Skeletal Muscle Metabolism in Duchenne and Becker Muscular Dystrophy—Implications for Therapies


Abstract
1. Introduction
2. The Interacting Nutritional and Pathological Aspects of MD
3. Normal Muscle Cellular Metabolism
4. Dystrophic Muscle Cellular Metabolism
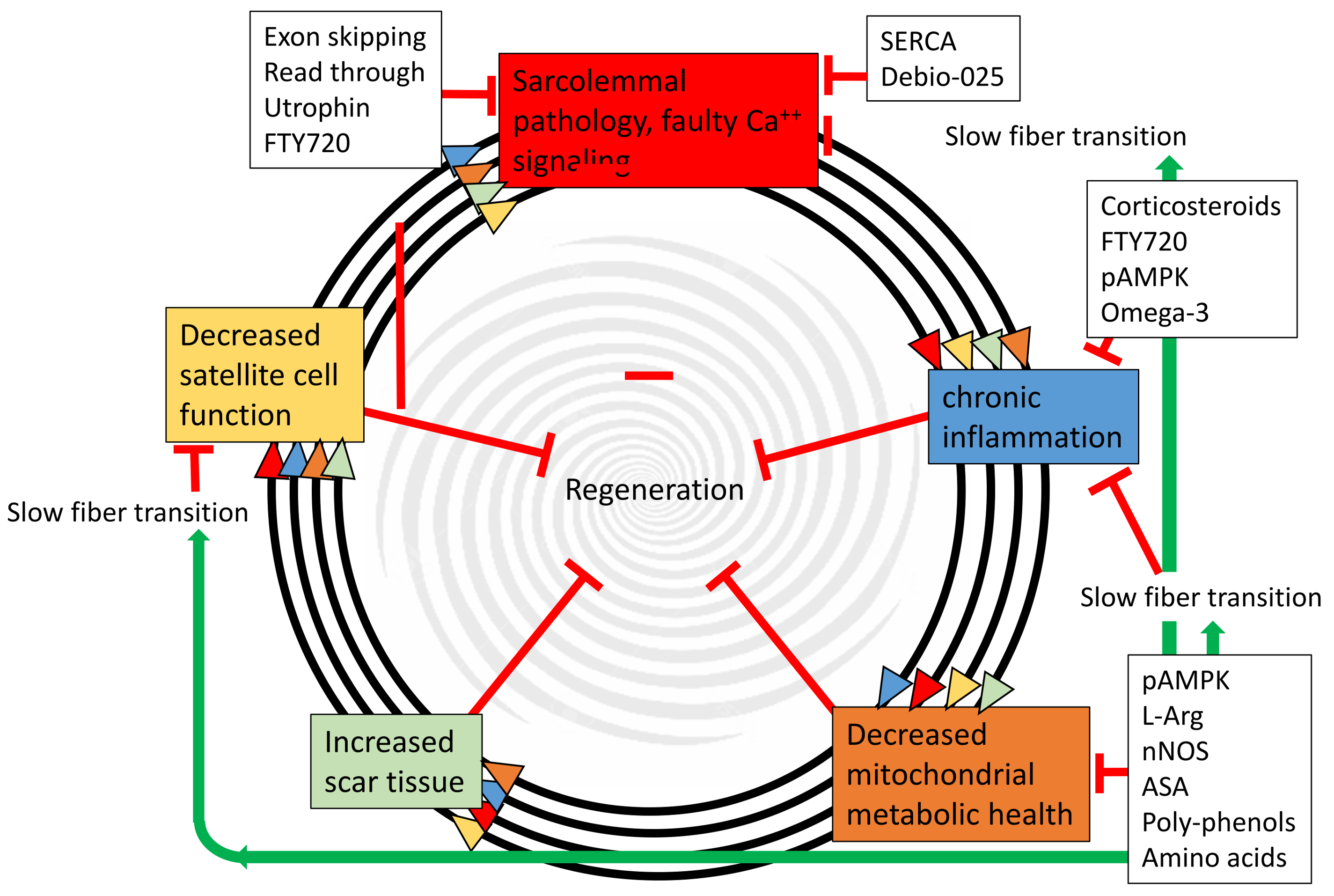
5. Possible Metabolic Therapeutic Avenues
6. Dietary Supplements
7. Polyphenol Supplement
8. Amino acid Supplements
9. Fatty acid Supplements
10. Exercise
11. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AICAR | 5-aminoimidazole-4-carboxamide riboside |
| ASA | 5-aminosalicylic acid |
| CK | creatine kinase |
| CN | central nuclei |
| DKO | double dystrophin and utrophin knockout mice |
| DMD | duchenne muscular dystrophy |
| EBD | Evans blue dye |
| Fnip-1 | Folliculin interacting protein 1 |
| MD | muscular dystrophy |
| MPTP | Mitochondrial permeability transition pore |
| nNOS | neuronal nitric oxide synthase |
| NO | nitric oxide |
| pAMPK | phosphorylated AMP activated protein kinase |
| PDE | phosphodiesterase |
| PGC1-α | Peroxisome proliferator-activated receptor γ coactivator 1-α |
| SERCA | sarco/endoplasmic reticulum Ca2+-ATPase |
| SIRT1 | NAD-dependent deacetylase sirtuin-1 |
| TGFβ | transforming growth factor beta 1 |
| Utr | utrophin |
References
- Whitmore, C.; Morgan, J. What do mouse models of muscular dystrophy tell us about the DAPC and its components? Int. J. Exp. Pathol. 2014, 95, 365–377. [Google Scholar] [CrossRef] [PubMed]
- Bulfield, G.; Siller, W.G.; Wight, P.A.; Moore, K.J. X chromosome-linked muscular dystrophy (mdx) in the mouse. Proc. Natl. Acad. Sci. USA 1984, 81, 1189–1192. [Google Scholar] [CrossRef] [PubMed]
- Dangain, J.; Vrbova, G. Muscle development in mdx mutant mice. Muscle Nerve 1984, 7, 700–704. [Google Scholar] [CrossRef] [PubMed]
- Deconinck, A.E.; Rafael, J.A.; Skinner, J.A.; Brown, S.C.; Potter, A.C.; Metzinger, L.; Watt, D.J.; Dickson, J.G.; Tinsley, J.M.; Davies, K.E. Utrophin-dystrophin-deficient mice as a model for Duchenne muscular dystrophy. Cell 1997, 90, 717–727. [Google Scholar] [CrossRef]
- Fukada, S.; Morikawa, D.; Yamamoto, Y.; Yoshida, T.; Sumie, N.; Yamaguchi, M.; Ito, T.; Miyagoe-Suzuki, Y.; Takeda, S.; Tsujikawa, K.; et al. Genetic background affects properties of satellite cells and mdx phenotypes. Am. J. Pathol. 2010, 176, 2414–2424. [Google Scholar] [CrossRef] [PubMed]
- Coley, W.D.; Bogdanik, L.; Vila, M.C.; Yu, Q.; Van Der Meulen, J.H.; Rayavarapu, S.; Novak, J.S.; Nearing, M.; Quinn, J.L.; Saunders, A.; et al. Effect of genetic background on the dystrophic phenotype in mdx mice. Hum. Mol. Genet. 2016, 25, 130–145. [Google Scholar] [CrossRef] [PubMed]
- Heydemann, A.; Ceco, E.; Lim, J.E.; Hadhazy, M.; Ryder, P.; Moran, J.L.; Beier, D.R.; Palmer, A.A.; McNally, E.M. Latent TGF-beta-binding protein 4 modifies muscular dystrophy in mice. J. Clin. Investig. 2009, 119, 3703–3712. [Google Scholar] [CrossRef] [PubMed]
- Heydemann, A.; Huber, J.M.; Demonbreun, A.; Hadhazy, M.; McNally, E.M. Genetic background influences muscular dystrophy. Neuromuscul. Disord. 2005, 15, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Roberts, N.W.; Holley-Cuthrell, J.; Gonzalez-Vega, M.; Mull, A.J.; Heydemann, A. Biochemical and Functional Comparisons of mdx and Sgcg(−/−) Muscular Dystrophy Mouse Models. BioMed Res. Int. 2015, 2015, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Tidball, J.G.; Villalta, S.A. Regulatory interactions between muscle and the immune system during muscle regeneration. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 298, R1173–R1187. [Google Scholar] [CrossRef] [PubMed]
- Mann, C.J.; Perdiguero, E.; Kharraz, Y.; Aguilar, S.; Pessina, P.; Serrano, A.L.; Munoz-Canoves, P. Aberrant repair and fibrosis development in skeletal muscle. Skelet. Muscle 2011, 1, 21. [Google Scholar] [CrossRef] [PubMed]
- Van Ruiten, H.J.; Straub, V.; Bushby, K.; Guglieri, M. Improving recognition of Duchenne muscular dystrophy: A retrospective case note review. Arch. Dis. Child. 2014, 99, 1074–1077. [Google Scholar] [CrossRef] [PubMed]
- Timpani, C.A.; Hayes, A.; Rybalka, E. Revisiting the dystrophin-ATP connection: How half a century of research still implicates mitochondrial dysfunction in Duchenne Muscular Dystrophy aetiology. Med. Hypotheses 2015, 85, 1021–1033. [Google Scholar] [CrossRef] [PubMed]
- Rodino-Klapac, L.R.; Janssen, P.M.; Shontz, K.M.; Canan, B.; Montgomery, C.L.; Griffin, D.; Heller, K.; Schmelzer, L.; Handy, C.; Clark, K.R.; et al. Micro-dystrophin and follistatin co-delivery restores muscle function in aged DMD model. Hum. Mol. Genet. 2013, 22, 4929–4937. [Google Scholar] [CrossRef] [PubMed]
- Heydemann, A. Severe murine limb-girdle muscular dystrophy type 2C pathology is diminished by FTY720 treatment. Muscle Nerve 2016, 56, 486–494. [Google Scholar] [CrossRef] [PubMed]
- Pereira, J.A.; Mauricio, A.F.; Marques, M.J.; Neto, H.S. Dual Therapy Deflazacort/Doxycyclyne Is Better Than Deflazacort Monotherapy to Alleviate Cardiomyopathy in Dystrophin-Deficient mdx Mice. J. Cardiovasc. Pharmacol. Ther. 2017, 22, 458–466. [Google Scholar] [CrossRef] [PubMed]
- Salera, S.; Menni, F.; Moggio, M.; Guez, S.; Sciacco, M.; Esposito, S. Nutritional Challenges in Duchenne Muscular Dystrophy. Nutrients 2017, 9, 594. [Google Scholar] [CrossRef] [PubMed]
- Pane, M.; Vasta, I.; Messina, S.; Sorleti, D.; Aloysius, A.; Sciarra, F.; Mangiola, F.; Kinali, M.; Ricci, E.; Mercuri, E. Feeding problems and weight gain in Duchenne muscular dystrophy. Eur. J. Paediatr. Neurol. 2006, 10, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.; Samuels, E.; Mullins, L. Nutrition Considerations in Duchenne Muscular Dystrophy. Nutr. Clin. Pract. 2015, 30, 511–521. [Google Scholar] [CrossRef] [PubMed]
- Pette, D.; Staron, R.S. Myosin isoforms, muscle fiber types, and transitions. Microsc. Res. Tech. 2000, 50, 500–509. [Google Scholar] [CrossRef]
- Westerblad, H.; Bruton, J.D.; Katz, A. Skeletal muscle: Energy metabolism, fiber types, fatigue and adaptability. Exp. Cell Res. 2010, 316, 3093–3099. [Google Scholar] [CrossRef] [PubMed]
- Ferraro, E.; Giammarioli, A.M.; Chiandotto, S.; Spoletini, I.; Rosano, G. Exercise-induced skeletal muscle remodeling and metabolic adaptation: Redox signaling and role of autophagy. Antioxid. Redox Signal. 2014, 21, 154–176. [Google Scholar] [CrossRef] [PubMed]
