabetes in the non-obese diabetic mouse. Diabetologia 1988, 31, 62–64. [Google Scholar] [PubMed]
- Elliott, R.B.; Wasmuth, W.H.; Bibby, N.J.; Hill, J.P. The role of β-casein variants in the induction of insulin-dependent diabetes in the non-obese diabetic mouse and humans. In Milk Protein Polymorphism; IDF Special Issue No. 9702; Food and Agriculture Organization: Brussels, Belgium, 1997; pp. 445–453. [Google Scholar]
- Johnsen, E.; Leknes, S.; Wilson, S.R.; Lundanes, E. Liquid chromatography-mass spectrometry platform for both small neurotransmitters and neuropeptides in blood, with automatic and robust solid phase extraction. Sci. Rep. 2015, 5, 9308. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Song, L.; Breitwieser, F.P.; Salzberg, S.L. Centrifuge: Rapid and sensitive classification of metagenomic sequences. Genome Res. 2016, 26, 1721–1729. [Google Scholar] [CrossRef] [PubMed]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed]
- Love, M.; Anders, S.; Huber, W. Differential Analysis of Count Data–The deseq2 Package. Genome Biol. 2014, 15, 10–1186. [Google Scholar]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Brusko, T.; Wasserfall, C.; McGrail, K.; Schatz, R.; Viener, H.L.; Schatz, D.; Haller, M.; Rockell, J.; Gottlieb, P.; Clare-Salzler, M.; et al. No alterations in the frequency of FOXP3+ regulatory T-cells in type 1 diabetes. Diabetes 2007, 56, 604–612. [Google Scholar] [CrossRef] [PubMed]
- Brusko, T.M.; Wasserfall, C.H.; Clare-Salzler, M.J.; Schatz, D.A.; Atkinson, M.A. Functional defects and the influence of age on the frequency of CD4+CD25+ T-cells in type 1 diabetes. Diabetes 2005, 54, 1407–1414. [Google Scholar] [CrossRef] [PubMed]
- Lindley, S.; Dayan, C.M.; Bishop, A.; Roep, B.O.; Peakman, M.; Tree, T.I. Defective suppressor function in CD4+CD25+ T-cells from patients with type 1 diabetes. Diabetes 2005, 54, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Ryba-Stanislawowska, M.; Rybarczyk-Kapturska, K.; Mysliwiec, M.; Mysliwska, J. Elevated levels of serum IL-12 and IL-18 are associated with lower frequencies of CD4+CD25highFOXP3+ regulatory t cells in young patients with type 1 diabetes. Inflammation 2014, 37, 1513–1520. [Google Scholar] [CrossRef] [PubMed]
- Haseda, F.; Imagawa, A.; Murase-Mishiba, Y.; Terasaki, J.; Hanafusa, T. CD4+CD45RA−FoxP3high activated regulatory T cells are functionally impaired and related to residual insulin-secreting capacity in patients with type 1 diabetes. Clin. Exp. Immunol. 2013, 173, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.D.; McGeer, A.; Schwartz, B.; Green, K.; Cann, D.; Simor, A.E.; Low, D.E. Invasive Group A Streptococcal Infections in Ontario, Canada. N. Engl. J. Med. 1996, 335, 547–554. [Google Scholar] [CrossRef] [PubMed]
- Hughes, J.M.; Wilson, M.E.; Wertheim, H.F.L.; Nghia, H.D.T.; Taylor, W.; Schultsz, C. Streptococcus suis: An Emerging Human Pathogen. Clin. Infect. Dis. 2009, 48, 617–625. [Google Scholar] [CrossRef]
- Vaarala, O.; Atkinson, M.A.; Neu, J. The “perfect storm” for type 1 diabetes: The complex interplay between intestinal microbiota, gut permeability, and mucosal immunity. Diabetes 2008, 57, 2555–2562. [Google Scholar] [CrossRef] [PubMed]
- Scott, F.W. Cow milk and insulin-dependent diabetes mellitus: Is there a relationship? Am. J. Clin. Nutr. 1990, 51, 489–491. [Google Scholar] [CrossRef] [PubMed]
