Chrysin Ameliorates Malfunction of Retinoid Visual Cycle through Blocking Activation of AGE-RAGE-ER Stress in Glucose-Stimulated Retinal Pigment Epithelial Cells and Diabetic Eyes

Abstract
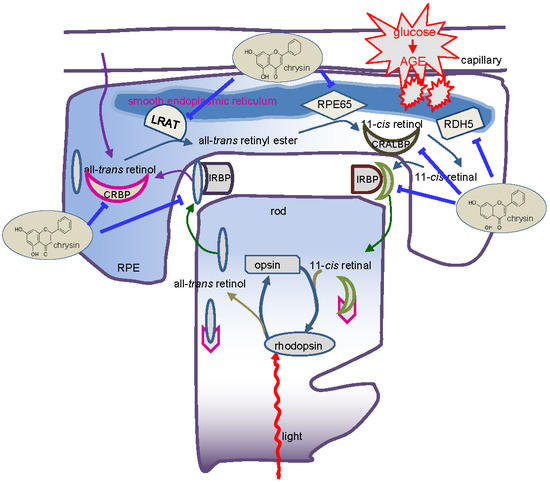
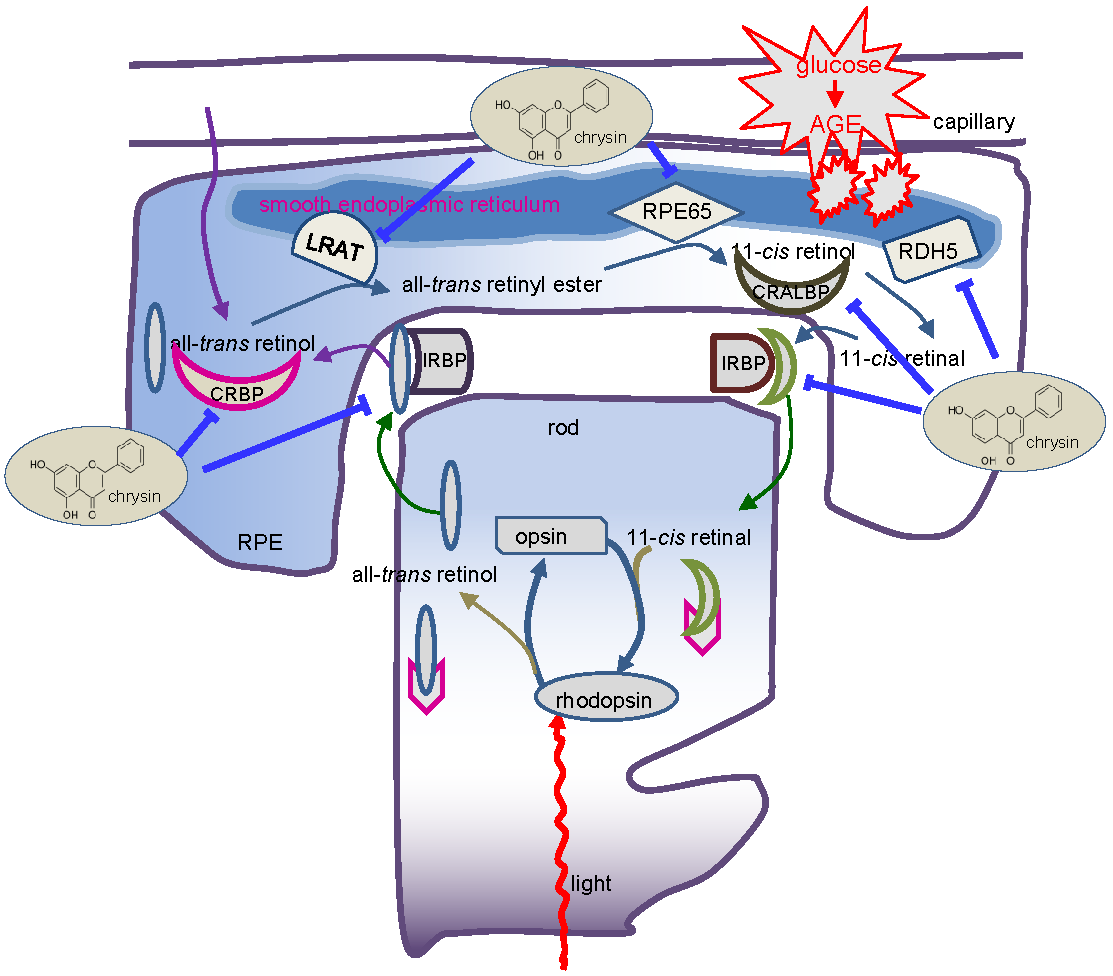{kind=link}
{kind=link}
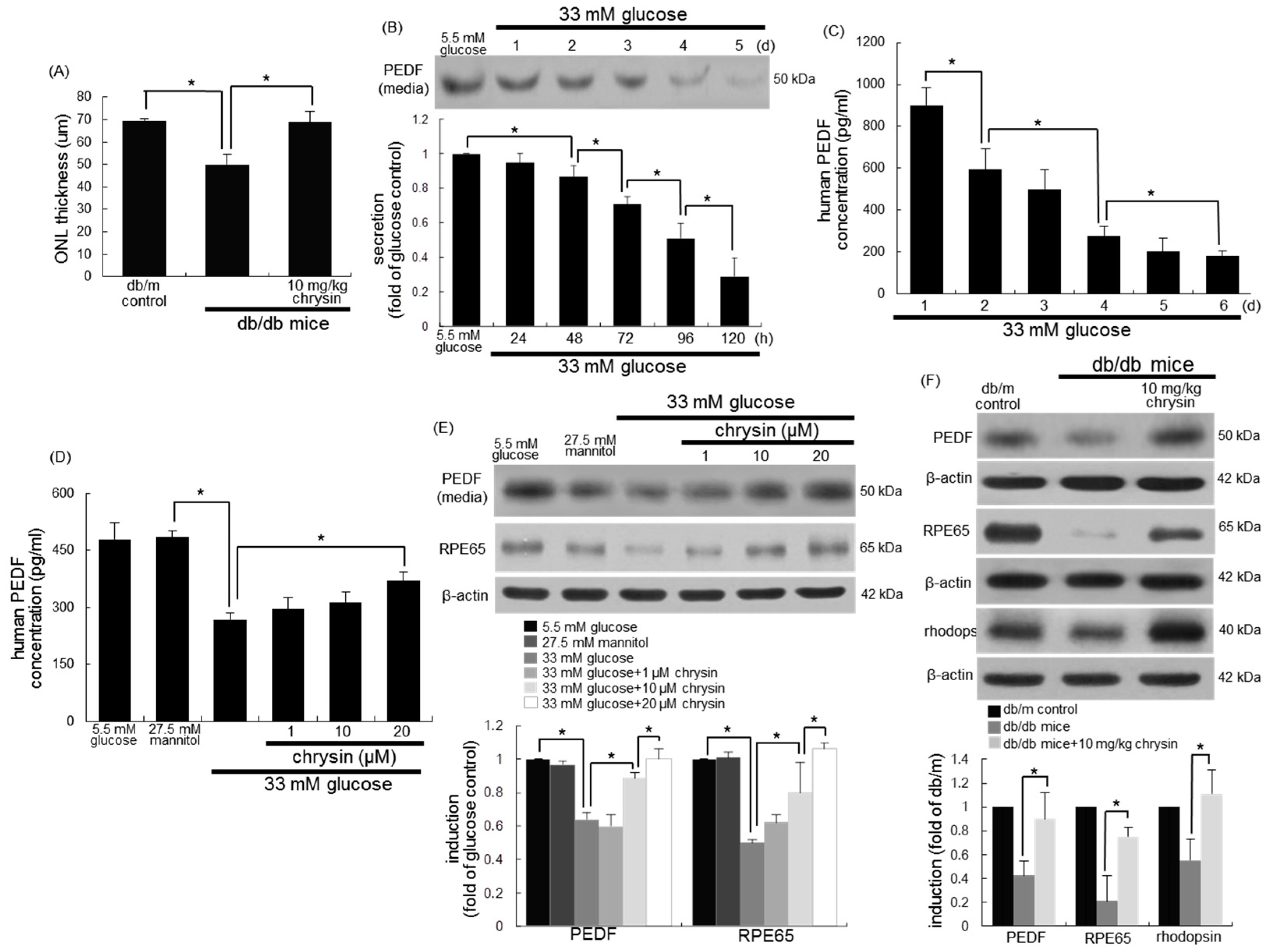{kind=link}
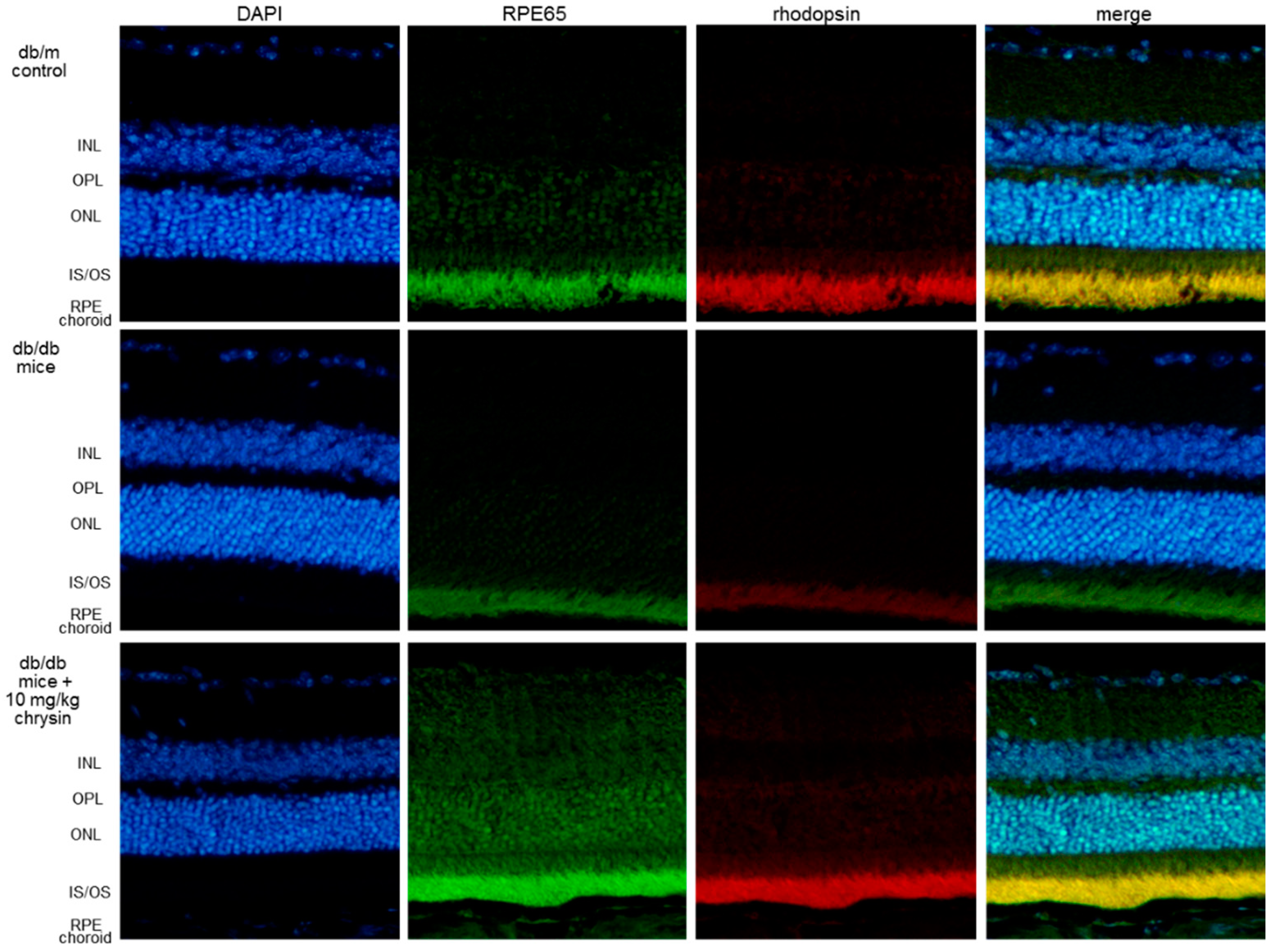{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
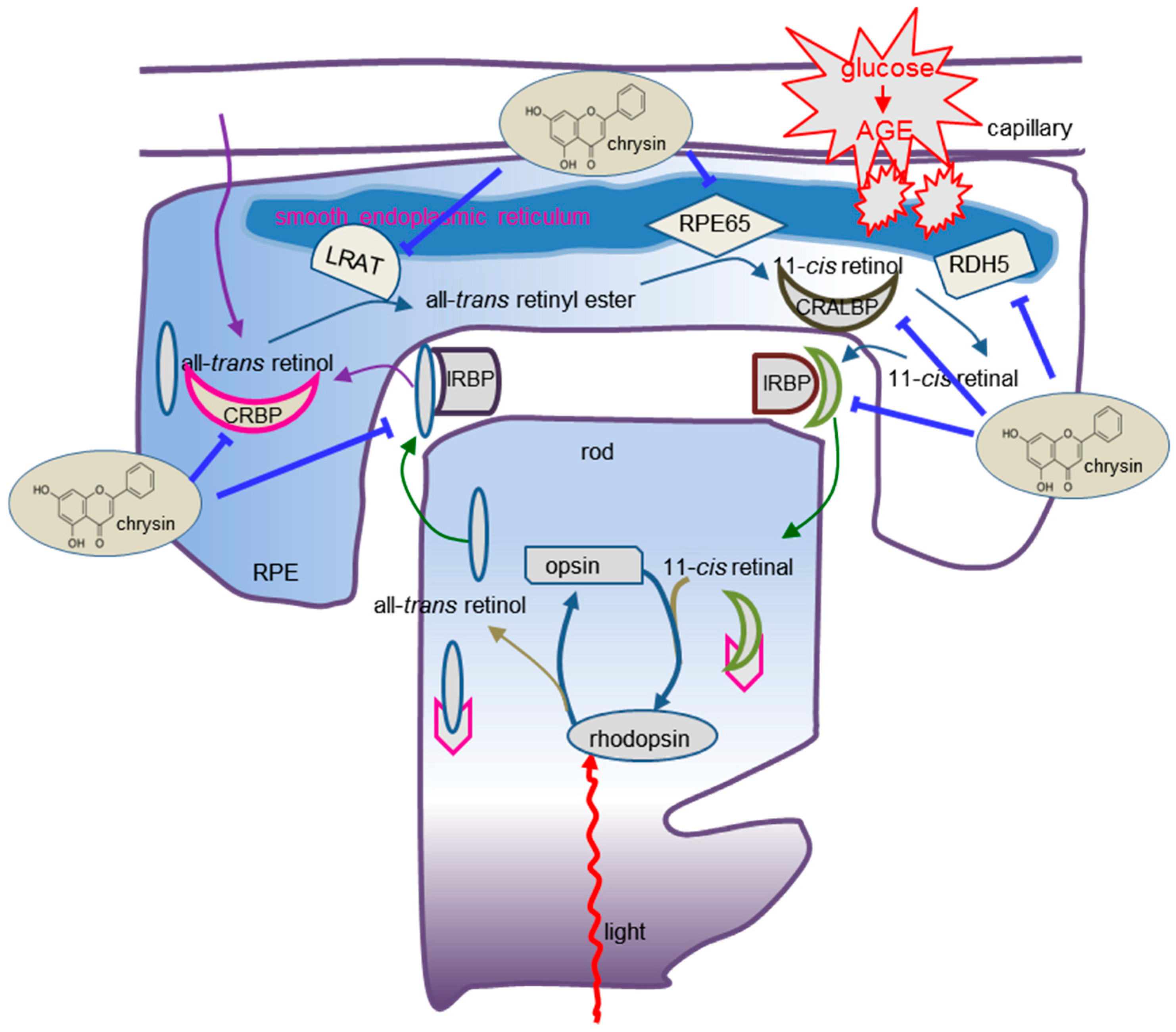
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. RPE Cell Culture
2.3. In Vivo Animal Experiments
2.4. Western Blot Analysis
2.5. Immunohistochemical Staining
2.6. Enzyme-Linked Immunosorbent Assay (ELISA)
2.7. Data Analysis
3. Results
3.1. Modulation of Production of VEGF and IGF-1 by Chrysin
3.2. Restoration of PEDF Production by Chrysin
3.3. Protective Effects of Chrysin on RPE65 Induction
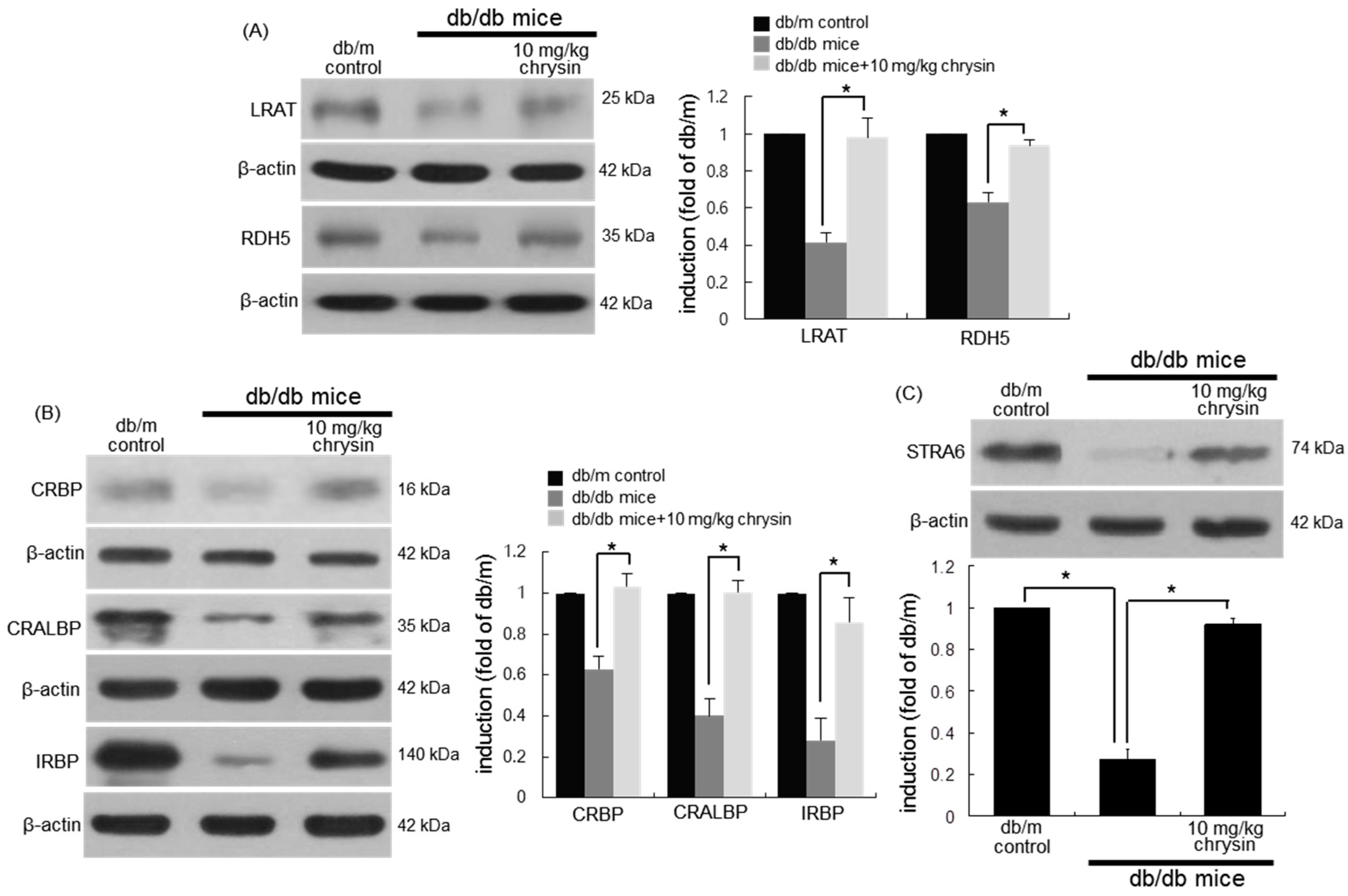
3.4. Protective Effects of Chrysin on Induction of Visual Cycle-Related Proteins
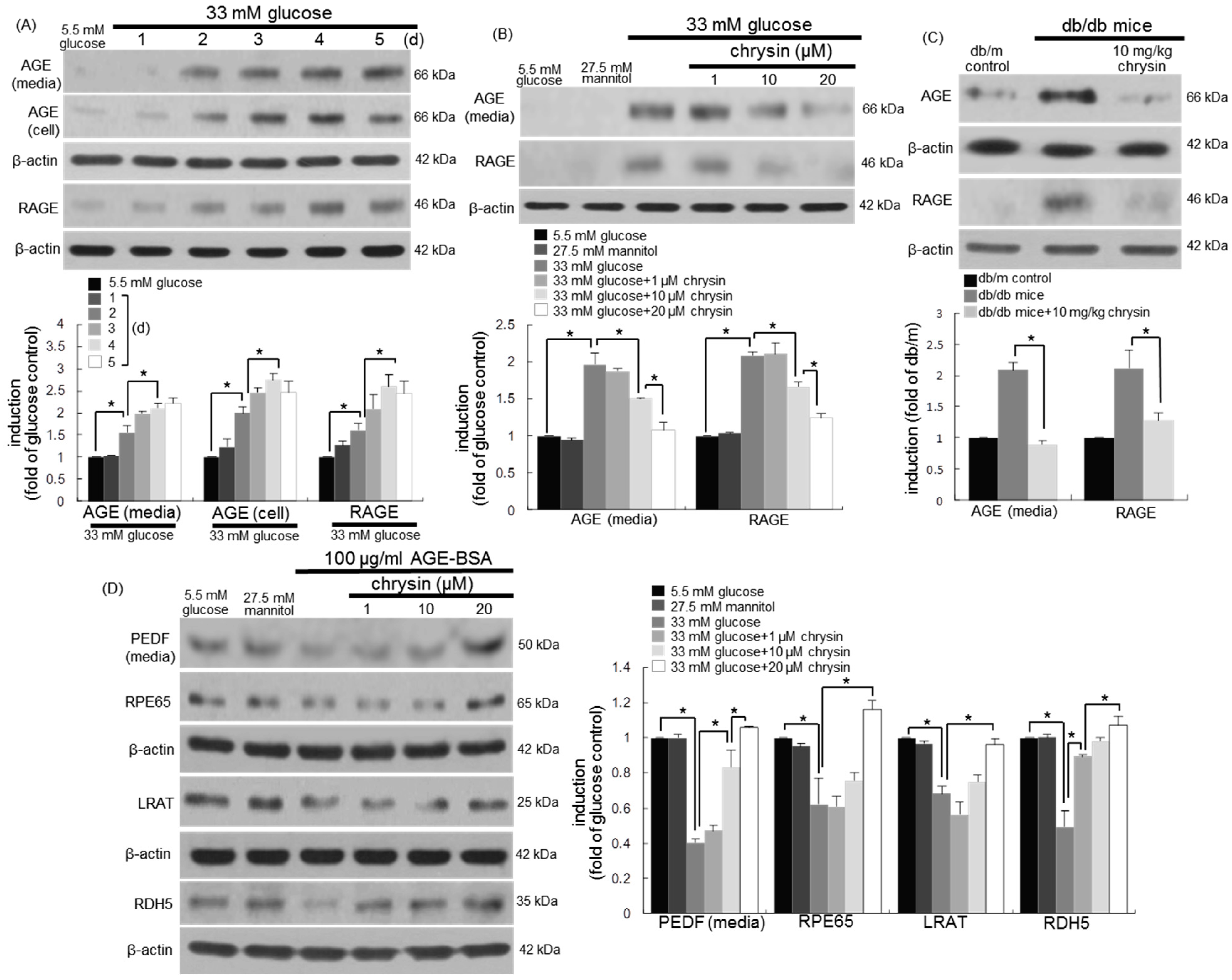
3.5. Blockade of AGE-Mediated Malfunction of Visual Cycle by Chrysin
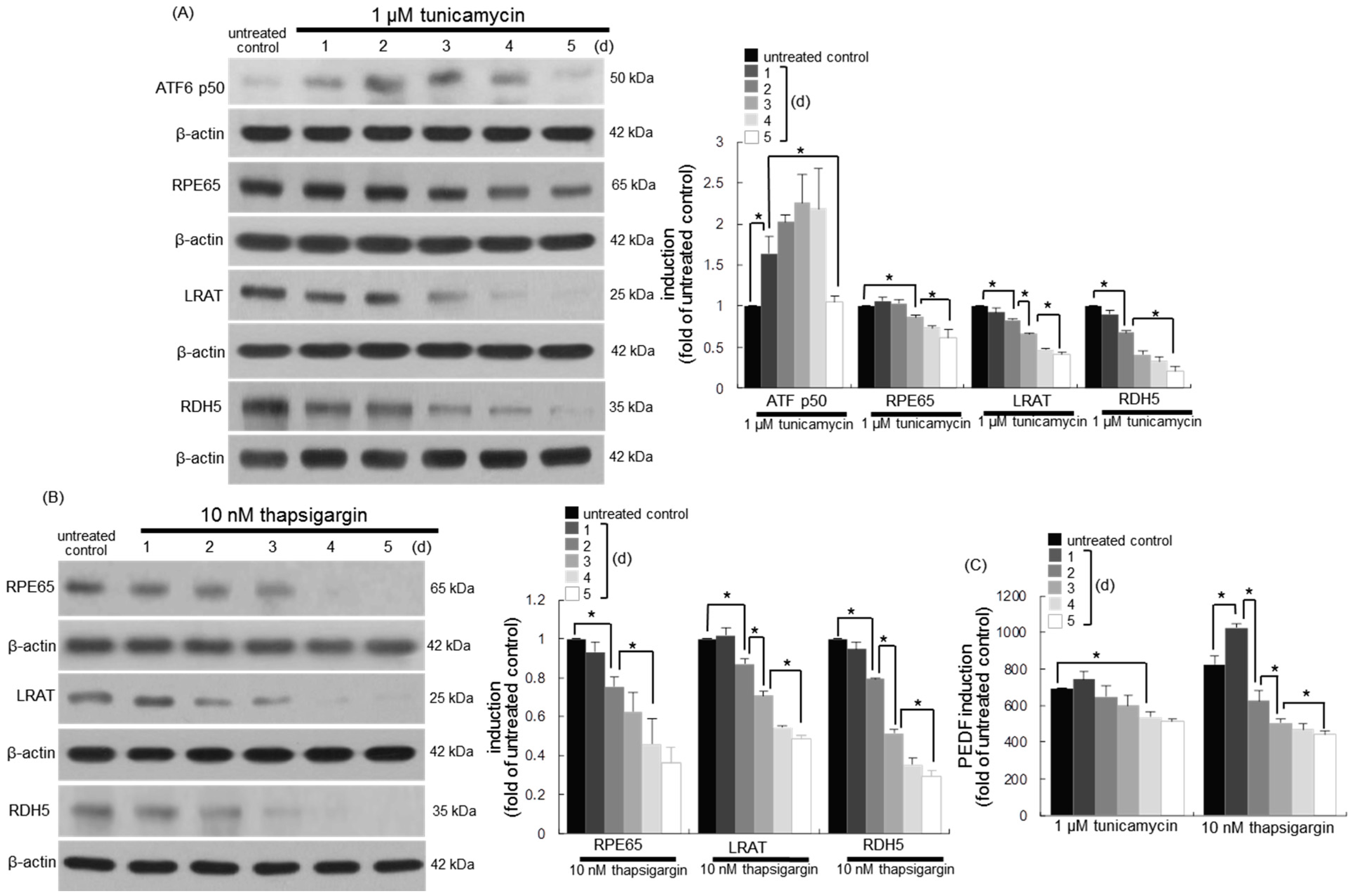
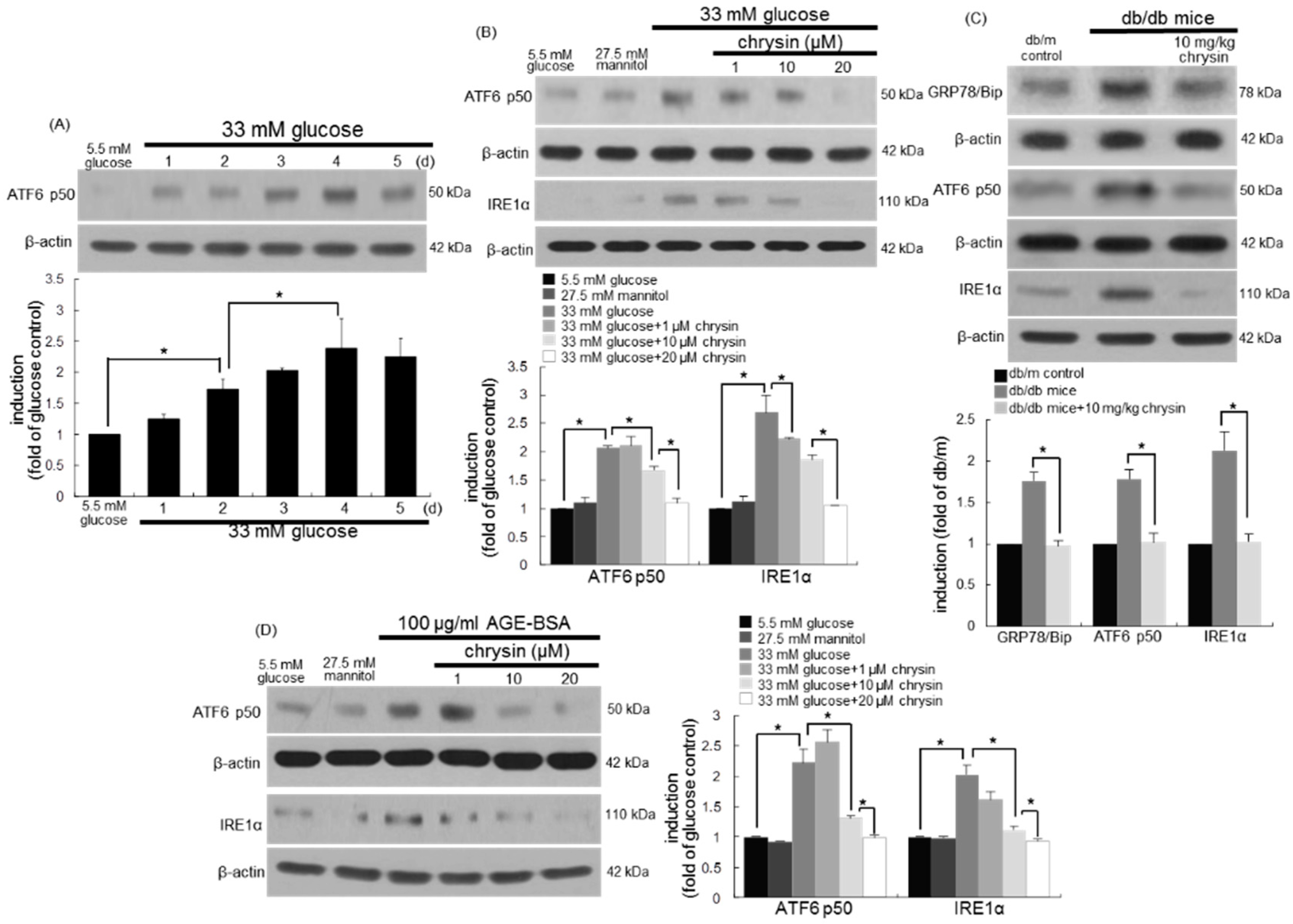
3.6. Involvement of ER Stress in Loss of Visual Cycle Proteins
3.7. Inhibition of ER Stress-Mediated Loss of Visual Cycle Proteins by Chrysin
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Roy, S.; Kern, T.S.; Song, B.; Stuebe, C. Mechanistic insights into pathological changes in the diabetic retina: Implications for targeting diabetic retinopathy. Am. J. Pathol. 2017, 187, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.R.; Gardner, T.W. Diabetic retinopathy: Research to clinical practice. Clin. Diabetes Endocrinol. 2017, 3, 9. [Google Scholar] [CrossRef] [PubMed]
- Wong, T.Y.; Cheung, C.M.; Larsen, M.; Sharma, S.; Simó, R. Diabetic retinopathy. Nat. Rev. Dis. Prim. 2016, 2, 16012. [Google Scholar] [CrossRef] [PubMed]