- Booth, F.W.; Babij, P.; Thomason, D.B.; Wong, T.S.; Morrison, P.R. Adaptation of muscle gene expression to changes in contractile activity. Adv. Myochem. 1987, 1, 205–216. [Google Scholar] [PubMed]
- Baldwin, K.M.; Klinkerfuss, G.H.; Terjung, R.L.; Mole, P.A.; Holloszy, J.O. Respiratory capacity of white, red, and intermediate muscle: Adaptative response to exercise. Am. J. Physiol. 1972, 222, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Webster, C.; Silberstein, L.; Hays, A.P.; Blau, H.M. Fast muscle fibers are preferentially affected in Duchenne muscular dystrophy. Cell 1988, 52, 503–513. [Google Scholar] [CrossRef]
- Sahlin, K. Muscle fatigue and lactic acid accumulation. Acta Physiol. Scand. Suppl. 1986, 556, 83–91. [Google Scholar] [PubMed]
- Baldwin, K.M.; Tipton, C.M. Work and metabolic patterns of fast and slow twitch skeletal muscle contracting in situ. Pflugers Arch. 1972, 334, 345–356. [Google Scholar] [CrossRef] [PubMed]
- Fluck, M. Functional, structural and molecular plasticity of mammalian skeletal muscle in response to exercise stimuli. J. Exp. Biol. 2006, 209, 2239–2248. [Google Scholar] [CrossRef] [PubMed]
- MacInnis, M.J.; Gibala, M.J. Physiological adaptations to interval training and the role of exercise intensity. J. Physiol. 2017, 595, 2915–2930. [Google Scholar] [CrossRef] [PubMed]
- Schiaffino, S.; Reggiani, C. Fiber types in mammalian skeletal muscles. Physiol. Rev. 2011, 91, 1447–1531. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liang, X.; Zhou, D.; Lai, L.; Xiao, L.; Liu, L.; Fu, T.; Kong, Y.; Zhou, Q.; Vega, R.B.; et al. Coupling of mitochondrial function and skeletal muscle fiber type by a miR-499/Fnip1/AMPK circuit. EMBO Mol. Med. 2016, 8, 1212–1228. [Google Scholar] [CrossRef] [PubMed]
- Moens, P.; Baatsen, P.H.; Marechal, G. Increased susceptibility of EDL muscles from mdx mice to damage induced by contractions with stretch. J. Muscle Res. Cell Motil. 1993, 14, 446–451. [Google Scholar] [CrossRef] [PubMed]
- Baker, P.E.; Kearney, J.A.; Gong, B.; Merriam, A.P.; Kuhn, D.E.; Porter, J.D.; Rafael-Fortney, J.A. Analysis of gene expression differences between utrophin/dystrophin-deficient vs. mdx skeletal muscles reveals a specific upregulation of slow muscle genes in limb muscles. Neurogenetics 2006, 7, 81–91. [Google Scholar] [PubMed]
- Selsby, J.T.; Morine, K.J.; Pendrak, K.; Barton, E.R.; Sweeney, H.L. Rescue of dystrophic skeletal muscle by PGC-1alpha involves a fast to slow fiber type shift in the mdx mouse. PLoS ONE 2012, 7, e30063. [Google Scholar] [CrossRef] [PubMed]
- Pant, M.; Sopariwala, D.H.; Bal, N.C.; Lowe, J.; Delfin, D.A.; Rafael-Fortney, J.; Periasamy, M. Metabolic dysfunction and altered mitochondrial dynamics in the utrophin-dystrophin deficient mouse model of duchenne muscular dystrophy. PLoS ONE 2015, 10, e0123875. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, K.; Ervasti, J.M.; Ohlendieck, K.; Kahl, S.D.; Campbell, K.P. Association of dystrophin-related protein with dystrophin-associated proteins in mdx mouse muscle. Nature 1992, 360, 588–591. [Google Scholar] [CrossRef] [PubMed]
- Miura, P.; Jasmin, B.J. Utrophin upregulation for treating Duchenne or Becker muscular dystrophy: How close are we? Trends Mol. Med. 2006, 12, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Anderson, E.J.; Neufer, P.D. Type II skeletal myofibers possess unique properties that potentiate mitochondrial H(2)O(2) generation. Am. J. Physiol. Cell Physiol. 2006, 290, C844–C851. [Google Scholar] [CrossRef] [PubMed]
- Zuo, L.; Pannell, B.K. Redox Characterization of Functioning Skeletal Muscle. Front. Physiol. 2015, 6, 338. [Google Scholar] [CrossRef] [PubMed]
- Ljubicic, V.; Jasmin, B.J. AMP-activated protein kinase at the nexus of therapeutic skeletal muscle plasticity in Duchenne muscular dystrophy. Trends Mol. Med. 2013, 19, 614–624. [Google Scholar] [CrossRef] [PubMed]
- Berhanu, T.K.; Holley-Cuthrell, J.; Roberts, N.W.; Mull, A.J.; Heydemann, A. Increased AMP-activated protein kinase in skeletal muscles of Murphy Roth Large mice and its potential role in altered metabolism. Physiol. Rep. 2014, 2, e00252. [Google Scholar] [CrossRef] [PubMed]
- Heydemann, A.; Swaggart, K.A.; Kim, G.H.; Holley-Cuthrell, J.; Hadhazy, M.; McNally, E.M. The superhealing MRL background improves muscular dystrophy. Skelet. Muscle 2012, 2, 26. [Google Scholar] [CrossRef] [PubMed]
- Luo, T.; Nocon, A.; Fry, J.; Sherban, A.; Rui, X.; Jiang, B.; Xu, X.J.; Han, J.; Yan, Y.; Yang, Q.; et al. AMPK Activation by Metformin Suppresses Abnormal Extracellular Matrix Remodeling in Adipose Tissue and Ameliorates Insulin Resistance in Obesity. Diabetes 2016, 65, 2295–2310. [Google Scholar] [CrossRef] [PubMed]
- Dial, A.G.; Ng, S.Y.; Manta, A.; Ljubicic, V. The Role of AMPK in Neuromuscular Biology and Disease. Trends Endocrinol. Metab. 2018, 29, 300–312. [Google Scholar] [CrossRef] [PubMed]
- Pauly, M.; Daussin, F.; Burelle, Y.; Li, T.; Godin, R.; Fauconnier, J.; Koechlin-Ramonatxo, C.; Hugon, G.; Lacampagne, A.; Coisy-Quivy, M.; et al. AMPK activation stimulates autophagy and ameliorates muscular dystrophy in the mdx mouse diaphragm. Am. J. Pathol. 2012, 181, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Rafael, J.A.; Townsend, E.R.; Squire, S.E.; Potter, A.C.; Chamberlain, J.S.; Davies, K.E. Dystrophin and utrophin influence fiber type composition and post-synaptic membrane structure. Hum. Mol. Genet. 2000, 9, 1357–1367. [Google Scholar] [CrossRef] [PubMed]
- Vila, M.C.; Rayavarapu, S.; Hogarth, M.W.; Van der Meulen, J.H.; Horn, A.; Defour, A.; Takeda, S.; Brown, K.J.; Hathout, Y.; Nagaraju, K.; et al. Mitochondria mediate cell membrane repair and contribute to Duchenne muscular dystrophy. Cell Death Differ. 2017, 24, 330–342. [Google Scholar] [CrossRef] [PubMed]
- Godin, R.; Daussin, F.; Matecki, S.; Li, T.; Petrof, B.J.; Burelle, Y. Peroxisome proliferator-activated receptor gamma coactivator1-gene alpha transfer restores mitochondrial biomass and improves mitochondrial calcium handling in post-necrotic mdx mouse skeletal muscle. J. Physiol. 2012, 590, 5487–5502. [Google Scholar] [CrossRef] [PubMed]
