Lactobacillus acidophilus DDS-1 Modulates the Gut Microbiota and Improves Metabolic Profiles in Aging Mice

 , ,
, ,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Methods
2.1. Ethics Statement
2.2. Bacterial Culture and Probiotic Feed Preparation
2.3. Animals and Probiotic Treatments
2.4. Fecal Sample Collection and Preparation
2.5. Microbiota Analysis Using 16S rRNA High-Throughput Sequencing
2.6. Fecal Metabolomics
2.7. Data Analysis and Multivariate Analysis
2.8. Statistical Analysis
3. Results
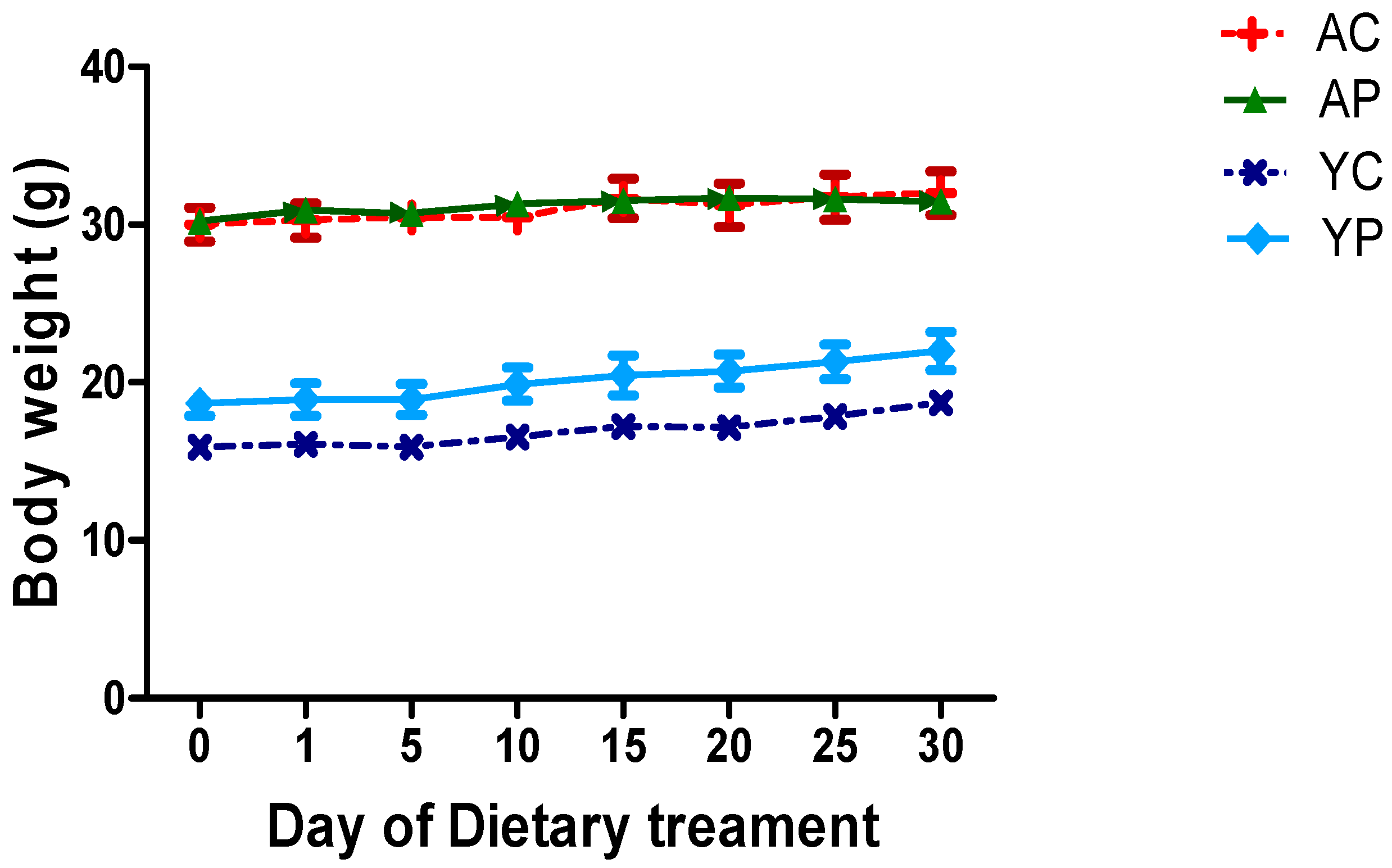
3.1. Body Weight During Days of Treatments
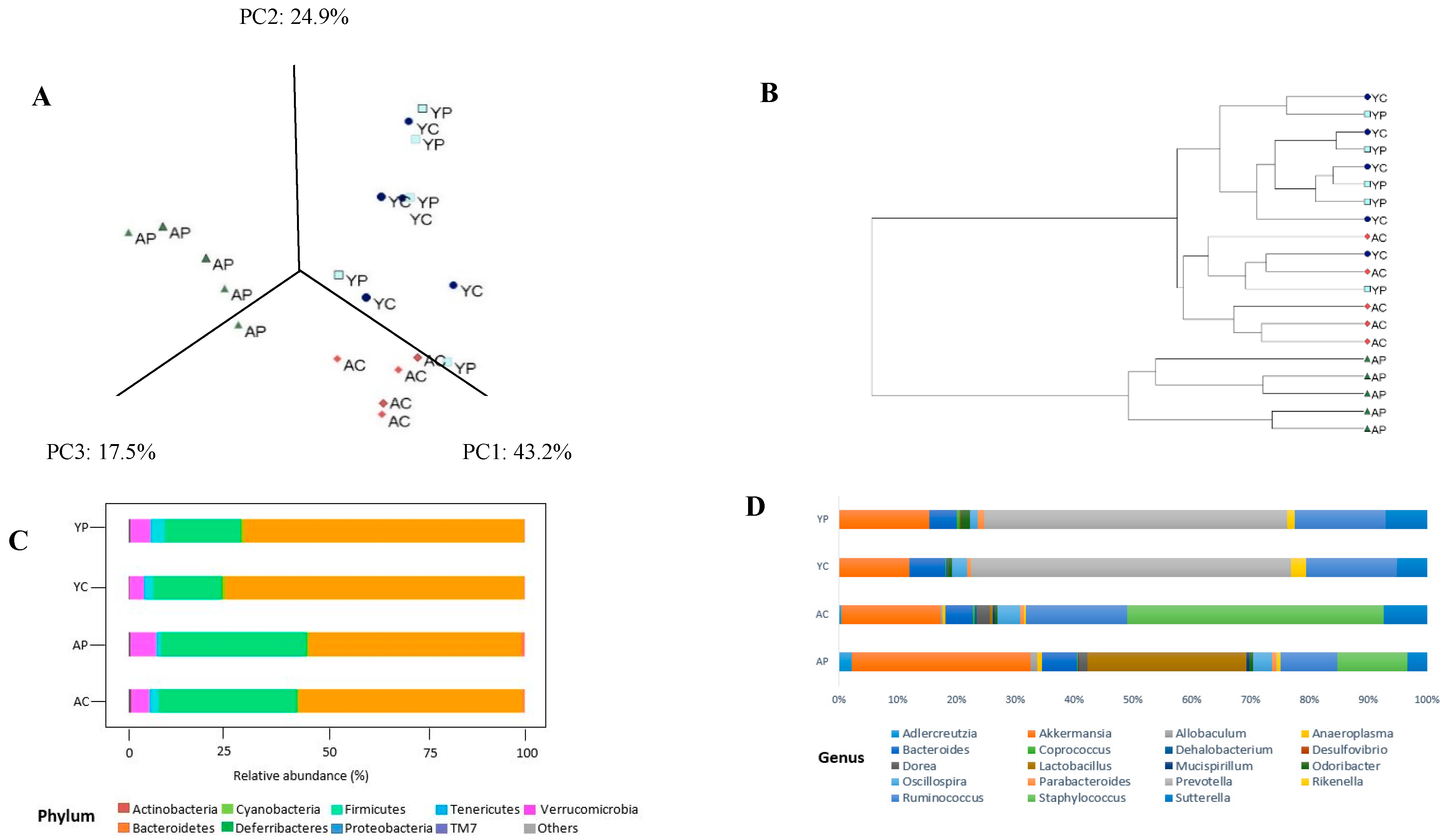
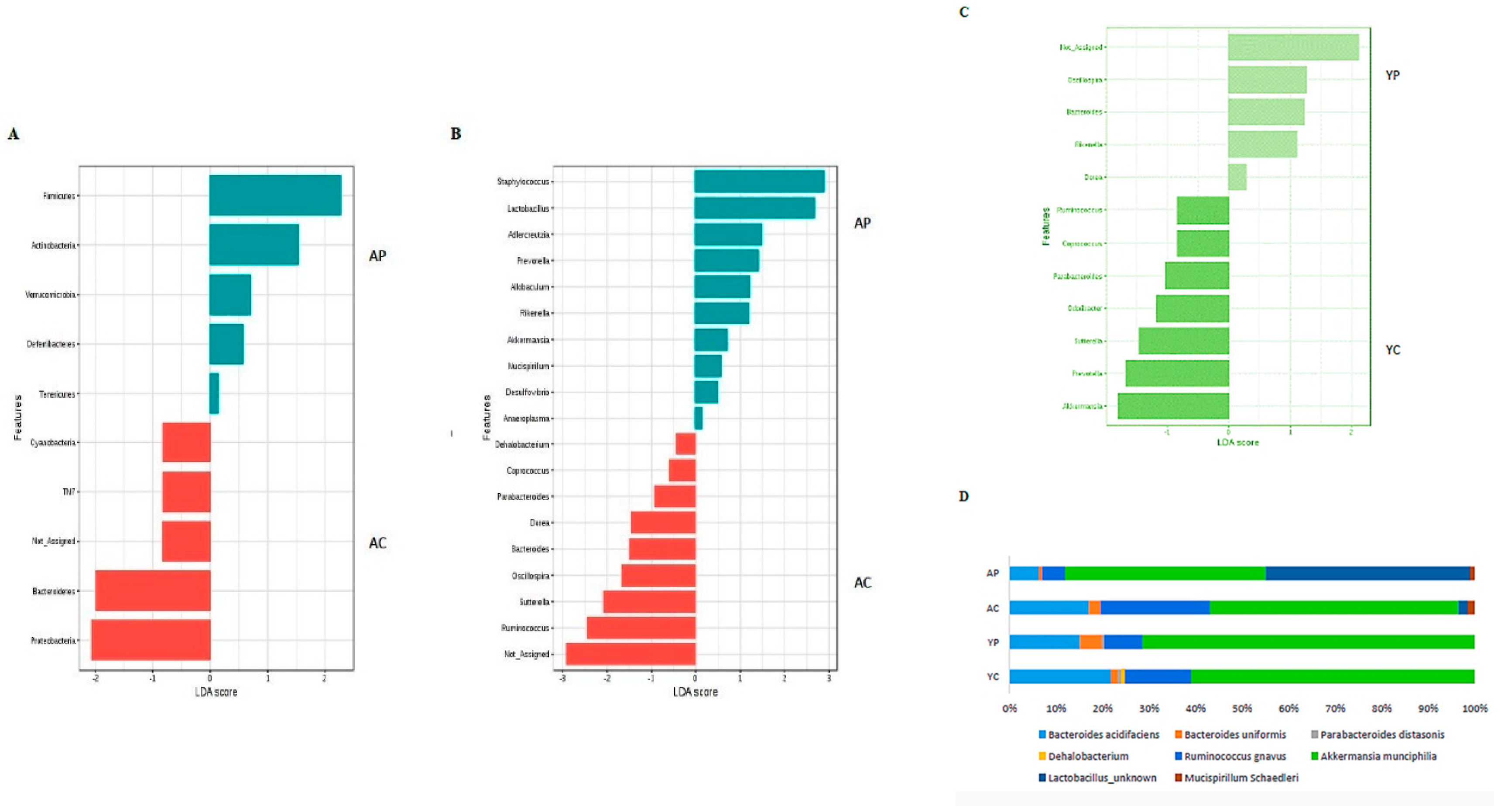
3.2. Gut Microbial Changes at the Phylum Level
3.3. Gut Microbial Changes at Genus and Species Level Among Four Groups
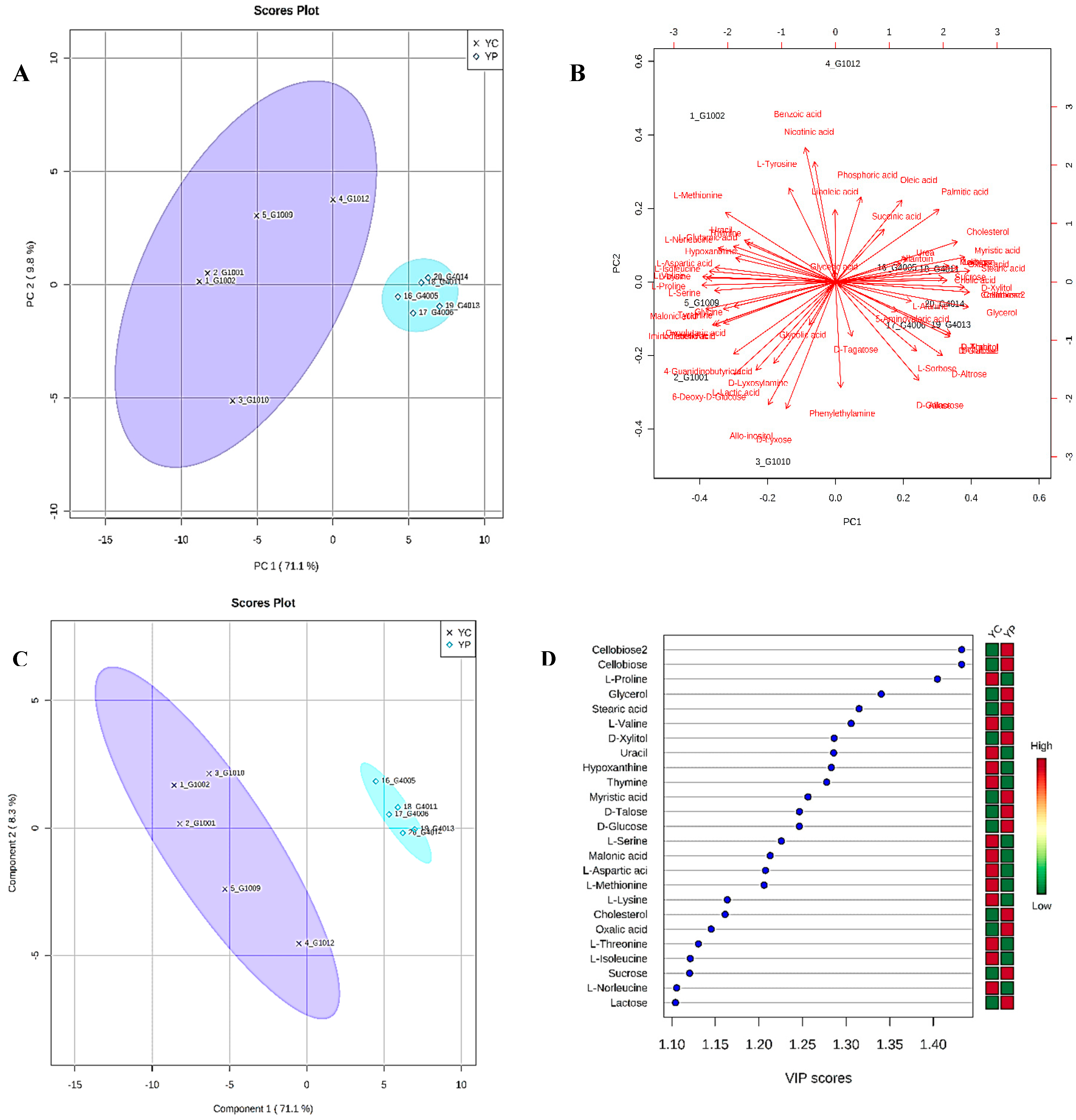
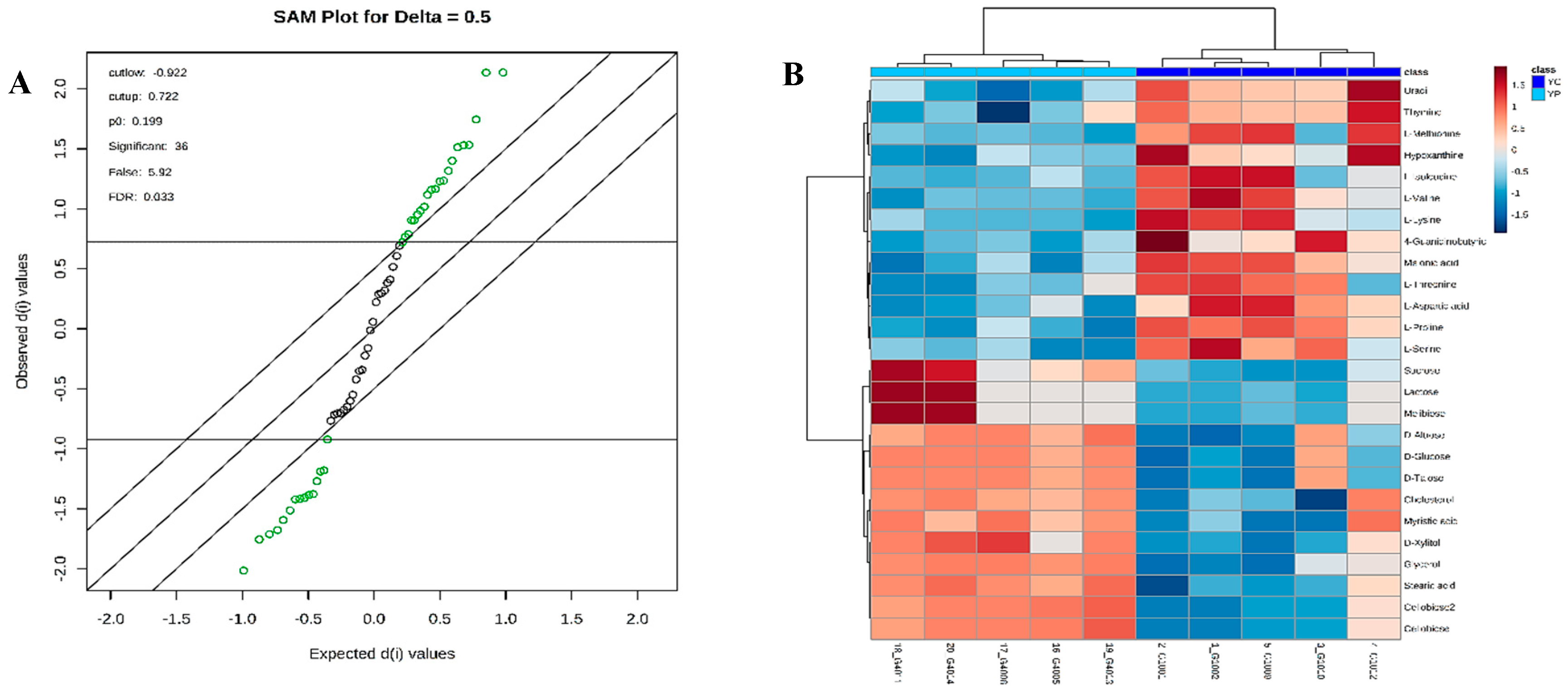
3.4. Metabolic Phenotyping of YC and YP Groups
3.5. Identification of Potential Fecal Metabolites Associated with the YP/YC Groups
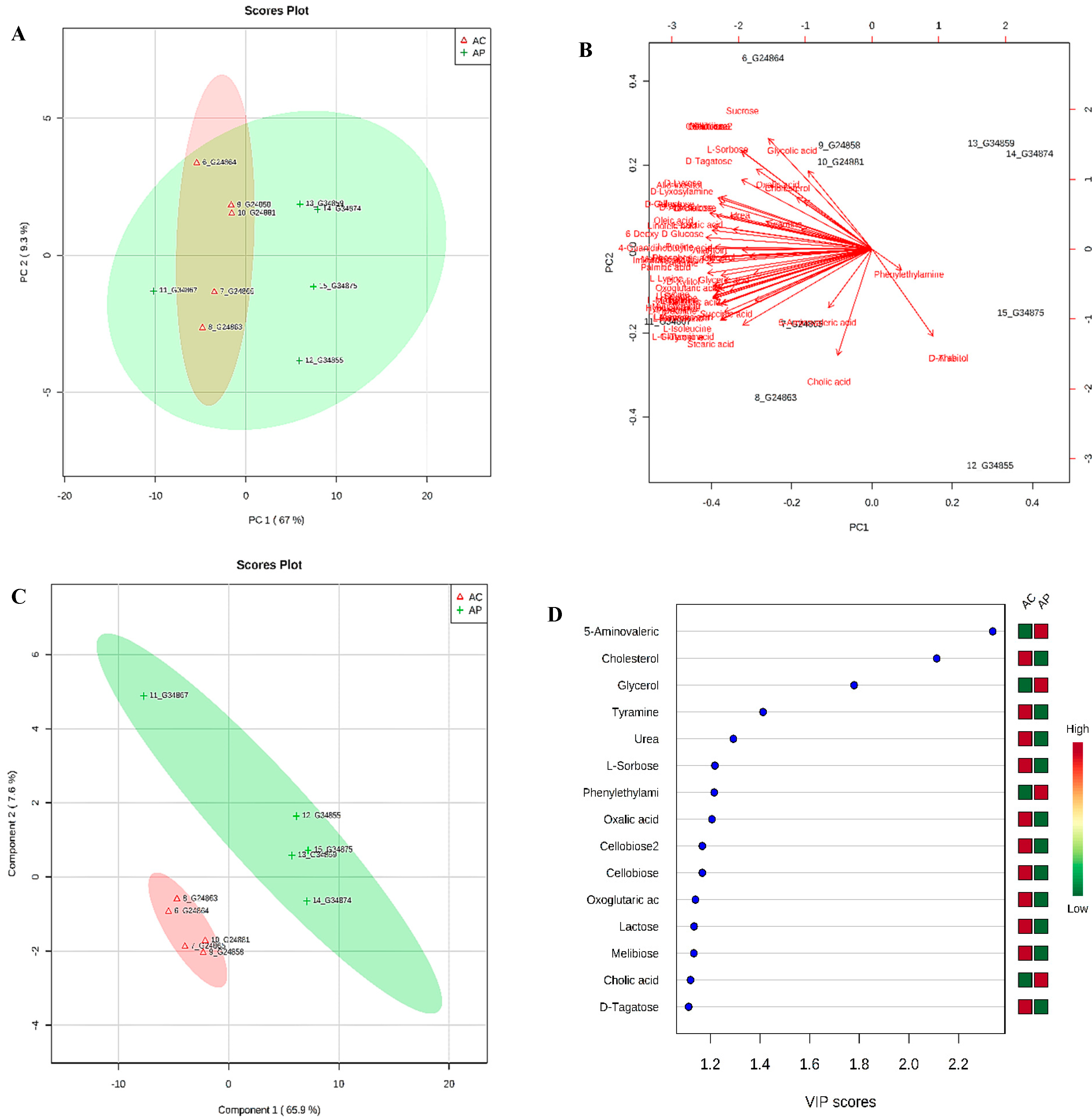
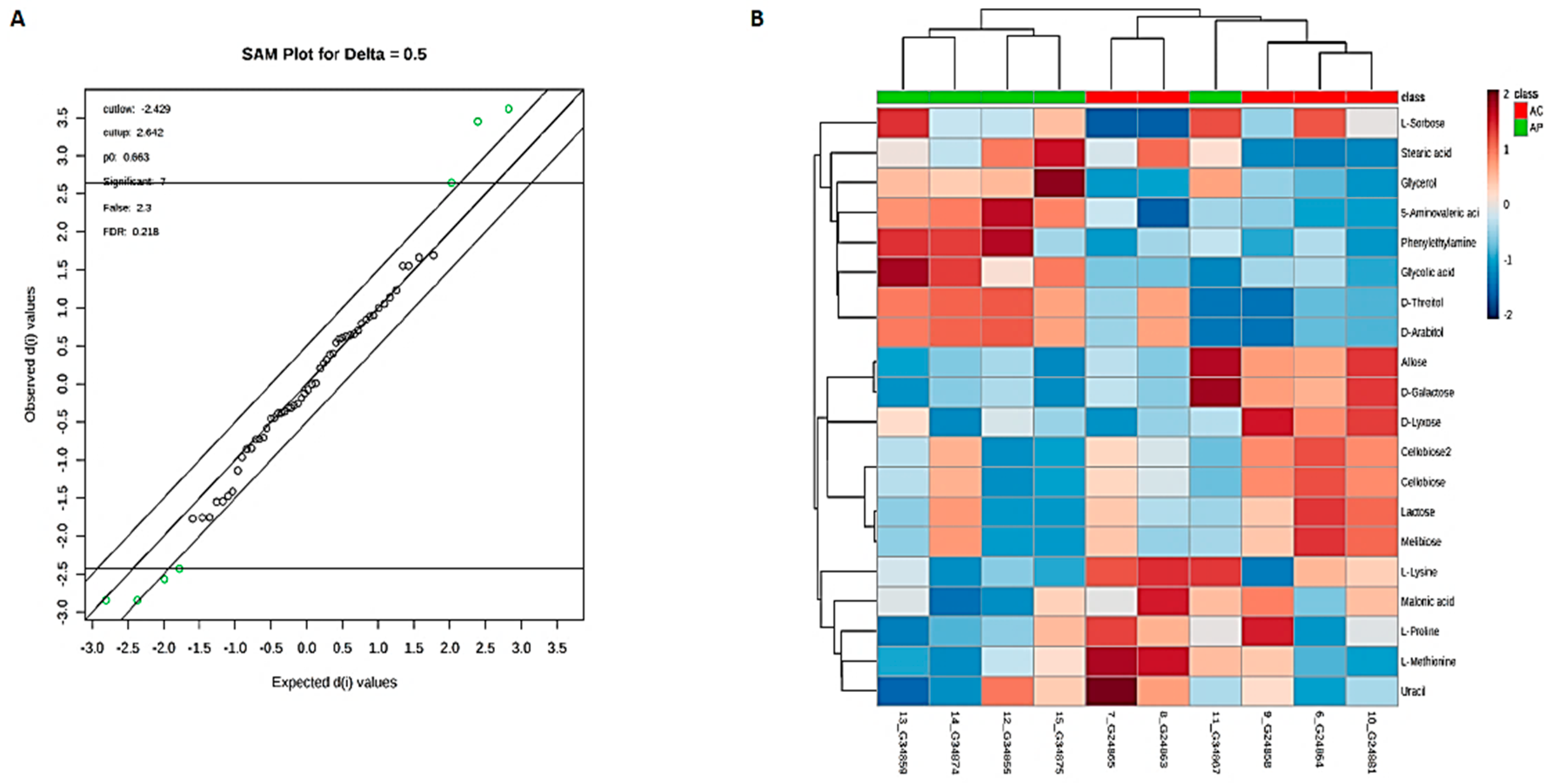
3.6. Metabolic Phenotyping of AC and AP Groups
3.7. Identification of Potential Fecal Metabolites Associated with the AP/AC Groups
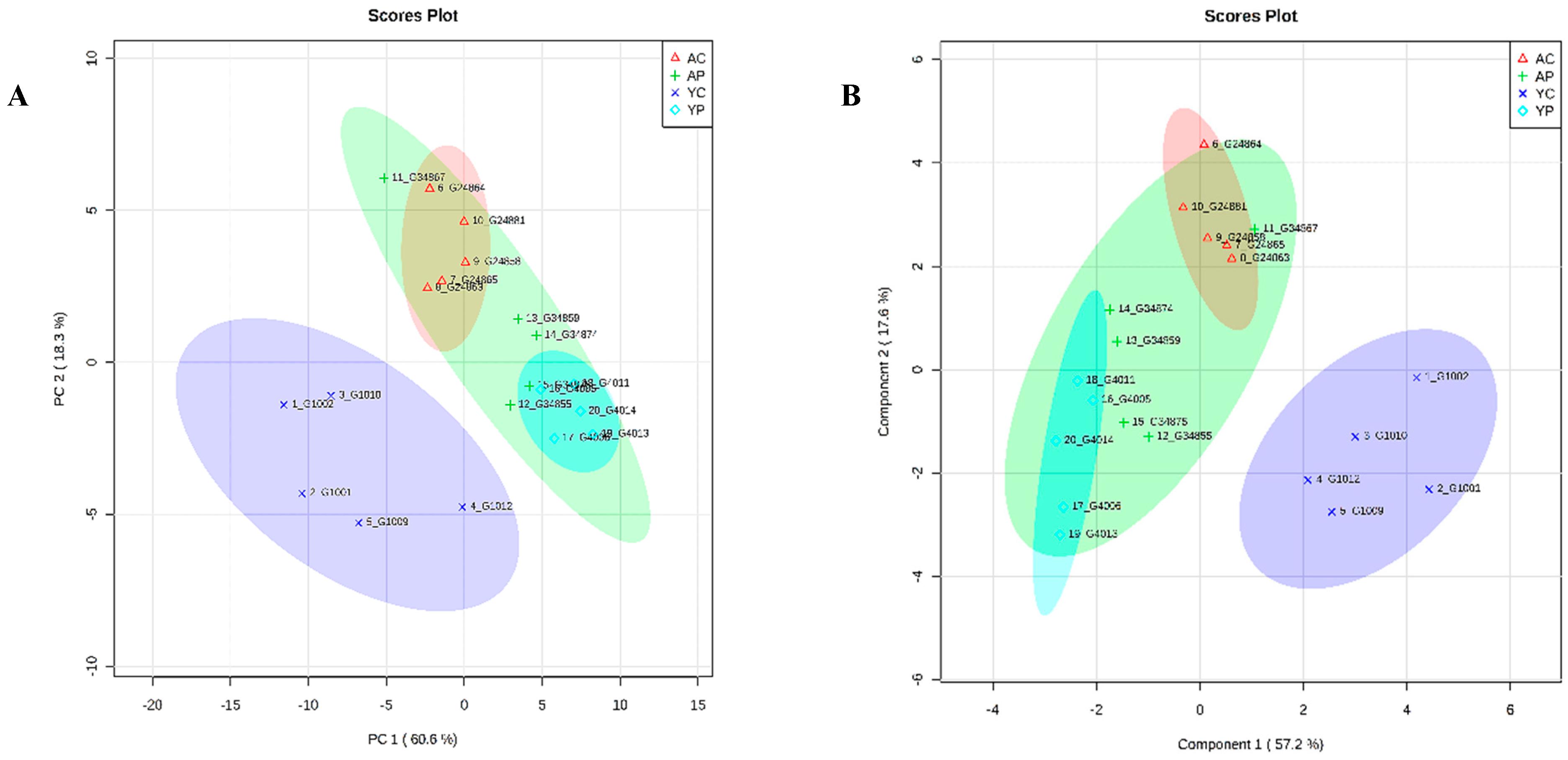
3.8. Comparative Metabolite Phenotyping in Young and Aging Groups
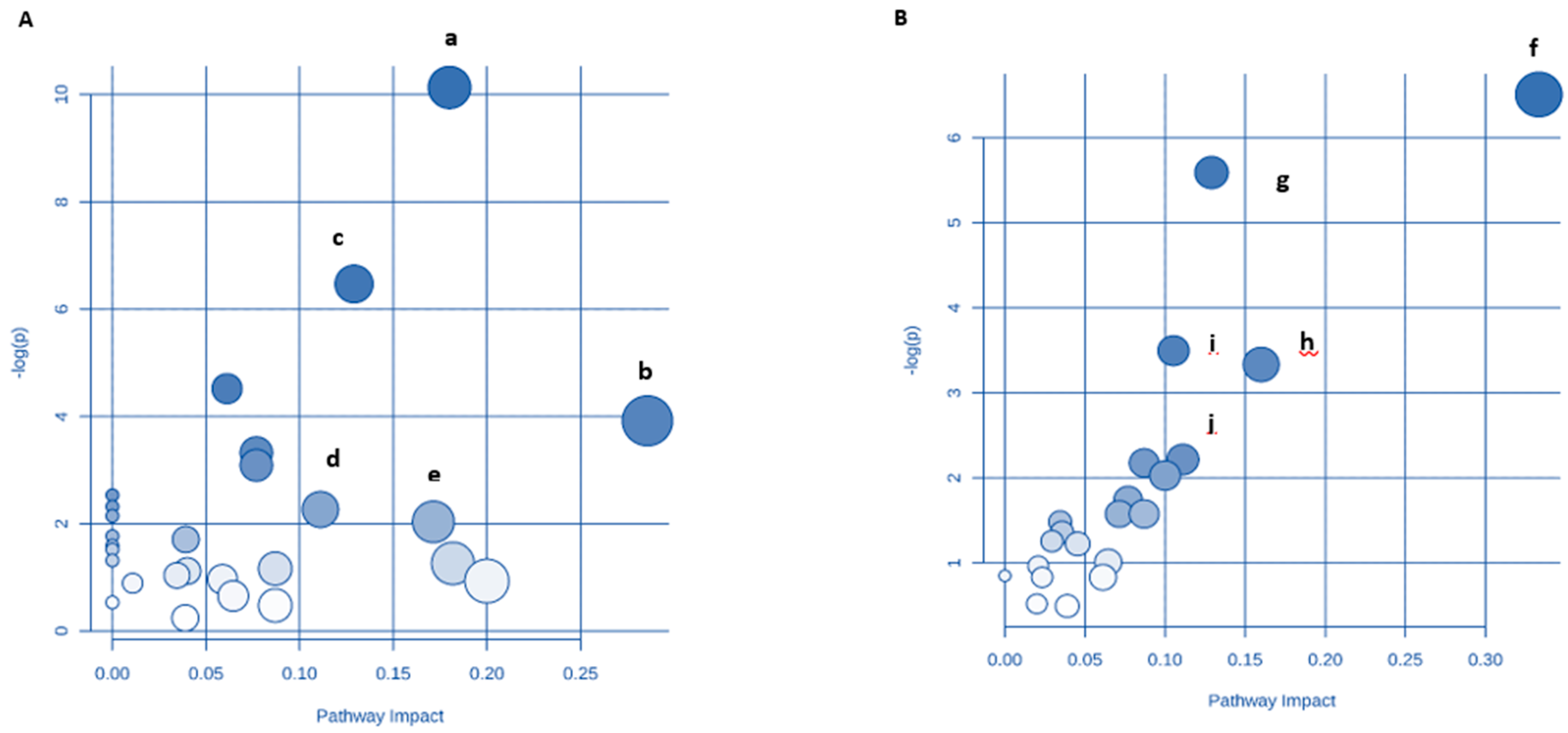
3.9. Identification of Key Metabolic Pathways Using MetPA
4. Discussion
4.1. Amino Acid and Protein Metabolism
4.2. Carbohydrate Metabolism
4.3. Butanoate Metabolism
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vemuri, R.; Gundamaraju, R.; Shastri, M.D.; Shukla, S.D.; Kalpurath, K.; Ball, M.; Tristram, S.; Shankar, E.M.; Ahuja, K.; Eri, R. Gut microbial changes, interactions, and their implications on human lifecycle: An ageing perspective. BioMed Res. Int. 2018, 2018. [Google Scholar] [CrossRef] [PubMed]
- Langille, M.G.; Meehan, C.J.; Koenig, J.E.; Dhanani, A.S.; Rose, R.A.; Howlett, S.E.; Beiko, R.G. Microbial shifts in the aging mouse gut. Microbiome 2014, 2, 50. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Jia, H.-M.; Zhou, C.; Yang, Y.; Sun, L.-L.; Zou, Z.-M. Urinary and fecal metabonomics study of the protective effect of chaihu-shu-gan-san on antibiotic-induced gut microbiota dysbiosis in rats. Sci. Rep. 2017, 7, 46551. [Google Scholar] [CrossRef] [PubMed]
- Biagi, E.; Nylund, L.; Candela, M.; Ostan, R.; Bucci, L.; Pini, E.; Nikkïla, J.; Monti, D.; Satokari, R.; Franceschi, C. Through ageing, and beyond: Gut microbiota and inflammatory status in seniors and centenarians. PLoS ONE 2010, 5, e10667. [Google Scholar] [CrossRef]