- Nentwich, M.M.; Ulbig, M.W. Diabetic retinopathy-ocular complications of diabetes mellitus. World J. Diabetes 2015, 6, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Crawford, T.N.; Alfaro, D.V., III; Kerrison, J.B.; Jablon, E.P. Diabetic retinopathy and angiogenesis. Curr. Diabetes Rev. 2009, 5, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Oshitari, T.; Hata, N.; Yamamoto, S. Endoplasmic reticulum stress and diabetic retinopathy. Vasc. Health Risk Manag. 2008, 4, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Trudeau, K.; Roy, S.; Tien, T.; Barrette, K.F. Mitochondrial dysfunction and endoplasmic reticulum stress in diabetic retinopathy: Mechanistic insights into high glucose-induced retinal cell death. Curr. Clin. Pharmacol. 2013, 8, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Wert, K.J.; Lin, J.H.; Tsang, S.H. General pathophysiology in retinal degeneration. Dev. Ophthalmol. 2014, 53, 33–43. [Google Scholar] [PubMed]
- Periyasamy, P.; Shinohara, T. Age-related cataracts: Role of unfolded protein response, Ca2+ mobilization, epigenetic DNA modifications, and loss of Nrf2/Keap1 dependent cytoprotection. Prog. Retin. Eye. Res. 2017, 60, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Boya, P.; Esteban-Martínez, L.; Serrano-Puebla, A.; Gómez-Sintes, R.; Villarejo-Zori, B. Autophagy in the eye: Development, degeneration, and aging. Prog. Retin. Eye Res. 2016, 55, 206–245. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.R.; Gardner, T.W. Diabetic retinopathy and diabetic macular edema. Dev. Ophthalmol. 2016, 55, 137–146. [Google Scholar] [PubMed]
- Wright, C.B.; Redmond, T.M.; Nickerson, J.M. A history of the classical visual cycle. Prog. Mol. Biol. Transl. Sci. 2015, 134, 433–448. [Google Scholar] [PubMed]