- Jahnke, V.E.; Van Der Meulen, J.H.; Johnston, H.K.; Ghimbovschi, S.; Partridge, T.; Hoffman, E.P.; Nagaraju, K. Metabolic remodeling agents show beneficial effects in the dystrophin-deficient mdx mouse model. Skelet. Muscle 2012, 2, 16. [Google Scholar] [CrossRef] [PubMed]
- Akhmedov, D.; Berdeaux, R. The effects of obesity on skeletal muscle regeneration. Front. Physiol. 2013, 4, 371. [Google Scholar] [CrossRef] [PubMed]
- Rybalka, E.; Timpani, C.A.; Stathis, C.G.; Hayes, A.; Cooke, M.B. Metabogenic and Nutriceutical Approaches to Address Energy Dysregulation and Skeletal Muscle Wasting in Duchenne Muscular Dystrophy. Nutrients 2015, 7, 9734–9767. [Google Scholar] [CrossRef] [PubMed]
- Austin, L.; de Niese, M.; McGregor, A.; Arthur, H.; Gurusinghe, A.; Gould, M.K. Potential oxyradical damage and energy status in individual muscle fibres from degenerating muscle diseases. Neuromuscul. Disord. 1992, 2, 27–33. [Google Scholar] [CrossRef]
- Cole, M.A.; Rafael, J.A.; Taylor, D.J.; Lodi, R.; Davies, K.E.; Styles, P. A quantitative study of bioenergetics in skeletal muscle lacking utrophin and dystrophin. Neuromuscul. Disord. 2002, 12, 247–257. [Google Scholar] [CrossRef]
- Kuznetsov, A.V.; Winkler, K.; Wiedemann, F.R.; von Bossanyi, P.; Dietzmann, K.; Kunz, W.S. Impaired mitochondrial oxidative phosphorylation in skeletal muscle of the dystrophin-deficient mdx mouse. Mol. Cell. Biochem. 1998, 183, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Camina, F.; Novo-Rodriguez, M.I.; Rodriguez-Segade, S.; Castro-Gago, M. Purine and carnitine metabolism in muscle of patients with Duchenne muscular dystrophy. Clin. Chim. Acta 1995, 243, 151–164. [Google Scholar] [CrossRef]
- Rybalka, E.; Timpani, C.A.; Cooke, M.B.; Williams, A.D.; Hayes, A. Defects in mitochondrial ATP synthesis in dystrophin-deficient mdx skeletal muscles may be caused by complex I insufficiency. PLoS ONE 2014, 9, e115763. [Google Scholar] [CrossRef] [PubMed]
- Masubuchi, N.; Shidoh, Y.; Kondo, S.; Takatoh, J.; Hanaoka, K. Subcellular localization of dystrophin isoforms in cardiomyocytes and phenotypic analysis of dystrophin-deficient mice reveal cardiac myopathy is predominantly caused by a deficiency in full-length dystrophin. Exp. Anim. 2013, 62, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Ueda, H.; Ueda, K.; Baba, T.; Ohno, S. Delta- and gamma-Sarcoglycan localization in the sarcoplasmic reticulum of skeletal muscle. J. Histochem. Cytochem. 2001, 49, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Kaprielian, R.R.; Stevenson, S.; Rothery, S.M.; Cullen, M.J.; Severs, N.J. Distinct patterns of dystrophin organization in myocyte sarcolemma and transverse tubules of normal and diseased human myocardium. Circulation 2000, 101, 2586–2594. [Google Scholar] [CrossRef] [PubMed]
- Dunn, J.F.; Radda, G.K. Total ion content of skeletal and cardiac muscle in the mdx mouse dystrophy: Ca2+ is elevated at all ages. J. Neurol. Sci. 1991, 103, 226–231. [Google Scholar] [CrossRef]
- Rando, T.A. Role of nitric oxide in the pathogenesis of muscular dystrophies: A “two hit” hypothesis of the cause of muscle necrosis. Microsc. Res. Tech. 2001, 55, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Menazza, S.; Blaauw, B.; Tiepolo, T.; Toniolo, L.; Braghetta, P.; Spolaore, B.; Reggiani, C.; Di Lisa, F.; Bonaldo, P.; Canton, M. Oxidative stress by monoamine oxidases is causally involved in myofiber damage in muscular dystrophy. Hum. Mol. Genet. 2010, 19, 4207–4215. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, L.F.; Laitano, O. Regulation of NADPH oxidases in skeletal muscle. Free Radic. Biol. Med. 2016, 98, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Sheu, S.S.; Wang, W.; Cheng, H.; Dirksen, R.T. Superoxide flashes: Illuminating new insights into cardiac ischemia/reperfusion injury. Future Cardiol. 2008, 4, 551–554. [Google Scholar] [CrossRef] [PubMed]
- Brand, M.D. The sites and topology of mitochondrial superoxide production. Exp. Gerontol. 2010, 45, 466–472. [Google Scholar] [CrossRef] [PubMed]
- Canton, M.; Menazza, S.; Di Lisa, F. Oxidative stress in muscular dystrophy: From generic evidence to specific sources and targets. J. Muscle Res. Cell Motil. 2014, 35, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Brenman, J.E.; Chao, D.S.; Xia, H.; Aldape, K.; Bredt, D.S. Nitric oxide synthase complexed with dystrophin and absent from skeletal muscle sarcolemma in Duchenne muscular dystrophy. Cell 1995, 82, 743–752. [Google Scholar] [CrossRef]
- Timpani, C.A.; Hayes, A.; Rybalka, E. Therapeutic strategies to address neuronal nitric oxide synthase deficiency and the loss of nitric oxide bioavailability in Duchenne Muscular Dystrophy. Orphanet J. Rare Dis. 2017, 12, 100. [Google Scholar] [CrossRef] [PubMed]
- Thomas, G.D.; Sander, M.; Lau, K.S.; Huang, P.L.; Stull, J.T.; Victor, R.G. Impaired metabolic modulation of alpha-adrenergic vasoconstriction in dystrophin-deficient skeletal muscle. Proc. Natl. Acad. Sci. USA 1998, 95, 15090–15095. [Google Scholar] [CrossRef] [PubMed]
- Torelli, S.; Brown, S.C.; Jimenez-Mallebrera, C.; Feng, L.; Muntoni, F.; Sewry, C.A. Absence of neuronal nitric oxide synthase (nNOS) as a pathological marker for the diagnosis of Becker muscular dystrophy with rod domain deletions. Neuropathol. Appl. Neurobiol. 2004, 30, 540–545. [Google Scholar] [CrossRef] [PubMed]
- Sander, M.; Chavoshan, B.; Harris, S.A.; Iannaccone, S.T.; Stull, J.T.; Thomas, G.D.; Victor, R.G. Functional muscle ischemia in neuronal nitric oxide synthase-deficient skeletal muscle of children with Duchenne muscular dystrophy. Proc. Natl. Acad. Sci. USA 2000, 97, 13818–13823. [Google Scholar] [CrossRef] [PubMed]
- Crosbie, R.H.; Straub, V.; Yun, H.Y.; Lee, J.C.; Rafael, J.A.; Chamberlain, J.S.; Dawson, V.L.; Dawson, T.M.; Campbell, K.P. Mdx muscle pathology is independent of nNOS perturbation. Hum. Mol. Genet. 1998, 7, 823–829. [Google Scholar] [CrossRef] [PubMed]
- Tidball, J.G.; Wehling-Henricks, M. The role of free radicals in the pathophysiology of muscular dystrophy. J. Appl. Physiol. 2007, 102, 1677–1686. [Google Scholar] [CrossRef] [PubMed]