- Dahl-Jorgensen, K.; Joner, G.; Hanssen, K.F. Relationship between cows’ milk consumption and incidence of IDDM in childhood. Diabetes Care 1991, 14, 1081–1083. [Google Scholar] [CrossRef] [PubMed]
- Pozzilli, P.; Signore, A.; Williams, A.J.; Beales, P.E. NOD mouse colonies around the world--recent facts and figures. Immunol. Today 1993, 14, 193–196. [Google Scholar] [CrossRef]
- Markle, J.G.; Frank, D.N.; Mortin-Toth, S.; Robertson, C.E.; Feazel, L.M.; Rolle-Kampczyk, U.; von Bergen, M.; McCoy, K.D.; Macpherson, A.J.; Danska, J.S. Sex differences in the gut microbiome drive hormone-dependent regulation of autoimmunity. Science 2013, 339, 1084–1088. [Google Scholar] [CrossRef] [PubMed]
- Pearson, J.A.; Wong, F.S.; Wen, L. The importance of the Non Obese Diabetic (NOD) mouse model in autoimmune diabetes. J. Autoimmun. 2016, 66, 76–88. [Google Scholar] [CrossRef] [PubMed]
- Jerram, S.T.; Dang, M.N.; Leslie, R.D. The Role of Epigenetics in Type 1 Diabetes. Curr. Diabetes Rep. 2017, 17, 89. [Google Scholar] [CrossRef] [PubMed]
- Dang, M.N.; Buzzetti, R.; Pozzilli, P. Epigenetics in autoimmune diseases with focus on type 1 diabetes. Diabetes/Metab. Res. Rev. 2013, 29, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Huypens, P.; Sass, S.; Wu, M.; Dyckhoff, D.; Tschop, M.; Theis, F.; Marschall, S.; de Angelis, M.H.; Beckers, J. Epigenetic germline inheritance of diet-induced obesity and insulin resistance. Nat. Genet. 2016, 48, 497–499. [Google Scholar] [CrossRef] [PubMed]
- Krautkramer, K.A.; Kreznar, J.H.; Romano, K.A.; Vivas, E.I.; Barrett-Wilt, G.A.; Rabaglia, M.E.; Keller, M.P.; Attie, A.D.; Rey, F.E.; Denu, J.M. Diet-Microbiota Interactions Mediate Global Epigenetic Programming in Multiple Host Tissues. Mol. Cell 2016, 64, 982–992. [Google Scholar] [CrossRef] [PubMed]
- Bonelli, M.; Savitskaya, A.; Steiner, C.W.; Rath, E.; Smolen, J.S.; Scheinecker, C. Phenotypic and functional analysis of CD4+CD25−Foxp3+ T cells in patients with systemic lupus erythematosus. J. Immunol. 2009, 182, 1689–1695. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.X.; Zhang, W.; Zhao, L.D.; Li, Y.; Zhang, F.C.; Tang, F.L.; He, W.; Zhang, X. Are CD4+CD25−Foxp3+ cells in untreated new-onset lupus patients regulatory T cells? Arthritis Res. Ther. 2009, 11, R153. [Google Scholar] [CrossRef] [PubMed]
- DuPage, M.; Bluestone, J.A. Harnessing the plasticity of CD4+ T cells to treat immune-mediated disease. Nat. Rev. Immunol. 2016, 16, 149–163. [Google Scholar] [CrossRef] [PubMed]
- Zelenay, S.; Lopes-Carvalho, T.; Caramalho, I.; Moraes-Fontes, M.F.; Rebelo, M.; Demengeot, J. Foxp3+ CD25−CD4 T cells constitute a reservoir of committed regulatory cells that regain CD25 expression upon homeostatic expansion. Proc. Natl. Acad. Sci. USA 2005, 102, 4091–4096. [Google Scholar] [CrossRef] [PubMed]
- Svedberg, J.; de Haas, J.; Leimenstoll, G.; Paul, F.; Teschemacher, H. Demonstration of beta-casomorphin immunoreactive materials in in vitro digests of bovine milk and in small intestine contents after bovine milk ingestion in adult humans. Peptides 1985, 6, 825–830. [Google Scholar] [CrossRef]
- Umbach, M.; Teschemacher, H.; Praetorius, K.; Hirschhauser, R.; Bostedt, H. Demonstration of a beta-casomorphin immunoreactive material in the plasma of newborn calves after milk intake. Regul. Pept. 1985, 12, 223–230. [Google Scholar] [CrossRef]