- Du, M.; Wu, M.; Fu, D.; Yang, S.; Chen, J.; Wilson, K.; Lyons, T.J. Effects of modified LDL and HDL on retinal pigment epithelial cells: A role in diabetic retinopathy? Diabetologia 2013, 56, 2318–2328. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.I.; Park, M.J.; Choi, J.H.; Lim, S.K.; Choi, H.J.; Park, S.H. Hyperglycemia-induced GLP-1R downregulation causes RPE cell apoptosis. Int. J. Biochem. Cell Biol. 2015, 59, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.C.; Snow, A.; LaBarbera, D.V.; Petrash, J.M. Aldose reductase inhibition alleviates hyperglycemic effects on human retinal pigment epithelial cells. Chem. Biol. Interact. 2015, 234, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.M.; Huang, D.Y.; Huang, Y.P.; Hsu, S.H.; Kang, L.Y.; Shen, C.M.; Lin, W.W. Methylglyoxal induces cell death through endoplasmic reticulum stress-associated ROS production and mitochondrial dysfunction. J. Cell. Mol. Med. 2016, 20, 1749–1760. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Conley, S.M.; Naash, M.I. RPE65: Role in the visual cycle, human retinal disease, and gene therapy. Ophthalmic. Genet. 2009, 30, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Kiser, P.D.; Golczak, M.; Maeda, A.; Palczewski, K. Key enzymes of the retinoid (visual) cycle in vertebrate retina. Biochim. Biophys. Acta. 2012, 1821, 137–151. [Google Scholar] [CrossRef] [PubMed]
- Travis, G.H.; Golczak, M.; Moise, A.R.; Palczewski, K. Diseases caused by defects in the visual cycle: Retinoids as potential therapeutic agents. Ann. Rev. Pharmacol. Toxicol. 2007, 47, 469. [Google Scholar] [CrossRef] [PubMed]
- Boyer, D.S.; Hopkins, J.J.; Sorof, J.; Ehrlich, J.S. Anti-vascular endothelial growth factor therapy for diabetic macular edema. Ther. Adv. Endocrinol. Metab. 2013, 4, 151–169. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, A.; Parriott, J.; Demirel, S.; Argo, C.; Sepah, Y.J.; Do, D.V.; Nguyen, Q.D. Nonbiological pharmacotherapies for the treatment of diabetic macular edema. Expert. Opin. Pharmacother. 2015, 16, 2625–2635. [Google Scholar] [CrossRef] [PubMed]