- Balon, T.W.; Nadler, J.L. Evidence that nitric oxide increases glucose transport in skeletal muscle. J. Appl. Physiol. 1997, 82, 359–363. [Google Scholar] [CrossRef] [PubMed]
- Wehling-Henricks, M.; Oltmann, M.; Rinaldi, C.; Myung, K.H.; Tidball, J.G. Loss of positive allosteric interactions between neuronal nitric oxide synthase and phosphofructokinase contributes to defects in glycolysis and increased fatigability in muscular dystrophy. Hum. Mol. Genet. 2009, 18, 3439–3451. [Google Scholar] [CrossRef] [PubMed]
- Giacomello, M.; Drago, I.; Pizzo, P.; Pozzan, T. Mitochondrial Ca2+ as a key regulator of cell life and death. Cell Death Differ. 2007, 14, 1267–1274. [Google Scholar] [CrossRef] [PubMed]
- Rubi, L.; Todt, H.; Kubista, H.; Koenig, X.; Hilber, K. Calcium current properties in dystrophin-deficient ventricular cardiomyocytes from aged mdx mice. Physiol. Rep. 2018, 6. [Google Scholar] [CrossRef] [PubMed]
- McCormack, J.G.; Denton, R.M. The role of intramitochondrial Ca2+ in the regulation of oxidative phosphorylation in mammalian tissues. Biochem. Soc. Trans. 1993, 21, 793–799. [Google Scholar] [CrossRef] [PubMed]
- Millay, D.P.; Goonasekera, S.A.; Sargent, M.A.; Maillet, M.; Aronow, B.J.; Molkentin, J.D. Calcium influx is sufficient to induce muscular dystrophy through a TRPC-dependent mechanism. Proc. Natl. Acad. Sci. USA 2009, 106, 19023–19028. [Google Scholar] [CrossRef] [PubMed]
- Burr, A.R.; Molkentin, J.D. Genetic evidence in the mouse solidifies the calcium hypothesis of myofiber death in muscular dystrophy. Cell Death Differ. 2015, 22, 1402–1412. [Google Scholar] [CrossRef] [PubMed]
- Baron, D.; Magot, A.; Ramstein, G.; Steenman, M.; Fayet, G.; Chevalier, C.; Jourdon, P.; Houlgatte, R.; Savagner, F.; Pereon, Y. Immune response and mitochondrial metabolism are commonly deregulated in DMD and aging skeletal muscle. PLoS ONE 2011, 6, e26952. [Google Scholar] [CrossRef] [PubMed]
- Gannoun-Zaki, L.; Fournier-Bidoz, S.; Le Cam, G.; Chambon, C.; Millasseau, P.; Leger, J.J.; Dechesne, C.A. Down-regulation of mitochondrial mRNAs in the mdx mouse model for Duchenne muscular dystrophy. FEBS Lett. 1995, 375, 268–272. [Google Scholar] [CrossRef]
- Mizunoya, W.; Okamoto, S.; Miyahara, H.; Akahoshi, M.; Suzuki, T.; Do, M.Q.; Ohtsubo, H.; Komiya, Y.; Qahar, M.; Waga, T.; et al. Fast-to-slow shift of muscle fiber-type composition by dietary apple polyphenols in rats: Impact of the low-dose supplementation. Anim. Sci. J. 2017, 88, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Hafner, P.; Bonati, U.; Erne, B.; Schmid, M.; Rubino, D.; Pohlman, U.; Peters, T.; Rutz, E.; Frank, S.; Neuhaus, C.; et al. Improved Muscle Function in Duchenne Muscular Dystrophy through L-Arginine and Metformin: An Investigator-Initiated, Open-Label, Single-Center, Proof-of-Concept-Study. PLoS ONE 2016, 11, e0147634. [Google Scholar] [CrossRef] [PubMed]
- Passaquin, A.C.; Renard, M.; Kay, L.; Challet, C.; Mokhtarian, A.; Wallimann, T.; Ruegg, U.T. Creatine supplementation reduces skeletal muscle degeneration and enhances mitochondrial function in mdx mice. Neuromuscul. Disord. 2002, 12, 174–182. [Google Scholar] [CrossRef]
- Mok, E.; Eleouet-Da Violante, C.; Daubrosse, C.; Gottrand, F.; Rigal, O.; Fontan, J.E.; Cuisset, J.M.; Guilhot, J.; Hankard, R. Oral glutamine and amino acid supplementation inhibit whole-body protein degradation in children with Duchenne muscular dystrophy. Am. J. Clin. Nutr. 2006, 83, 823–828. [Google Scholar] [CrossRef] [PubMed]
- Mok, E.; Letellier, G.; Cuisset, J.M.; Denjean, A.; Gottrand, F.; Alberti, C.; Hankard, R. Lack of functional benefit with glutamine versus placebo in Duchenne muscular dystrophy: A randomized crossover trial. PLoS ONE 2009, 4, e5448. [Google Scholar] [CrossRef] [PubMed]
- Gordon, B.S.; Lowe, D.A.; Kostek, M.C. Exercise increases utrophin protein expression in the mdx mouse model of Duchenne muscular dystrophy. Muscle Nerve 2014, 49, 915–918. [Google Scholar] [CrossRef] [PubMed]
- Morici, G.; Frinchi, M.; Pitruzzella, A.; Di Liberto, V.; Barone, R.; Pace, A.; Di Felice, V.; Belluardo, N.; Cappello, F.; Mudo, G.; et al. Mild Aerobic Exercise Training Hardly Affects the Diaphragm of mdx Mice. J. Cell. Physiol. 2017, 232, 2044–2052. [Google Scholar] [CrossRef] [PubMed]
- Hyzewicz, J.; Tanihata, J.; Kuraoka, M.; Nitahara-Kasahara, Y.; Beylier, T.; Ruegg, U.T.; Vater, A.; Takeda, S. Low-Intensity Training and the C5a Complement Antagonist NOX-D21 Rescue the mdx Phenotype through Modulation of Inflammation. Am. J. Pathol. 2017, 187, 1147–1161. [Google Scholar] [CrossRef] [PubMed]
- Timmons, J.A.; Larsson, O.; Jansson, E.; Fischer, H.; Gustafsson, T.; Greenhaff, P.L.; Ridden, J.; Rachman, J.; Peyrard-Janvid, M.; Wahlestedt, C.; et al. Human muscle gene expression responses to endurance training provide a novel perspective on Duchenne muscular dystrophy. FASEB J. 2005, 19, 750–760. [Google Scholar] [CrossRef] [PubMed]
- De Lateur, B.J.; Giaconi, R.M. Effect on maximal strength of submaximal exercise in Duchenne muscular dystrophy. Am. J. Phys. Med. 1979, 58, 26–36. [Google Scholar] [PubMed]
- Markert, C.D.; Ambrosio, F.; Call, J.A.; Grange, R.W. Exercise and Duchenne muscular dystrophy: Toward evidence-based exercise prescription. Muscle Nerve 2011, 43, 464–478. [Google Scholar] [CrossRef] [PubMed]
- Vignos, P.J., Jr. Physical models of rehabilitation in neuromuscular disease. Muscle Nerve 1983, 6, 323–338. [Google Scholar] [CrossRef] [PubMed]
- Jansen, M.; van Alfen, N.; Geurts, A.C.; de Groot, I.J. Assisted bicycle training delays functional deterioration in boys with Duchenne muscular dystrophy: The randomized controlled trial “no use is disuse”. Neurorehabilit. Neural Repair 2013, 27, 816–827. [Google Scholar] [CrossRef] [PubMed]
- Ljubicic, V.; Jasmin, B.J. Metformin increases peroxisome proliferator-activated receptor gamma Co-activator-1alpha and utrophin a expression in dystrophic skeletal muscle. Muscle Nerve 2015, 52, 139–142. [Google Scholar] [CrossRef] [PubMed]
- Ljubicic, V.; Miura, P.; Burt, M.; Boudreault, L.; Khogali, S.; Lunde, J.A.; Renaud, J.M.; Jasmin, B.J. Chronic AMPK activation evokes the slow, oxidative myogenic program and triggers beneficial adaptations in mdx mouse skeletal muscle. Hum. Mol. Genet. 2011, 20, 3478–3493. [Google Scholar] [CrossRef] [PubMed]