- Barbé, F.; Le Feunteun, S.; Rémond, D.; Ménard, O.; Jardin, J.; Henry, G.; Laroche, B.; Dupont, D. Tracking the in vivo release of bioactive peptides in the gut during digestion: Mass spectrometry peptidomic characterization of effluents collected in the gut of dairy matrix fed mini-pigs. Food Res. Int. 2014, 63, 147–156. [Google Scholar] [CrossRef]
- Brooke-Taylor, S.; Dwyer, K.; Woodford, K.; Kost, N. Systematic Review of the Gastrointestinal Effects of A1 Compared with A2 beta-Casein. Adv. Nutr. 2017, 8, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Ul Haq, M.R.; Kapila, R.; Sharma, R.; Saliganti, V.; Kapila, S. Comparative evaluation of cow beta-casein variants (A1/A2) consumption on Th2-mediated inflammatory response in mouse gut. Eur. J. Nutr. 2014, 53, 1039–1049. [Google Scholar] [CrossRef] [PubMed]
- Barnett, M.P.; McNabb, W.C.; Roy, N.C.; Woodford, K.B.; Clarke, A.J. Dietary A1 beta-casein affects gastrointestinal transit time, dipeptidyl peptidase-4 activity, and inflammatory status relative to A2 beta-casein in Wistar rats. Int. J. Food Sci. Nutr. 2014, 65, 720–727. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Lin, X.; Zhao, F.; Shi, X.; Li, H.; Li, Y.; Zhu, W.; Xu, X.; Li, C.; Zhou, G. Meat, dairy and plant proteins alter bacterial composition of rat gut bacteria. Sci. Rep. 2015, 5, 15220. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Lin, X.; Li, H.; Li, Y.; Shi, X.; Zhao, F.; Xu, X.; Li, C.; Zhou, G. Intake of Meat Proteins Substantially Increased the Relative Abundance of Genus Lactobacillus in Rat Feces. PLoS ONE 2016, 11, e0152678. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.F.; Majid, N.A.; Yamaoka, Y.; Lee, Y.Y. Food Allergy and Helicobacter pylori Infection: A Systematic Review. Front. Microbiol. 2016, 7, 368. [Google Scholar] [CrossRef] [PubMed]
- Kolho, K.L.; Haapaniemi, A.; Haahtela, T.; Rautelin, H. Helicobacter pylori and specific immunoglobulin E antibodies to food allergens in children. J. Pediatr. Gastroenterol. Nutr. 2005, 40, 180–183. [Google Scholar] [CrossRef] [PubMed]
- Pozzilli, P. Beta-casein in cow’s milk: A major antigenic determinant for type 1 diabetes? J. Endocrinol. Investig. 1999, 22, 562–567. [Google Scholar] [CrossRef]
- Inman, L.R.; McAllister, C.T.; Chen, L.; Hughes, S.; Newgard, C.B.; Kettman, J.R.; Unger, R.H.; Johnson, J.H. Autoantibodies to the GLUT-2 glucose transporter of beta cells in insulin-dependent diabetes mellitus of recent onset. Proc. Natl. Acad. Sci. USA 1993, 90, 1281–1284. [Google Scholar] [CrossRef] [PubMed]
- Monetini, L.; Barone, F.; Stefanini, L.; Petrone, A.; Walk, T.; Jung, G.; Thorpe, R.; Pozzilli, P.; Cavallo, M.G. Establishment of T cell lines to bovine beta-casein and beta-casein-derived epitopes in patients with type 1 diabetes. J. Endocrinol. 2003, 176, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Kar, S.K.; Jansman, A.J.M.; Benis, N.; Ramiro-Garcia, J.; Schokker, D.; Kruijt, L.; Stolte, E.H.; Taverne-Thiele, J.J.; Smits, M.A.; Wells, J.M. Dietary protein sources differentially affect microbiota, mTOR activity and transcription of mTOR signaling pathways in the small intestine. PLoS ONE 2017, 12, e0188282. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Deng, Y.; Feng, Y.; Long, D.; Ma, K.; Wang, X.; Zhao, M.; Lu, L.; Lu, Q. Epigenetic regulation in B-cell maturation and its dysregulation in autoimmunity. Cell. Mol. Immunol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Dalgleish, D.G.; Corredig, M. The structure of the casein micelle of milk and its changes during processing. Annu. Rev. Food Sci. Technol. 2012, 3, 449–467. [Google Scholar] [CrossRef] [PubMed]