- Obrosova, I.G.; Kador, P.F. Aldose reductase/polyol inhibitors for diabetic retinopathy. Curr. Pharm. Biotechnol. 2011, 12, 373–385. [Google Scholar] [CrossRef] [PubMed]
- Falkenstein, I.A.; Cheng, L.; Wong-Staal, F.; Tammewar, A.M.; Barron, E.C.; Silva, G.A.; Li, Q.X.; Yu, D.; Hysell, M.; Liu, G.; et al. Toxicity and intraocular properties of a novel long-acting anti-proliferative and anti-angiogenic compound IMS2186. Curr. Eye Res. 2008, 33, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Jo, H.; Jung, S.H.; Yim, H.B.; Lee, S.J.; Kang, K.D. The effect of baicalin in a mouse model of retinopathy of prematurity. BMB Rep. 2015, 48, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.P.; Sun, L.; Yu, H.S.; Liang, L.P.; Li, W.; Ding, H.; Song, X.B.; Zhang, L.J. The Pharmacological effects of lutein and zeaxanthin on visual disorders and cognition diseases. Molecules 2017, 22, 610. [Google Scholar] [CrossRef] [PubMed]
- Neelam, K.; Goenadi, C.J.; Lun, K.; Yip, C.C.; Au Eong, K.G. Putative protective role of lutein and zeaxanthin in diabetic retinopathy. Br. J. Ophthalmol. 2017, 101, 551–558. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Qin, H.; Shi, Q.; Zhang, Y.; Zhou, F.; Wu, H.; Ding, S.; Niu, Z.; Lu, Y.; Shen, P. Chrysin attenuates inflammation by regulating M1/M2 status via activating PPARγ. Biochem. Pharmacol. 2014, 89, 503–514. [Google Scholar] [CrossRef] [PubMed]
- Nabavi, S.F.; Braidy, N.; Habtemariam, S.; Orhan, I.E.; Daglia, M.; Manayi, A.; Gortzi, O.; Nabavi, S.M. Neuroprotective effects of chrysin: From chemistry to medicine. Neurochem. Int. 2015, 90, 224–231. [Google Scholar] [CrossRef] [PubMed]
- Samarghandian, S.; Farkhondeh, T.; Azimi-Nezhad, M. Protective effects of chrysin against drugs and toxic agents. Dose Response 2017, 15. [Google Scholar] [CrossRef] [PubMed]