- Fahlström, A.; Yu, Q.; Ulfhake, B. Behavioral changes in aging female c57bl/6 mice. Neurobiol. Aging 2011, 32, 1868–1880. [Google Scholar] [CrossRef] [PubMed]
- Krych, Ł.; Nielsen, D.S.; Hansen, A.K.; Hansen, C.H.F. Gut microbial markers are associated with diabetes onset, regulatory imbalance, and ifn-γ level in nod mice. Gut Microbes 2015, 6, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Vemuri, R.; Gundamaraju, R.; Eri, R. Role of lactic acid probiotic bacteria in ibd. Curr. Pharm. Des. 2017, 23, 2352–2355. [Google Scholar] [CrossRef] [PubMed]
- Schneeberger, M.; Everard, A.; Gómez-Valadés, A.G.; Matamoros, S.; Ramírez, S.; Delzenne, N.M.; Gomis, R.; Claret, M.; Cani, P.D. Akkermansia muciniphila inversely correlates with the onset of inflammation, altered adipose tissue metabolism and metabolic disorders during obesity in mice. Sci. Rep. 2015, 5, 16643. [Google Scholar] [CrossRef] [PubMed]
- Sybille, T.; June, Z.; Michael, K.; Roy, M.; Maria, L.M. The intestinal microbiota in aged mice is modulated by dietary resistant starch and correlated with improvements in host responses. FEMS Microbiol. Ecol. 2013, 83, 299–309. [Google Scholar]
- Azzu, V.; Valencak, T.G. Energy metabolism and ageing in the mouse: A mini-review. Gerontology 2017, 63, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Valdes, A.M.; Glass, D.; Spector, T.D. Omics technologies and the study of human ageing. Nat. Rev. Genet. 2013, 14, 601. [Google Scholar] [CrossRef] [PubMed]
- Rampelli, S.; Candela, M.; Turroni, S.; Biagi, E.; Collino, S.; Franceschi, C.; O’Toole, P.W.; Brigidi, P. Functional metagenomic profiling of intestinal microbiome in extreme ageing. Aging (Albany N. Y.) 2013, 5, 902. [Google Scholar] [CrossRef] [PubMed]
- Son, N.; Hur, H.J.; Sung, M.J.; Kim, M.-S.; Hwang, J.-T.; Park, J.H.; Yang, H.J.; Kwon, D.Y.; Yoon, S.H.; Chung, H.Y. Liquid chromatography–mass spectrometry-based metabolomic analysis of livers from aged rats. J. Proteome Res. 2012, 11, 2551–2558. [Google Scholar] [CrossRef] [PubMed]