- Ljubicic, V.; Burt, M.; Lunde, J.A.; Jasmin, B.J. Resveratrol induces expression of the slow, oxidative phenotype in mdx mouse muscle together with enhanced activity of the SIRT1-PGC-1alpha axis. Am. J. Physiol. Cell Physiol. 2014, 307, C66–C82. [Google Scholar] [CrossRef] [PubMed]
- Hori, Y.S.; Kuno, A.; Hosoda, R.; Tanno, M.; Miura, T.; Shimamoto, K.; Horio, Y. Resveratrol ameliorates muscular pathology in the dystrophic mdx mouse, a model for Duchenne muscular dystrophy. J. Pharmacol. Exp. Ther. 2011, 338, 784–794. [Google Scholar] [CrossRef] [PubMed]
- Reyes, N.L.; Banks, G.B.; Tsang, M.; Margineantu, D.; Gu, H.; Djukovic, D.; Chan, J.; Torres, M.; Liggitt, H.D.; Hirenallur, S.D.; et al. Fnip1 regulates skeletal muscle fiber type specification, fatigue resistance, and susceptibility to muscular dystrophy. Proc. Natl. Acad. Sci. USA 2015, 112, 424–429. [Google Scholar] [CrossRef] [PubMed]
- Rafael, J.A.; Tinsley, J.M.; Potter, A.C.; Deconinck, A.E.; Davies, K.E. Skeletal muscle-specific expression of a utrophin transgene rescues utrophin-dystrophin deficient mice. Nat. Genet. 1998, 19, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Tinsley, J.; Deconinck, N.; Fisher, R.; Kahn, D.; Phelps, S.; Gillis, J.M.; Davies, K. Expression of full-length utrophin prevents muscular dystrophy in mdx mice. Nat. Med. 1998, 4, 1441–1444. [Google Scholar] [CrossRef] [PubMed]
- Tinsley, J.M.; Fairclough, R.J.; Storer, R.; Wilkes, F.J.; Potter, A.C.; Squire, S.E.; Powell, D.S.; Cozzoli, A.; Capogrosso, R.F.; Lambert, A.; et al. Daily treatment with SMTC1100, a novel small molecule utrophin upregulator, dramatically reduces the dystrophic symptoms in the mdx mouse. PLoS ONE 2011, 6, e19189. [Google Scholar] [CrossRef] [PubMed]
- Landisch, R.M.; Kosir, A.M.; Nelson, S.A.; Baltgalvis, K.A.; Lowe, D.A. Adaptive and nonadaptive responses to voluntary wheel running by mdx mice. Muscle Nerve 2008, 38, 1290–1303. [Google Scholar] [CrossRef] [PubMed]
- Ricotti, V.; Spinty, S.; Roper, H.; Hughes, I.; Tejura, B.; Robinson, N.; Layton, G.; Davies, K.; Muntoni, F.; Tinsley, J. Safety, Tolerability, and Pharmacokinetics of SMT C1100, a 2-Arylbenzoxazole Utrophin Modulator, following Single- and Multiple-Dose Administration to Pediatric Patients with Duchenne Muscular Dystrophy. PLoS ONE 2016, 11, e0152840. [Google Scholar] [CrossRef] [PubMed]
- Al-Rewashdy, H.; Ljubicic, V.; Lin, W.; Renaud, J.M.; Jasmin, B.J. Utrophin A is essential in mediating the functional adaptations of mdx mouse muscle following chronic AMPK activation. Hum. Mol. Genet. 2015, 24, 1243–1255. [Google Scholar] [CrossRef] [PubMed]
- Shiao, T.; Fond, A.; Deng, B.; Wehling-Henricks, M.; Adams, M.E.; Froehner, S.C.; Tidball, J.G. Defects in neuromuscular junction structure in dystrophic muscle are corrected by expression of a NOS transgene in dystrophin-deficient muscles, but not in muscles lacking alpha- and beta1-syntrophins. Hum. Mol. Genet. 2004, 13, 1873–1884. [Google Scholar] [CrossRef] [PubMed]
- Tidball, J.G.; Wehling-Henricks, M. Expression of a NOS transgene in dystrophin-deficient muscle reduces muscle membrane damage without increasing the expression of membrane-associated cytoskeletal proteins. Mol. Genet. Metab. 2004, 82, 312–320. [Google Scholar] [CrossRef] [PubMed]
- Wehling, M.; Spencer, M.J.; Tidball, J.G. A nitric oxide synthase transgene ameliorates muscular dystrophy in mdx mice. J. Cell Biol. 2001, 155, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Barton, E.R.; Morris, L.; Kawana, M.; Bish, L.T.; Toursel, T. Systemic administration of L-arginine benefits mdx skeletal muscle function. Muscle Nerve 2005, 32, 751–760. [Google Scholar] [CrossRef] [PubMed]
- Ascah, A.; Khairallah, M.; Daussin, F.; Bourcier-Lucas, C.; Godin, R.; Allen, B.G.; Petrof, B.J.; Des Rosiers, C.; Burelle, Y. Stress-induced opening of the permeability transition pore in the dystrophin-deficient heart is attenuated by acute treatment with sildenafil. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H144–H153. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Durrant, D.; Salloum, F.N.; Xi, L.; Kukreja, R.C. PDE5 inhibitors as therapeutics for heart disease, diabetes and cancer. Pharmacol. Ther. 2015, 147, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Leung, D.G.; Herzka, D.A.; Thompson, W.R.; He, B.; Bibat, G.; Tennekoon, G.; Russell, S.D.; Schuleri, K.H.; Lardo, A.C.; Kass, D.A.; et al. Sildenafil does not improve cardiomyopathy in Duchenne/Becker muscular dystrophy. Ann. Neurol. 2014, 76, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Witting, N.; Kruuse, C.; Nyhuus, B.; Prahm, K.P.; Citirak, G.; Lundgaard, S.J.; von Huth, S.; Vejlstrup, N.; Lindberg, U.; Krag, T.O.; et al. Effect of sildenafil on skeletal and cardiac muscle in Becker muscular dystrophy. Ann. Neurol. 2014, 76, 550–557. [Google Scholar] [CrossRef] [PubMed]
- Handschin, C.; Kobayashi, Y.M.; Chin, S.; Seale, P.; Campbell, K.P.; Spiegelman, B.M. PGC-1alpha regulates the neuromuscular junction program and ameliorates Duchenne muscular dystrophy. Genes Dev. 2007, 21, 770–783. [Google Scholar] [CrossRef] [PubMed]
- Hollinger, K.; Selsby, J.T. PGC-1alpha gene transfer improves muscle function in dystrophic muscle following prolonged disease progress. Exp. Physiol. 2015, 100, 1145–1158. [Google Scholar] [CrossRef] [PubMed]
- Bonsett, C.A.; Rudman, A. The dystrophin connection—ATP? Med. Hypotheses 1992, 38, 139–154. [Google Scholar] [CrossRef]
- Thomson, W.H.; Smith, I. Allopurinol in Duchenne’s muscular dystrophy. N. Engl. J. Med. 1978, 299, 101. [Google Scholar] [PubMed]
- Thomson, W.H.; Smith, I. X-linked recessive (Duchenne) muscular dystrophy (DMD) and purine metabolism: Effects of oral allopurinol and adenylate. Metabolism 1978, 27, 151–163. [Google Scholar] [CrossRef]
- Tamari, H.; Ohtani, Y.; Higashi, A.; Miyoshino, S.; Matsuda, I. Xanthine oxidase inhibitor in Duchenne muscular dystrophy. Brain Dev. 1982, 4, 137–143. [Google Scholar] [CrossRef]
- Abou-Samra, M.; Boursereau, R.; Lecompte, S.; Noel, L.; Brichard, S.M. Potential Therapeutic Action of Adiponectin in Duchenne Muscular Dystrophy. Am. J. Pathol. 2017, 187, 1577–1585. [Google Scholar] [CrossRef] [PubMed]