- Walle, T.; Otake, Y.; Brubaker, J.A.; Walle, U.K.; Halushka, P.V. Disposition and metabolism of the flavonoid chrysin in normal volunteers. Br. J. Clin. Pharmacol. 2001, 51, 143–146. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.K.; Park, S.H.; Kim, Y.H.; Lee, E.J.; Antika, L.D.; Kim, D.Y.; Choi, Y.J.; Kang, Y.H. Dietary compound chrysin inhibits retinal neovascularization with abnormal capillaries in db/db mice. Nutrients 2016, 8, 782. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.K.; Park, S.H.; Choi, Y.J.; Shin, D.; Kang, Y.H. Chrysin inhibits diabetic renal tubulointerstitial fibrosis through blocking epithelial to mesenchymal transition. J. Mol. Med. 2015, 93, 759–772. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson-Berka, J.L.; Wraight, C.; Werther, G. The role of growth hormone, insulin-like growth factor and somatostatin in diabetic retinopathy. Curr. Med. Chem. 2006, 13, 3307–3317. [Google Scholar] [CrossRef] [PubMed]
- Sall, J.W.; Klisovic, D.D.; O’Dorisio, M.S.; Katz, S.E. Somatostatin inhibits IGF-1 mediated induction of VEGF in human retinal pigment epithelial cells. Exp. Eye Res. 2004, 79, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Mori, K.; Duh, E.; Gehlbach, P.; Ando, A.; Takahashi, K.; Pearlman, J.; Mori, K.; Yang, H.S.; Zack, D.J.; Ettyreddy, D.; et al. Pigment epithelium-derived factor inhibits retinal and choroidal neovascularization. J. Cell. Physiol. 2001, 188, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Parker, R.O.; Crouch, R.K. Retinol dehydrogenases (RDHs) in the visual cycle. Exp. Eye Res. 2010, 91, 788–792. [Google Scholar] [CrossRef] [PubMed]
- Simó, R.; Carrasco, E.; García-Ramírez, M.; Hernández, C. Angiogenic and antiangiogenic factors in proliferative diabetic retinopathy. Curr. Diabetes Rev. 2006, 2, 71–98. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, Q.D.; De Falco, S.; Behar-Cohen, F.; Lam, W.C.; Li, X.; Reichhart, N.; Ricci, F.; Pluim, J.; Li, W.W. Placental growth factor and its potential role in diabetic retinopathy and other ocular neovascular diseases. Acta Ophthalmol. 2018, 96, e1–e9. [Google Scholar] [CrossRef] [PubMed]