- Deda, O.; Gika, H.G.; Taitzoglou, I.; Raikos, Ν.; Theodoridis, G. Impact of exercise and aging on rat urine and blood metabolome. An lc-ms based metabolomics longitudinal study. Metabolites 2017, 7, 10. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Cheon, H.-S.; Song, J.-C.; Yun, S.-M.; Park, S.I.; Jeon, J.-P. Aging-related changes in mouse serum glycerophospholipid profiles. Osong Public Health Res. Perspect. 2014, 5, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Zhao, X.; Zhao, J.; Zhang, H.; Zhai, Q.; Narbad, A.; Chen, W. A mixture of lactobacillus species isolated from traditional fermented foods promote recovery from antibiotic-induced intestinal disruption in mice. J. Appl. Microbiol. 2018, 124, 842–854. [Google Scholar] [CrossRef] [PubMed]
- Vemuri, R.; Gundamaraju, R.; Shinde, T.; Eri, R. Therapeutic interventions for gut dysbiosis and related disorders in the elderly: Antibiotics, probiotics or faecal microbiota transplantation? Benef. Microbes 2017, 8, 179–192. [Google Scholar] [CrossRef] [PubMed]
- Vemuri, R.; Shinde, T.; Shastri, M.D.; Perera, A.P.; Tristram, S.; Martoni, C.J.; Gundamaraju, R.; Ahuja, K.D.; Ball, M.; Eri, R. A human origin strain lactobacillus acidophilus dds-1 exhibits superior in vitro probiotic efficacy in comparison to plant or dairy origin probiotics. Int. J. Med. Sci. 2018, 15, 840–848. [Google Scholar] [CrossRef] [PubMed]
- Nagata, S.; Asahara, T.; Wang, C.; Suyama, Y.; Chonan, O.; Takano, K.; Daibou, M.; Takahashi, T.; Nomoto, K.; Yamashiro, Y. The effectiveness of lactobacillus beverages in controlling infections among the residents of an aged care facility: A randomized placebo-controlled double-blind trial. Ann. Nutr. Metab. 2016, 68, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Van den Nieuwboer, M.; Klomp-Hogeterp, A.; Verdoorn, S.; Metsemakers-Brameijer, L.; Vriend, T.; Claassen, E.; Larsen, O. Improving the bowel habits of elderly residents in a nursing home using probiotic fermented milk. Benef. Microbes 2015, 6, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Rampelli, S.; Candela, M.; Severgnini, M.; Biagi, E.; Turroni, S.; Roselli, M.; Carnevali, P.; Donini, L.; Brigidi, P. A probiotics-containing biscuit modulates the intestinal microbiota in the elderly. J. Nutr. Health Aging 2013, 17, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Pellino, G.; Sciaudone, G.; Candilio, G.; Camerlingo, A.; Marcellinaro, R.; De Fatico, S.; Rocco, F.; Canonico, S.; Riegler, G.; Selvaggi, F. Early postoperative administration of probiotics versus placebo in elderly patients undergoing elective colorectal surgery: A double-blind randomized controlled trial. BMC Surg. 2013, 13, S57. [Google Scholar] [CrossRef] [PubMed]