- Lecompte, S.; Abou-Samra, M.; Boursereau, R.; Noel, L.; Brichard, S.M. Skeletal muscle secretome in Duchenne muscular dystrophy: A pivotal anti-inflammatory role of adiponectin. Cell. Mol. Life Sci. 2017, 74, 2487–2501. [Google Scholar] [CrossRef] [PubMed]
- Kyrychenko, V.; Polakova, E.; Janicek, R.; Shirokova, N. Mitochondrial dysfunctions during progression of dystrophic cardiomyopathy. Cell Calcium 2015, 58, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Reutenauer, J.; Dorchies, O.M.; Patthey-Vuadens, O.; Vuagniaux, G.; Ruegg, U.T. Investigation of Debio 025, a cyclophilin inhibitor, in the dystrophic mdx mouse, a model for Duchenne muscular dystrophy. Br. J. Pharmacol. 2008, 155, 574–584. [Google Scholar] [CrossRef] [PubMed]
- Millay, D.P.; Sargent, M.A.; Osinska, H.; Baines, C.P.; Barton, E.R.; Vuagniaux, G.; Sweeney, H.L.; Robbins, J.; Molkentin, J.D. Genetic and pharmacologic inhibition of mitochondrial-dependent necrosis attenuates muscular dystrophy. Nat. Med. 2008, 14, 442–447. [Google Scholar] [CrossRef] [PubMed]
- Ljubicic, V.; Khogali, S.; Renaud, J.M.; Jasmin, B.J. Chronic AMPK stimulation attenuates adaptive signaling in dystrophic skeletal muscle. Am. J. Physiol. Cell Physiol. 2012, 302, C110–C121. [Google Scholar] [CrossRef] [PubMed]
- Bueno Junior, C.R.; Pantaleao, L.C.; Voltarelli, V.A.; Bozi, L.H.; Brum, P.C.; Zatz, M. Combined effect of AMPK/PPAR agonists and exercise training in mdx mice functional performance. PLoS ONE 2012, 7, e45699. [Google Scholar] [CrossRef] [PubMed]
- Barker, R.G.; Horvath, D.; van der Poel, C.; Murphy, R.M. Benefits of Prenatal Taurine Supplementation in Preventing the Onset of Acute Damage in the Mdx Mouse. PLoS Curr. 2017, 9. [Google Scholar] [CrossRef]
- Fiaccavento, R.; Carotenuto, F.; Vecchini, A.; Binaglia, L.; Forte, G.; Capucci, E.; Maccari, A.M.; Minieri, M.; Di Nardo, P. An omega-3 fatty acid-enriched diet prevents skeletal muscle lesions in a hamster model of dystrophy. Am. J. Pathol. 2010, 177, 2176–2184. [Google Scholar] [CrossRef] [PubMed]
- Apolinario, L.M.; De Carvalho, S.C.; Santo Neto, H.; Marques, M.J. Long-Term Therapy With Omega-3 Ameliorates Myonecrosis and Benefits Skeletal Muscle Regeneration in Mdx Mice. Anat. Rec. 2015, 298, 1589–1596. [Google Scholar] [CrossRef] [PubMed]
- Mauricio, A.F.; de Carvalho, S.C.; Santo Neto, H.; Marques, M.J. Effects of dietary omega-3 on dystrophic cardiac and diaphragm muscles as evaluated by 1H magnetic resonance spectroscopy: Metabolic profile and calcium-related proteins. Clin. Nutr. ESPEN 2017, 20, 60–67. [Google Scholar] [CrossRef] [PubMed]
- De Carvalho, S.C.; Matsumura, C.Y.; Santo Neto, H.; Marques, M.J. Identification of plasma interleukins as biomarkers for deflazacort and omega-3 based Duchenne muscular dystrophy therapy. Cytokine 2018, 102, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, S.C.; Apolinario, L.M.; Matheus, S.M.; Santo Neto, H.; Marques, M.J. EPA protects against muscle damage in the mdx mouse model of Duchenne muscular dystrophy by promoting a shift from the M1 to M2 macrophage phenotype. J. Neuroimmunol. 2013, 264, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Cruz, M.; Cruz-Guzman, O.D.R.; Almeida-Becerril, T.; Solis-Serna, A.D.; Atilano-Miguel, S.; Sanchez-Gonzalez, J.R.; Barbosa-Cortes, L.; Ruiz-Cruz, E.D.; Huicochea, J.C.; Cardenas-Conejo, A.; et al. Potential therapeutic impact of omega-3 long chain-polyunsaturated fatty acids on inflammation markers in Duchenne muscular dystrophy: A double-blind, controlled randomized trial. Clin. Nutr. 2017. [Google Scholar] [CrossRef] [PubMed]
- Cotan, D.; Paz, M.V.; Alcocer-Gomez, E.; Garrido-Maraver, J.; Oropesa-Avila, M.; de la Mata, M.; Pavon, A.D.; de Lavera, I.; Galan, F.; Ybot-Gonzalez, P.; et al. AMPK as A Target in Rare Diseases. Curr. Drug Targets 2016, 17, 921–931. [Google Scholar] [CrossRef] [PubMed]
- Squire, S.; Raymackers, J.M.; Vandebrouck, C.; Potter, A.; Tinsley, J.; Fisher, R.; Gillis, J.M.; Davies, K.E. Prevention of pathology in mdx mice by expression of utrophin: Analysis using an inducible transgenic expression system. Hum. Mol. Genet. 2002, 11, 3333–3344. [Google Scholar] [CrossRef] [PubMed]
- Garbincius, J.F.; Michele, D.E. Dystrophin-glycoprotein complex regulates muscle nitric oxide production through mechanoregulation of AMPK signaling. Proc. Natl. Acad. Sci. USA 2015, 112, 13663–13668. [Google Scholar] [CrossRef] [PubMed]
- Thakur, S.; Viswanadhapalli, S.; Kopp, J.B.; Shi, Q.; Barnes, J.L.; Block, K.; Gorin, Y.; Abboud, H.E. Activation of AMP-activated protein kinase prevents TGF-beta1-induced epithelial-mesenchymal transition and myofibroblast activation. Am. J. Pathol. 2015, 185, 2168–2180. [Google Scholar] [CrossRef] [PubMed]
- Heydemann, A. The super super-healing MRL mouse strain. Front. Biol. 2012, 7, 522–538. [Google Scholar] [CrossRef] [PubMed]
- Mantuano, P.; Sanarica, F.; Conte, E.; Morgese, M.G.; Capogrosso, R.F.; Cozzoli, A.; Fonzino, A.; Quaranta, A.; Rolland, J.F.; De Bellis, M.; et al. Effect of a long-term treatment with metformin in dystrophic mdx mice: A reconsideration of its potential clinical interest in Duchenne muscular dystrophy. Biochem. Pharmacol. 2018, 154, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Bagley, J.R. Fibre type-specific hypertrophy mechanisms in human skeletal muscle: Potential role of myonuclear addition. J. Physiol. 2014, 592, 5147–5148. [Google Scholar] [CrossRef] [PubMed]
- Fitts, R.H.; Widrick, J.J. Muscle mechanics: Adaptations with exercise-training. Exerc. Sport Sci. Rev. 1996, 24, 427–473. [Google Scholar] [CrossRef] [PubMed]
- Kotelnikova, E.; Shkrob, M.A.; Pyatnitskiy, M.A.; Ferlini, A.; Daraselia, N. Novel approach to meta-analysis of microarray datasets reveals muscle remodeling-related drug targets and biomarkers in Duchenne muscular dystrophy. PLoS Comput. Biol. 2012, 8, e1002365. [Google Scholar] [CrossRef] [PubMed]
- Mendes, K.L.; Lelis, D.F.; Santos, S.H.S. Nuclear sirtuins and inflammatory signaling pathways. Cytokine Growth Factor Rev. 2017, 38, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Handschin, C.; Spiegelman, B.M. The role of exercise and PGC1alpha in inflammation and chronic disease. Nature 2008, 454, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Hyzewicz, J.; Ruegg, U.T.; Takeda, S. Comparison of Experimental Protocols of Physical Exercise for mdx Mice and Duchenne Muscular Dystrophy Patients. J. Neuromuscul. Dis. 2015, 2, 325–342. [Google Scholar] [CrossRef] [PubMed]