- Peddada, K.V.; Brown, A.; Verma, V.; Nebbioso, M. Therapeutic potential of curcumin in major retinal pathologies. Int. Ophthalmol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Liao, R.; Yan, F.; Zeng, Z.; Wang, H.; Qiu, K.; Xu, J.; Zheng, W. Insulin-like growth factor-1 activates PI3K/Akt signalling to protect human retinal pigment epithelial cells from amiodarone-induced oxidative injury. Br. J. Pharmacol. 2018, 175, 125–139. [Google Scholar] [CrossRef] [PubMed]
- Hussain, R.M.; Gregori, N.Z.; Ciulla, T.A.; Lam, B.L. Pharmacotherapy of retinal disease with visual cycle modulators. Expert. Opin. Pharmacother. 2018, 19, 471–481. [Google Scholar] [CrossRef] [PubMed]
- Ha, J.H.; Shil, P.K.; Zhu, P.; Gu, L.; Li, Q.; Chung, S. Ocular inflammation and endoplasmic reticulum stress are attenuated by supplementation with grape polyphenols in human retinal pigmented epithelium cells and in C57BL/6 mice. J. Nutr. 2014, 144, 799–806. [Google Scholar] [CrossRef] [PubMed]
- Bian, M.; Zhang, Y.; Du, X.; Xu, J.; Cui, J.; Gu, J.; Zhu, W.; Zhang, T.; Chen, Y. Apigenin-7-diglucuronide protects retinas against bright light-induced photoreceptor degeneration through the inhibition of retinal oxidative stress and inflammation. Brain Res. 2017, 1663, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Hytti, M.; Szabó, D.; Piippo, N.; Korhonen, E.; Honkakoski, P.; Kaarniranta, K.; Petrovski, G.; Kauppinen, A. Two dietary polyphenols, fisetin and luteolin, reduce inflammation but augment DNA damage-induced toxicity in human RPE cells. J. Nutr. Biochem. 2017, 42, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A.; Mishra, M. Oxidative stress, mitochondrial damage and diabetic retinopathy. Biochim. Biophys. Acta. 2015, 1852, 2474–2483. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Curtis, T.M.; Stitt, A.W. Advanced glycation end products and diabetic retinopathy. Curr. Med. Chem. 2013, 20, 3234–3240. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Z.F.; Zhang, Q.; Liu, X.Z. Protective effects of curcumin on retinal Müller cell in early diabetic rats. Int. J. Ophthalmol. 2013, 6, 422–424. [Google Scholar] [PubMed]