- Allen, S.; Wareham, K.; Wang, D.; Bradley, C.; Sewell, B.; Hutchings, H.; Harris, W.; Dhar, A.; Brown, H.; Foden, A. Pwe-008 placide: Probiotics in the prevention of antibiotic associated diarrhoea (aad) and clostridium difficile associated diarrhoea (cdd) in elderly patients admitted to hospital–results of a large multi-centre rct in the uk. Gut 2013, 62, A133. [Google Scholar] [CrossRef]
- Pakdaman, M.N.; Udani, J.K.; Molina, J.P.; Shahani, M. The effects of the dds-1 strain of lactobacillus on symptomatic relief for lactose intolerance-a randomized, double-blind, placebo-controlled, crossover clinical trial. Nutr. J. 2015, 15, 56. [Google Scholar] [CrossRef] [PubMed]
- Hulshof, L.; van’t Land, B.; Sprikkelman, A.B.; Garssen, J. Role of microbial modulation in management of atopic dermatitis in children. Nutrients 2017, 9, 854. [Google Scholar] [CrossRef] [PubMed]
- Gerasimov, S.V.; Vasjuta, V.V.; Myhovych, O.O.; Bondarchuk, L.I. Probiotic supplement reduces atopic dermatitis in preschool children. Am. J. Clin. Dermatol. 2010, 11, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Gerasimov, S.; Ivantsiv, V.; Bobryk, L.; Tsitsura, O.; Dedyshin, L.; Guta, N.; Yandyo, B. Role of short-term use of l. Acidophilus dds-1 and b. Lactis uabla-12 in acute respiratory infections in children: A randomized controlled trial. Eur. J. Clin. Nutr. 2016, 70, 463. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yang, B.; Zhang, A.; Sun, H.; Yan, G. Potential drug targets on insomnia and intervention effects of jujuboside a through metabolic pathway analysis as revealed by uplc/esi-synapt-hdms coupled with pattern recognition approach. J. Proteom. 2012, 75, 1411–1427. [Google Scholar] [CrossRef] [PubMed]
- Kuo, S.-M.; Merhige, P.M.; Hagey, L.R. The effect of dietary prebiotics and probiotics on body weight, large intestine indices, and fecal bile acid profile in wild type and il10−/−mice. PLoS ONE 2013, 8, e60270. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Kobert, K.; Flouri, T.; Stamatakis, A. Pear: A fast and accurate illumina paired-end read merger. Bioinformatics 2013, 30, 614–620. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Uparse: Highly accurate otu sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996. [Google Scholar] [CrossRef] [PubMed]
- Kuczynski, J.; Stombaugh, J.; Walters, W.A.; González, A.; Caporaso, J.G.; Knight, R. Using qiime to analyze 16s rrna gene sequences from microbial communities. Curr. Protoc. Microbiol. 2012, 27, 1E–5. [Google Scholar]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. Uchime improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [PubMed]