- Frinchi, M.; Macaluso, F.; Licciardi, A.; Perciavalle, V.; Coco, M.; Belluardo, N.; Morici, G.; Mudo, G. Recovery of damaged skeletal muscle in mdx mice through low-intensity endurance exercise. Int. J. Sports Med. 2014, 35, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Hayes, A.; Williams, D.A. Beneficial effects of voluntary wheel running on the properties of dystrophic mouse muscle. J. Appl. Physiol. 1996, 80, 670–679. [Google Scholar] [CrossRef] [PubMed]
- Call, J.A.; McKeehen, J.N.; Novotny, S.A.; Lowe, D.A. Progressive resistance voluntary wheel running in the mdx mouse. Muscle Nerve 2010, 42, 871–880. [Google Scholar] [CrossRef] [PubMed]
- Smythe, G.M.; White, J.D. Voluntary wheel running in dystrophin-deficient (mdx) mice: Relationships between exercise parameters and exacerbation of the dystrophic phenotype. PLoS Curr. 2011, 3, RRN1295. [Google Scholar] [CrossRef] [PubMed]
- Fry, C.S.; Noehren, B.; Mula, J.; Ubele, M.F.; Westgate, P.M.; Kern, P.A.; Peterson, C.A. Fibre type-specific satellite cell response to aerobic training in sedentary adults. J. Physiol. 2014, 592, 2625–2635. [Google Scholar] [CrossRef] [PubMed]
- Shefer, G.; Rauner, G.; Yablonka-Reuveni, Z.; Benayahu, D. Reduced satellite cell numbers and myogenic capacity in aging can be alleviated by endurance exercise. PLoS ONE 2010, 5, e13307. [Google Scholar] [CrossRef] [PubMed]
- Kurosaka, M.; Naito, H.; Ogura, Y.; Kojima, A.; Goto, K.; Katamoto, S. Effects of voluntary wheel running on satellite cells in the rat plantaris muscle. J. Sports Sci. Med. 2009, 8, 51–57. [Google Scholar] [PubMed]
- Abreu, P.; Mendes, S.V.; Ceccatto, V.M.; Hirabara, S.M. Satellite cell activation induced by aerobic muscle adaptation in response to endurance exercise in humans and rodents. Life Sci. 2017, 170, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Gibson, M.C.; Schultz, E. Age-related differences in absolute numbers of skeletal muscle satellite cells. Muscle Nerve 1983, 6, 574–580. [Google Scholar] [CrossRef] [PubMed]
- Nederveen, J.P.; Joanisse, S.; Seguin, C.M.; Bell, K.E.; Baker, S.K.; Phillips, S.M.; Parise, G. The effect of exercise mode on the acute response of satellite cells in old men. Acta Physiol. 2015, 215, 177–190. [Google Scholar] [CrossRef] [PubMed]
- Hafner, P.; Bonati, U.; Rubino, D.; Gocheva, V.; Zumbrunn, T.; Gueven, N.; Fischer, D. Treatment with L-citrulline and metformin in Duchenne muscular dystrophy: Study protocol for a single-centre, randomised, placebo-controlled trial. Trials 2016, 17, 389. [Google Scholar] [CrossRef] [PubMed]
- Boon, H.; Bosselaar, M.; Praet, S.F.; Blaak, E.E.; Saris, W.H.; Wagenmakers, A.J.; McGee, S.L.; Tack, C.J.; Smits, P.; Hargreaves, M.; et al. Intravenous AICAR administration reduces hepatic glucose output and inhibits whole body lipolysis in type 2 diabetic patients. Diabetologia 2008, 51, 1893–1900. [Google Scholar] [CrossRef] [PubMed]
- Bastin, J.; Djouadi, F. Resveratrol and Myopathy. Nutrients 2016, 8, 254. [Google Scholar] [CrossRef] [PubMed]
- Fuller, H.R.; Humphrey, E.L.; Morris, G.E. Naturally occurring plant polyphenols as potential therapies for inherited neuromuscular diseases. Future Med. Chem. 2013, 5, 2091–2101. [Google Scholar] [CrossRef] [PubMed]
- Capogrosso, R.F.; Cozzoli, A.; Mantuano, P.; Camerino, G.M.; Massari, A.M.; Sblendorio, V.T.; De Bellis, M.; Tamma, R.; Giustino, A.; Nico, B.; et al. Assessment of resveratrol, apocynin and taurine on mechanical-metabolic uncoupling and oxidative stress in a mouse model of duchenne muscular dystrophy: A comparison with the gold standard, alpha-methyl prednisolone. Pharmacol. Res. 2016, 106, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Gordon, B.S.; Delgado-Diaz, D.C.; Carson, J.; Fayad, R.; Wilson, L.B.; Kostek, M.C. Resveratrol improves muscle function but not oxidative capacity in young mdx mice. Can. J. Physiol. Pharmacol. 2014, 92, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Tinsley, J.; Robinson, N.; Davies, K.E. Safety, tolerability, and pharmacokinetics of SMT C1100, a 2-arylbenzoxazole utrophin modulator, following single- and multiple-dose administration to healthy male adult volunteers. J. Clin. Pharmacol. 2015, 55, 698–707. [Google Scholar] [CrossRef] [PubMed]
- Wehling-Henricks, M.; Jordan, M.C.; Roos, K.P.; Deng, B.; Tidball, J.G. Cardiomyopathy in dystrophin-deficient hearts is prevented by expression of a neuronal nitric oxide synthase transgene in the myocardium. Hum. Mol. Genet. 2005, 14, 1921–1933. [Google Scholar] [CrossRef] [PubMed]
- Warner, L.E.; DelloRusso, C.; Crawford, R.W.; Rybakova, I.N.; Patel, J.R.; Ervasti, J.M.; Chamberlain, J.S. Expression of Dp260 in muscle tethers the actin cytoskeleton to the dystrophin-glycoprotein complex and partially prevents dystrophy. Hum. Mol. Genet. 2002, 11, 1095–1105. [Google Scholar] [CrossRef] [PubMed]
- Adamo, C.M.; Dai, D.F.; Percival, J.M.; Minami, E.; Willis, M.S.; Patrucco, E.; Froehner, S.C.; Beavo, J.A. Sildenafil reverses cardiac dysfunction in the mdx mouse model of Duchenne muscular dystrophy. Proc. Natl. Acad. Sci. USA 2010, 107, 19079–19083. [Google Scholar] [CrossRef] [PubMed]
- Hoppeler, H.; Fluck, M. Plasticity of skeletal muscle mitochondria: Structure and function. Med. Sci. Sports Exerc. 2003, 35, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Feng, X.; Li, Q.; Wang, Y.; Li, Q.; Hua, M. Adiponectin, TNF-alpha and inflammatory cytokines and risk of type 2 diabetes: A systematic review and meta-analysis. Cytokine 2016, 86, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Abou-Samra, M.; Lecompte, S.; Schakman, O.; Noel, L.; Many, M.C.; Gailly, P.; Brichard, S.M. Involvement of adiponectin in the pathogenesis of dystrophinopathy. Skelet. Muscle 2015, 5, 25. [Google Scholar] [CrossRef] [PubMed]
- Wissing, E.R.; Millay, D.P.; Vuagniaux, G.; Molkentin, J.D. Debio-025 is more effective than prednisone in reducing muscular pathology in mdx mice. Neuromuscul. Disord. 2010, 20, 753–760. [Google Scholar] [CrossRef] [PubMed]