- Rosa, M.D.; Distefano, G.; Gagliano, C.; Rusciano, D.; Malaguarnera, L. Autophagy in diabetic retinopathy. Curr. Neuropharmacol. 2016, 14, 810–825. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Tang, L.; Chen, B. Oxidative stress: Implications for the development of diabetic retinopathy and antioxidant therapeutic perspectives. Oxid. Med. Cell. Longev. 2014, 2014, 752387. [Google Scholar] [CrossRef] [PubMed]
- Maeda, S.; Matsui, T.; Ojima, A.; Takeuchi, M.; Yamagishi, S. Sulforaphane inhibits advanced glycation end product-induced pericyte damage by reducing expression of receptor for advanced glycation end products. Nutr. Res. 2014, 34, 807–813. [Google Scholar] [CrossRef] [PubMed]
- Elmasry, K.; Ibrahim, A.S.; Saleh, H.; Elsherbiny, N.; Elshafey, S.; Hussein, K.A.; Al-Shabrawey, M. Role of endoplasmic reticulum stress in 12/15-lipoxygenase-induced retinal microvascular dysfunction in a mouse model of diabetic retinopathy. Diabetologia 2018, 61, 1220–1232. [Google Scholar] [CrossRef] [PubMed]
- Minasyan, L.; Sreekumar, P.G.; Hinton, D.R.; Kannan, R. Protective mechanisms of the mitochondrial-derived peptide humanin in oxidative and endoplasmic reticulum stress in RPE cells. Oxid. Med. Cell. Longev. 2017, 2017, 1675230. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Cai, X.; Xia, Q.; Yao, K.; Chen, J.; Zhang, Y.; Naranmandura, H.; Liu, X.; Wu, Y. Involvement of endoplasmic reticulum stress in all-trans-retinal-induced retinal pigment epithelium degeneration. Toxicol. Sci. 2015, 143, 196–208. [Google Scholar] [CrossRef] [PubMed]
- Osada, H.; Okamoto, T.; Kawashima, H.; Toda, E.; Miyake, S.; Nagai, N.; Kobayashi, S.; Tsubota, K.; Ozawa, Y. Neuroprotective effect of bilberry extract in a murine model of photo-stressed retina. PLoS ONE 2017, 12, e0178627. [Google Scholar] [CrossRef] [PubMed]