- Huson, D.H.; Mitra, S. Introduction to the analysis of environmental sequences: Metagenomics with megan. In Evolutionary Genomics; Springer: Berlin, Germany, 2012; pp. 415–429. [Google Scholar]
- Dhariwal, A.; Chong, J.; Habib, S.; King, I.L.; Agellon, L.B.; Xia, J. Microbiomeanalyst: A web-based tool for comprehensive statistical, visual and meta-analysis of microbiome data. Nucleic Acids Res. 2017, 45, W180–W188. [Google Scholar] [CrossRef] [PubMed]
- Beale, D.J.; Marney, D.; Marlow, D.R.; Morrison, P.D.; Dunn, M.S.; Key, C.; Palombo, E.A. Metabolomic analysis of cryptosporidium parvum oocysts in water: A proof of concept demonstration. Environ. Pollut. 2013, 174, 201–203. [Google Scholar] [CrossRef] [PubMed]
- Karpe, A.V.; Beale, D.J.; Harding, I.H.; Palombo, E.A. Optimization of degradation of winery-derived biomass waste by ascomycetes. J. Chem. Technol. Biotechnol. 2015, 90, 1793–1801. [Google Scholar] [CrossRef]
- Beale, D.; Morrison, P.; Key, C.; Palombo, E. Metabolic profiling of biofilm bacteria known to cause microbial influenced corrosion. Water Sci. Technol. 2014, 69, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Sansone, S.A.; Fan, T.; Goodacre, R.; Griffin, J.L.; Hardy, N.W.; Kaddurah-Daouk, R.; Kristal, B.S.; Lindon, J.; Mendes, P.; Morrison, N.; et al. The metabolomics standards initiative. Nat. Biotechnol. 2007, 25, 846–848. [Google Scholar] [PubMed]
- French, K.E.; Harvey, J.; McCullagh, J.S. Targeted and untargeted metabolic profiling of wild grassland plants identifies antibiotic and anthelmintic compounds targeting pathogen physiology, metabolism and reproduction. Sci. Rep. 2018, 8, 1695. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zhang, X.; Wang, Y.; Li, Y.; Chen, Y.; Zheng, H.; Ma, F.; Ma, C.W.; Lu, B.; Xie, Z. Metabonomic strategy for the detection of metabolic effects of probiotics combined with prebiotic supplementation in weaned rats. RSC Adv. 2018, 8, 5042–5057. [Google Scholar] [CrossRef]
- Sun, H.; Zhang, A.; Yan, G.; Piao, C.; Li, W.; Sun, C.; Wu, X.; Li, X.; Chen, Y.; Wang, X. Metabolomic analysis of key regulatory metabolites in hcv-infected tree shrews. Mol. Cell. Proteom. 2012, 12, 710–719. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, K.; Matsuda, F.; Shimizu, H. Metabolome analysis of saccharomyces cerevisiae and optimization of culture medium for s-adenosyl-l-methionine production. AMB Express 2016, 6, 38. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D. Human gut microbiome: Hopes, threats and promises. Gut 2018, gutjnl-2018-316723. [Google Scholar] [CrossRef] [PubMed]