- De Kruif, C.G.; Huppertz, T. Casein micelles: Size distribution in milks from individual cows. J. Agric. Food Chem. 2012, 60, 4649–4655. [Google Scholar] [CrossRef] [PubMed]
- Raynes, J.K.; Day, L.; Augustin, M.A.; Carver, J.A. Structural differences between bovine A(1) and A(2) beta-casein alter micelle self-assembly and influence molecular chaperone activity. J. Dairy Sci. 2015, 98, 2172–2182. [Google Scholar] [CrossRef] [PubMed]
- Gerstein, H.C.; VanderMeulen, J. The relationship between cow’s milk exposure and type 1 diabetes. Diabet. Med. 1996, 13, 23–29. [Google Scholar] [CrossRef]
- Virtanen, S.M.; Hypponen, E.; Laara, E.; Vahasalo, P.; Kulmala, P.; Savola, K.; Rasanen, L.; Aro, A.; Knip, M.; Akerblom, H.K. Cow’s milk consumption, disease-associated autoantibodies and type 1 diabetes mellitus: A follow-up study in siblings of diabetic children. Childhood Diabetes in Finland Study Group. Diabet. Med. 1998, 15, 730–738. [Google Scholar] [CrossRef]
- Birgisdottir, B.E.; Hill, J.P.; Harris, D.P.; Thorsdottir, I. Variation in consumption of cow milk proteins and lower incidence of Type 1 diabetes in Iceland vs the other 4 Nordic countries. Diabetes Nutr. Metab. 2002, 15, 240–245. [Google Scholar] [PubMed]
- Beales, P.E.; Elliott, R.B.; Flohe, S.; Hill, J.P.; Kolb, H.; Pozzilli, P.; Wang, G.S.; Wasmuth, H.; Scott, F.W. A multi-centre, blinded international trial of the effect of A(1) and A(2) beta-casein variants on diabetes incidence in two rodent models of spontaneous Type I diabetes. Diabetologia 2002, 45, 1240–1246. [Google Scholar] [CrossRef] [PubMed]
- Butalia, S.; Kaplan, G.G.; Khokhar, B.; Rabi, D.M. Environmental Risk Factors and Type 1 Diabetes: Past, Present, and Future. Can. J. Diabetes 2016, 40, 586–593. [Google Scholar] [CrossRef] [PubMed]

 ) and A2 (
) and A2 ( ) beta-casein fed female mice. BGLs of female mice assessed in (c) glucose and (d) insulin tolerance tests. (e) distribution of insulitis in islets represented as a percentage of islet infiltration in 10-week old female mice fed either A1 or A2 beta-casein supplemented diets. Islets were scored blindly from individual mice. Islet inflammation was calculated on a scale from 0 to 4. 0—islets devoid of mononuclear cells; 1—minimal (<10%) focal islet infiltrate; 2—peri-islet infiltrate in <25% of islet circumference; 3—peri-islet infiltrate in >25% but <50% intra-islet and 4—>50% intra-islet infiltration.
) and A2 () beta-casein fed female mice. BGLs of female mice assessed in (c) glucose and (d) insulin tolerance tests. (e) distribution of insulitis in islets represented as a percentage of islet infiltration in 10-week old female mice fed either A1 or A2 beta-casein supplemented diets. Islets were scored blindly from individual mice. Islet inflammation was calculated on a scale from 0 to 4. 0—islets devoid of mononuclear cells; 1—minimal (<10%) focal islet infiltrate; 2—peri-islet infiltrate in <25% of islet circumference; 3—peri-islet infiltrate in >25% but <50% intra-islet and 4—>50% intra-islet infiltration.