- Karthikeyan, B.; Harini, L.; Krishnakumar, V.; Kannan, V.R.; Sundar, K.; Kathiresan, T. Insights on the involvement of (-)-epigallocatechin gallate in ER stress-mediated apoptosis in age-related macular degeneration. Apoptosis 2017, 22, 72–85. [Google Scholar] [CrossRef] [PubMed]
- Griciuc, A.; Aron, L.; Ueffing, M. ER stress in retinal degeneration: A target for rational therapy? Trends Mol. Med. 2011, 17, 442–451. [Google Scholar] [CrossRef] [PubMed]








© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, M.-K.; Lee, E.-J.; Kim, Y.-H.; Kim, D.Y.; Oh, H.; Kim, S.-I.; Kang, Y.-H. Chrysin Ameliorates Malfunction of Retinoid Visual Cycle through Blocking Activation of AGE-RAGE-ER Stress in Glucose-Stimulated Retinal Pigment Epithelial Cells and Diabetic Eyes. Nutrients 2018, 10, 1046. https://doi.org/10.3390/nu10081046
Kang M-K, Lee E-J, Kim Y-H, Kim DY, Oh H, Kim S-I, Kang Y-H. Chrysin Ameliorates Malfunction of Retinoid Visual Cycle through Blocking Activation of AGE-RAGE-ER Stress in Glucose-Stimulated Retinal Pigment Epithelial Cells and Diabetic Eyes. Nutrients. 2018; 10(8):1046. https://doi.org/10.3390/nu10081046
Chicago/Turabian StyleKang, Min-Kyung, Eun-Jung Lee, Yun-Ho Kim, Dong Yeon Kim, Hyeongjoo Oh, Soo-Il Kim, and Young-Hee Kang. 2018. "Chrysin Ameliorates Malfunction of Retinoid Visual Cycle through Blocking Activation of AGE-RAGE-ER Stress in Glucose-Stimulated Retinal Pigment Epithelial Cells and Diabetic Eyes" Nutrients 10, no. 8: 1046. https://doi.org/10.3390/nu10081046
APA StyleKang, M.-K., Lee, E.-J., Kim, Y.-H., Kim, D. Y., Oh, H., Kim, S.-I., & Kang, Y.-H. (2018). Chrysin Ameliorates Malfunction of Retinoid Visual Cycle through Blocking Activation of AGE-RAGE-ER Stress in Glucose-Stimulated Retinal Pigment Epithelial Cells and Diabetic Eyes. Nutrients, 10(8), 1046. https://doi.org/10.3390/nu10081046