- Robinson, A.M.; Gondalia, S.V.; Karpe, A.V.; Eri, R.; Beale, D.J.; Morrison, P.D.; Palombo, E.A.; Nurgali, K. Fecal microbiota and metabolome in a mouse model of spontaneous chronic colitis: Relevance to human inflammatory bowel disease. Inflamm. Bowel Dis. 2016, 22, 2767–2787. [Google Scholar] [CrossRef] [PubMed]
- Welly, R.J.; Liu, T.-W.; Zidon, T.M.; Rowles III, J.L.; Park, Y.-M.; Smith, T.N.; Swanson, K.S.; Padilla, J.; Vieira-Potter, V.J. Comparison of diet vs. Exercise on metabolic function & gut microbiota in obese rats. Med. Sci. Sports Exerc. 2016, 48, 1688. [Google Scholar] [PubMed]
- Lee, H.; Lee, Y.; Kim, J.; An, J.; Lee, S.; Kong, H.; Song, Y.; Lee, C.-K.; Kim, K. Modulation of the gut microbiota by metformin improves metabolic profiles in aged obese mice. Gut Microbes 2018, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.B.; Yassour, M.; Sauk, J.; Garner, A.; Jiang, X.; Arthur, T.; Lagoudas, G.K.; Vatanen, T.; Fornelos, N.; Wilson, R. A novel ruminococcus gnavus clade enriched in inflammatory bowel disease patients. Genome Med. 2017, 9, 103. [Google Scholar] [CrossRef] [PubMed]
- Vital, M.; Howe, A.C.; Tiedje, J.M. Revealing the bacterial butyrate synthesis pathways by analyzing (meta) genomic data. MBio 2014, 5, e00889-14. [Google Scholar] [CrossRef] [PubMed]
- Neis, E.P.; Dejong, C.H.; Rensen, S.S. The role of microbial amino acid metabolism in host metabolism. Nutrients 2015, 7, 2930–2946. [Google Scholar] [CrossRef] [PubMed]
- Vernocchi, P.; Del Chierico, F.; Quagliariello, A.; Ercolini, D.; Lucidi, V.; Putignani, L. A metagenomic and in silico functional prediction of gut microbiota profiles may concur in discovering new cystic fibrosis patient-targeted probiotics. Nutrients 2017, 9, 1342. [Google Scholar] [CrossRef] [PubMed]
- Hertz, L. The glutamate–glutamine (gaba) cycle: Importance of late postnatal development and potential reciprocal interactions between biosynthesis and degradation. Front. Endocrinol. (Lausanne) 2013, 4, 59. [Google Scholar] [CrossRef] [PubMed]
- Fisher, D.Y.; Fukagawa, N.K. Protein and amino acid metabolism in the elderly. In Methods for Investigation of Amino Acid and Protein Metabolism; Routledge: Abingdon, UK, 2017; pp. 167–175. [Google Scholar]
- Munro, H.N.; Gersovitz, M.; Young, V.R. Human aging: Protein and amino acid metabolism and implications for protein and amino acid requirements. In Nutritional Approaches to Aging Research; CRC Press: Boca Raton, FL, USA, 2018; pp. 58–93. [Google Scholar]