) beta-casein fed female mice. BGLs of female mice assessed in (c) glucose and (d) insulin tolerance tests. (e) distribution of insulitis in islets represented as a percentage of islet infiltration in 10-week old female mice fed either A1 or A2 beta-casein supplemented diets. Islets were scored blindly from individual mice. Islet inflammation was calculated on a scale from 0 to 4. 0—islets devoid of mononuclear cells; 1—minimal (<10%) focal islet infiltrate; 2—peri-islet infiltrate in <25% of islet circumference; 3—peri-islet infiltrate in >25% but <50% intra-islet and 4—>50% intra-islet infiltration.
) and A2 () beta-casein fed female mice. BGLs of female mice assessed in (c) glucose and (d) insulin tolerance tests. (e) distribution of insulitis in islets represented as a percentage of islet infiltration in 10-week old female mice fed either A1 or A2 beta-casein supplemented diets. Islets were scored blindly from individual mice. Islet inflammation was calculated on a scale from 0 to 4. 0—islets devoid of mononuclear cells; 1—minimal (<10%) focal islet infiltrate; 2—peri-islet infiltrate in <25% of islet circumference; 3—peri-islet infiltrate in >25% but <50% intra-islet and 4—>50% intra-islet infiltration.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ingredients (g/100 g) | A1A1 Skim Milk Diet | A2A2 Skim Milk Diet |
|---|---|---|
| Sucrose | 25.753 | 25.753 |
| Skim milk powder (as supplement) | 60.528 | 60.529 |
| Instruction | Standard mixing | |
| COPHA hydrogenated vegetable oil | 1.366 | 1.366 |
| Palm oil | 3.709 | 3.709 |
| Safflower oil (High Linoleic) | 0.787 | 0.787 |
| Flax oil | 0.462 | 0.462 |
| Instructions | Standard mixing | |
| Cellulose | 5.000 | 5.000 |
| Instructions | Standard mixing | |
| dl Methionine | 0.904 | 0.904 |
| A1N_93_Trace minerals | 0.140 | 0.140 |
| Salt (Fine sodium chloride) | 0.100 | 0.100 |
| AIN_93_Vitamins | 1.000 | 1.000 |
| Choline chloride 75% w/w | 0.250 | 0.250 |
| Red food colour | 0.001 | - |
| Instructions | Standard mixing | |
| Total | 100.000 | 100.000 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chia, J.S.J.; McRae, J.L.; Enjapoori, A.K.; Lefèvre, C.M.; Kukuljan, S.; Dwyer, K.M. Dietary Cows’ Milk Protein A1 Beta-Casein Increases the Incidence of T1D in NOD Mice. Nutrients 2018, 10, 1291. https://doi.org/10.3390/nu10091291
Chia JSJ, McRae JL, Enjapoori AK, Lefèvre CM, Kukuljan S, Dwyer KM. Dietary Cows’ Milk Protein A1 Beta-Casein Increases the Incidence of T1D in NOD Mice. Nutrients. 2018; 10(9):1291. https://doi.org/10.3390/nu10091291
Chicago/Turabian StyleChia, Joanne S. J., Jennifer L. McRae, Ashwantha Kumar Enjapoori, Christophe M. Lefèvre, Sonja Kukuljan, and Karen M. Dwyer. 2018. "Dietary Cows’ Milk Protein A1 Beta-Casein Increases the Incidence of T1D in NOD Mice" Nutrients 10, no. 9: 1291. https://doi.org/10.3390/nu10091291
APA StyleChia, J. S. J., McRae, J. L., Enjapoori, A. K., Lefèvre, C. M., Kukuljan, S., & Dwyer, K. M. (2018). Dietary Cows’ Milk Protein A1 Beta-Casein Increases the Incidence of T1D in NOD Mice. Nutrients, 10(9), 1291. https://doi.org/10.3390/nu10091291