- Waldrop, M.A.; Gumienny, F.; El Husayni, S.; Frank, D.E.; Weiss, R.B.; Flanigan, K.M. Low-level dystrophin expression attenuating the dystrophinopathy phenotype. Neuromuscul. Disord. 2018, 28, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, E.P.; Kunkel, L.M.; Angelini, C.; Clarke, A.; Johnson, M.; Harris, J.B. Improved diagnosis of Becker muscular dystrophy by dystrophin testing. Neurology 1989, 39, 1011–1017. [Google Scholar] [CrossRef] [PubMed]
- El Refaey, M.; Xu, L.; Gao, Y.; Canan, B.D.; Adesanya, T.M.A.; Warner, S.C.; Akagi, K.; Symer, D.E.; Mohler, P.J.; Ma, J.; et al. In Vivo Genome Editing Restores Dystrophin Expression and Cardiac Function in Dystrophic Mice. Circ. Res. 2017, 121, 923–929. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Target | Rx | Mice | Humans | References |
|---|---|---|---|---|
| Dietary supplements | Apple polyphenols | Increases slow fibers | [83] | |
| Arginine & Metformin | 16 weeks of co-treatment improved clinical scores in normal volunteers | [84] | ||
| Creatine | Early treatment reduced mdx pathology | [85] | ||
| Glutamine | 10 treatment days did not alter protein degradation compared to amino acid controls | [86] | ||
| 4 months of treatment did not improve DMD pathology | [87] | |||
| Increase pAMPK | Exercise | Increases slow fiber types, PGC-1α, SIRT1 | Increases slow fiber types, PGC-1α, SIRT1 | Reviewed in [22] |
| Increases utrophin in skeletal muscles after 12 weeks of voluntary wheel running | [88] | |||
| Low intensity training improved mdx phenotype through inflammation inhibition in skeletal muscle and diaphragm. | [89,90] | |||
| Exercise increased utrophin in skeletal muscles of normal people | [91] | |||
| Submaximal exercise increases function without causing increased pathology | [92] | |||
| DMD exercise reviews | [93,94] | |||
| Assisted bicycle training maintains function without causing increased pathology | [95] | |||
| Metformin | Increased PGC1α, utrophin. | [96] | ||
| Metformin with l-arginine | 16 weeks of treatment caused a trend to improved oxidative stress and function | [84] | ||
| AICAR | Increased slow fiber types, PGC-1α, SIRT1 | [97] | ||
| Restored calcium-induced PTP opening to normal levels in diaphragm. | [45] | |||
| Resveratrol | Modest pathology decline, increased Utr | [98,99] | ||
| Transgenic overexpression of Mir-499 | Reduced pathology | [31] | ||
| Inhibit Fnip1 | Reduced pathology | [100] | ||
| Through breeding | Reduced fibrosis and functional decline | [41] | ||
| Increase utrophin | Transgenically | Significantly decreases pathology | [101,102] | |
| SMT C1100 | Significantly decreases pathology | [103] | ||
| Safe in healthy volunteers | [104] | |||
| Tolerated in DMD patients, high degree of variability in serum SMT C1100 levels | [105] | |||
| AICAR | Increases utrophin | [106] | ||
| Metformin | Increases utrophin | [96] | ||
| Increase nNOS/NO | Transgenically | Decreased pathology | [68,107,108,109] | |
| l-Arginine | Reduced pathology | [110] | ||
| PDE inhibitors, sildenafil | Reduced pathology in skeletal, including diaphragm and cardiac muscles. Increased slow fiber transition. | Acute treatment reduced exercise-associated ischemia. | [111] reviewed in [68,112] | |
| Two phase 3 trials have recently been completed but failed to demonstrate improvements, chronic treatment. | [113,114] | |||
| Increase PGC1α | Transgenic | Reduced pathology, increased slow fibers, mitochondria and Utr. Decreased CN, EBD and CK. | [34,48,115,116] | |
| Support ATP generation ROS inhibitor | ASA | Improved ATP content and multiple functional parameters | [117] | |
| Allopurinol | Initial increased creatine phosphate and ATP, and most patients retained benefits after 22 months. | [118,119] | ||
| Unclear benefit after 27 months of treatment. | [120] | |||
| Increase adiponectin | Transgenic overexpression of adiponectin | Increased slow fibers Increased utrophin Decreased pathology | [121] | |
| Treated DMD myotubes with adiponectin | Decreased inflammation and upregulated utrophin | [122] | ||
| Transition pore inhibitors | Cyclosporine A | Restored normal redox state in isolated mdx cell mitochondria | [123] | |
| PGC1α transgene | Normalized MPTP opening kinetics. | [48] | ||
| AICAR | Normalized MPTP opening kinetics in the diaphragm. | [45] | ||
| Sildenafil | Normalized MPTP opening kinetics in hearts. | [111] | ||
| Debio 025 | 2 weeks of oral treatment, some structural and functional improvements in diaphragm and skeletal muscles. | [124] | ||
| Genetically targeting cyclophilin D, or Debio 025 | Reduces myofiber necrosis and pathology in Lama2 and delta-sarcolglycan deficient mice | [125] | ||
| Co-therapies | 30 days AICAR followed by exercise | Paradoxically AICAR blunted the beneficial effects of exercise | [126] | |
| AMPK/PPARγ agonists | Histological and functional improvements | [49] | ||
| Exercise with AMPK/PPARγ agonists | Functional improvement in the combination group | [127] |
| Targeted Molecular Pathway | Therapy |
|---|---|
| Improve sarcolemmal strength, dystrophin expression | Omega-3 FTY720 Utrophin upregulation Exon skipping Cell based |
| Decrease inflammation | Corticosteroids NF-κb inhibition TGFβ inhibition Antioxidants |
| Improve mitochondrial function | AICAR Metformin Exercise Antioxidants |
| Decrease fibrosis | Losartan TGFβ inhibition |
| Improve satellite cell functions | Cell based |
© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heydemann, A. Skeletal Muscle Metabolism in Duchenne and Becker Muscular Dystrophy—Implications for Therapies. Nutrients 2018, 10, 796. https://doi.org/10.3390/nu10060796
Heydemann A. Skeletal Muscle Metabolism in Duchenne and Becker Muscular Dystrophy—Implications for Therapies. Nutrients. 2018; 10(6):796. https://doi.org/10.3390/nu10060796
Chicago/Turabian StyleHeydemann, Ahlke. 2018. "Skeletal Muscle Metabolism in Duchenne and Becker Muscular Dystrophy—Implications for Therapies" Nutrients 10, no. 6: 796. https://doi.org/10.3390/nu10060796
APA StyleHeydemann, A. (2018). Skeletal Muscle Metabolism in Duchenne and Becker Muscular Dystrophy—Implications for Therapies. Nutrients, 10(6), 796. https://doi.org/10.3390/nu10060796