- Bauchart-Thevret, C.; Stoll, B.; Burrin, D.G. Intestinal metabolism of sulfur amino acids. Nutr. Res. Rev. 2009, 22, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Bogicevic, B.; Berthoud, H.; Portmann, R.; Bavan, T.; Meile, L.; Irmler, S. Cysteine biosynthesis in lactobacillus casei: Identification and characterization of a serine acetyltransferase. FEMS Microbiol. Lett. 2016, 363, 4. [Google Scholar] [CrossRef] [PubMed]
- Hausmann, C.D.; Ibba, M. Aminoacyl-trna synthetase complexes: Molecular multitasking revealed. FEMS Microbiol. Rev. 2008, 32, 705–721. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, T.K.; Kapil, C.; Khan, S.; Jairajpuri, M.A.; Sharma, V.; Santoni, D.; Silvestrini, F.; Pizzi, E.; Sharma, A. A genomic glimpse of aminoacyl-trna synthetases in malaria parasite plasmodium falciparum. BMC Genomics 2009, 10, 644. [Google Scholar] [CrossRef] [PubMed]
- Park, S.G.; Schimmel, P.; Kim, S. Aminoacyl trna synthetases and their connections to disease. Proc. Natl. Acad. Sci. USA 2008. [Google Scholar] [CrossRef] [PubMed]
- Jafarnejad, S.M.; Kim, S.-H.; Sonenberg, N. Aminoacylation of proteins: New targets for the old arsenal. Cell Metab. 2018, 27, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Kong, J.; Fang, P.; Madoux, F.; Spicer, T.P.; Scampavia, L.; Kim, S.; Guo, M. High-throughput screening for protein synthesis inhibitors targeting aminoacyl-trna synthetases. SLAS Discov. 2018, 23, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Finkel, T. The metabolic regulation of aging. Nat. Med. 2015, 21, 1416. [Google Scholar] [CrossRef] [PubMed]
- De Las Heras, J.; Aldámiz-Echevarría, L.; Martínez-Chantar, M.-L.; Delgado, T.C. An update on the use of benzoate, phenylacetate and phenylbutyrate ammonia scavengers for interrogating and modifying liver nitrogen metabolism and its implications in urea cycle disorders and liver disease. Expert Opin. Drug Metab. Toxicol. 2017, 13, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Yudkoff, M. Interactions in the metabolism of glutamate and the branched-chain amino acids and ketoacids in the cns. Neurochem. Res. 2017, 42, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Garibotto, G.; Verzola, D.; Vettore, M.; Tessari, P. The contribution of muscle, kidney, and splanchnic tissues to leucine transamination in humans. Can. J Physiol. Pharmacol. 2017, 96, 382–387. [Google Scholar] [CrossRef] [PubMed]
- Waisbren, S.E.; Cuthbertson, D.; Burgard, P.; Holbert, A.; McCarter, R.; Cederbaum, S.; Consortium, U.C.D. Biochemical markers and neuropsychological functioning in distal urea cycle disorders. Pharmacol. Res. J Inherit. Metab. Dis. 2018, 41, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Davila-Gay, A.-M.; Blachier, F.; Gotteland, M.; Andriamihaja, M.; Benetti, P.-H.; Sanz, Y.; Tomé, D. Intestinal luminal nitrogen metabolism: Role of the gut microbiota and consequences for the host. Pharmacol. Res. 2013, 68, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Coelho, A.I.; Berry, G.T.; Rubio-Gozalbo, M.E. Galactose metabolism and health. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 422–427. [Google Scholar] [CrossRef] [PubMed]
- Hobbs, M.E.; Williams, H.J.; Hillerich, B.; Almo, S.C.; Raushel, F.M. L-galactose metabolism in bacteroides vulgatus from the human gut microbiota. Biochemistry 2014, 53, 4661–4670. [Google Scholar] [CrossRef] [PubMed]
- Gundamaraju, R.; Vemuri, R.; Eri, R.; M Ishiki, H.; Coy-Barrera, E.; Sastry Yarla, N.; Golzio dos Santos, S.; Feitosa Alves, M.; Barbosa Filho, M.; FFM Diniz, M. Metabolomics as a functional tool in screening gastro intestinal diseases: Where are we in high throughput screening? Comb. Chem. High Throughput Screen. 2017, 20, 247–254. [Google Scholar] [CrossRef] [PubMed]









© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vemuri, R.; Shinde, T.; Gundamaraju, R.; Gondalia, S.V.; Karpe, A.V.; Beale, D.J.; Martoni, C.J.; Eri, R. Lactobacillus acidophilus DDS-1 Modulates the Gut Microbiota and Improves Metabolic Profiles in Aging Mice. Nutrients 2018, 10, 1255. https://doi.org/10.3390/nu10091255
Vemuri R, Shinde T, Gundamaraju R, Gondalia SV, Karpe AV, Beale DJ, Martoni CJ, Eri R. Lactobacillus acidophilus DDS-1 Modulates the Gut Microbiota and Improves Metabolic Profiles in Aging Mice. Nutrients. 2018; 10(9):1255. https://doi.org/10.3390/nu10091255
Chicago/Turabian StyleVemuri, Ravichandra, Tanvi Shinde, Rohit Gundamaraju, Shakuntla V. Gondalia, Avinash V. Karpe, David J. Beale, Christopher J. Martoni, and Rajaraman Eri. 2018. "Lactobacillus acidophilus DDS-1 Modulates the Gut Microbiota and Improves Metabolic Profiles in Aging Mice" Nutrients 10, no. 9: 1255. https://doi.org/10.3390/nu10091255
APA StyleVemuri, R., Shinde, T., Gundamaraju, R., Gondalia, S. V., Karpe, A. V., Beale, D. J., Martoni, C. J., & Eri, R. (2018). Lactobacillus acidophilus DDS-1 Modulates the Gut Microbiota and Improves Metabolic Profiles in Aging Mice. Nutrients, 10(9), 1255. https://doi.org/10.3390/nu10091255