High-Glucose or -Fructose Diet Cause Changes of the Gut Microbiota and Metabolic Disorders in Mice without Body Weight Change


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Animals and Diets
2.3. Oral Glucose Tolerance Tests and Insulin Tolerance Tests
2.4. Intestinal Permeability
2.5. Gut Microbiota Analysis
2.6. Blood Serum Analysis
2.7. Western Blotting
2.8. Histological Analysis
2.9. Statistical Analysis
3. Results
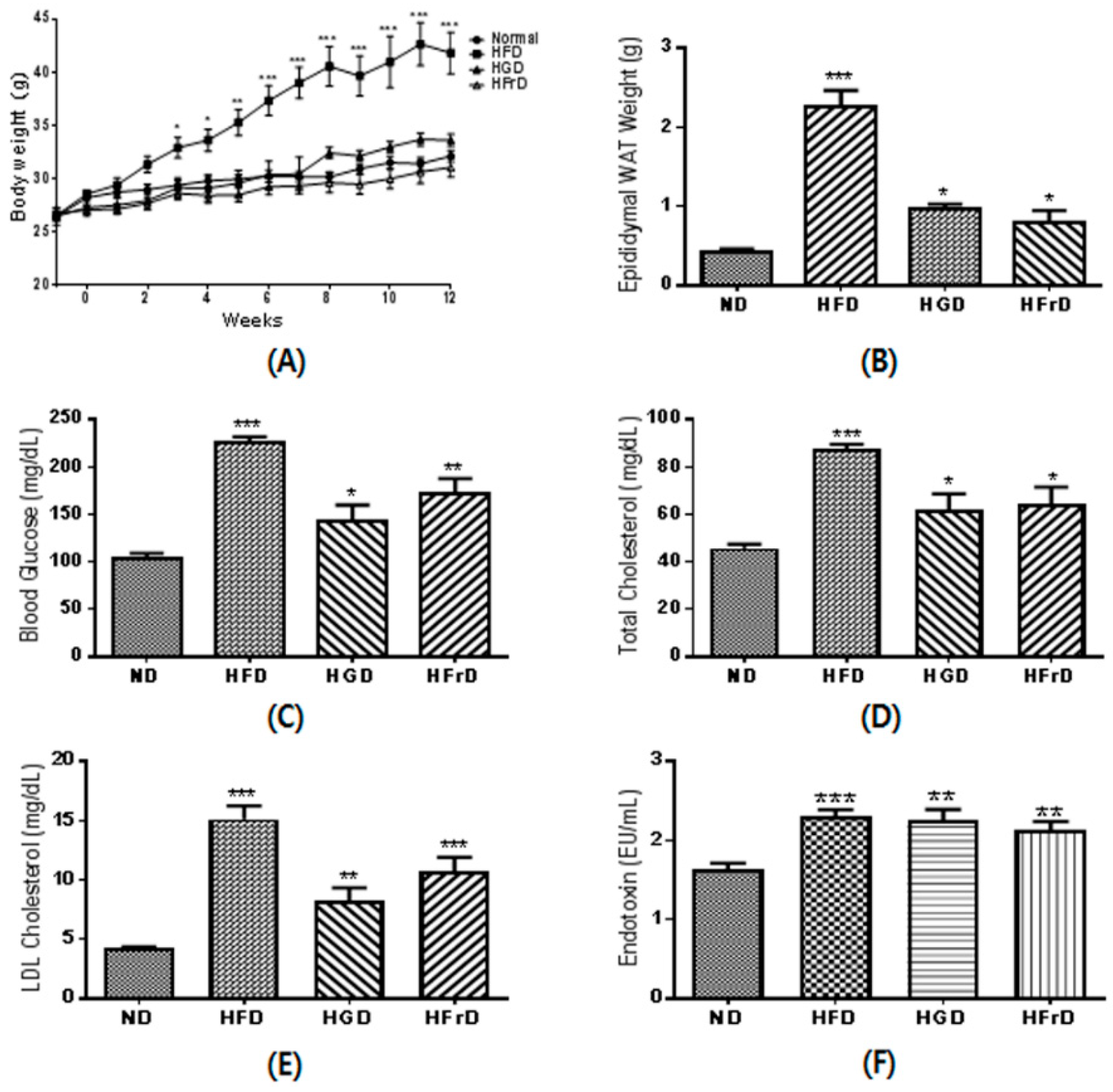
3.1. Effects of Diet on Body Weight and Metabolic Parameters
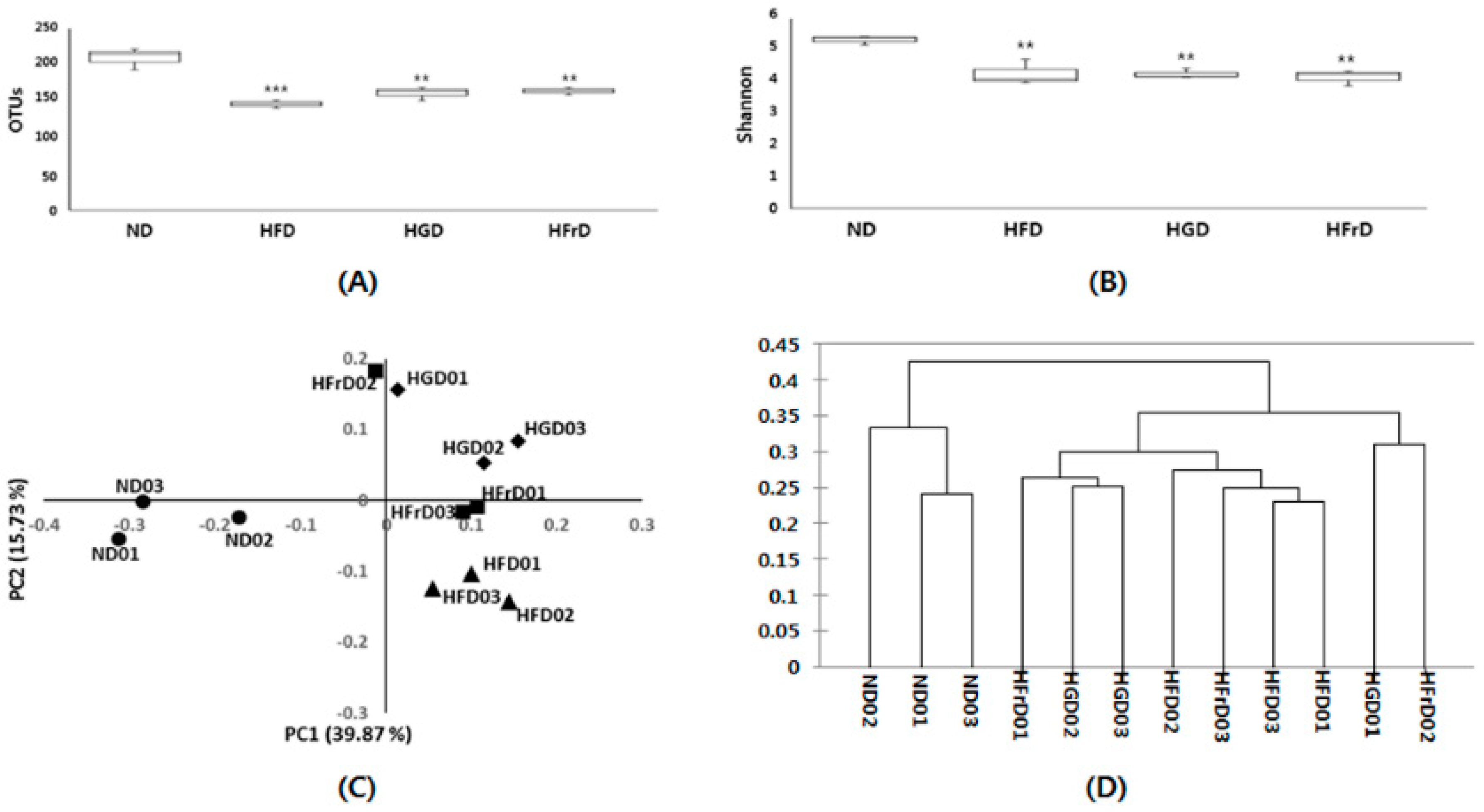
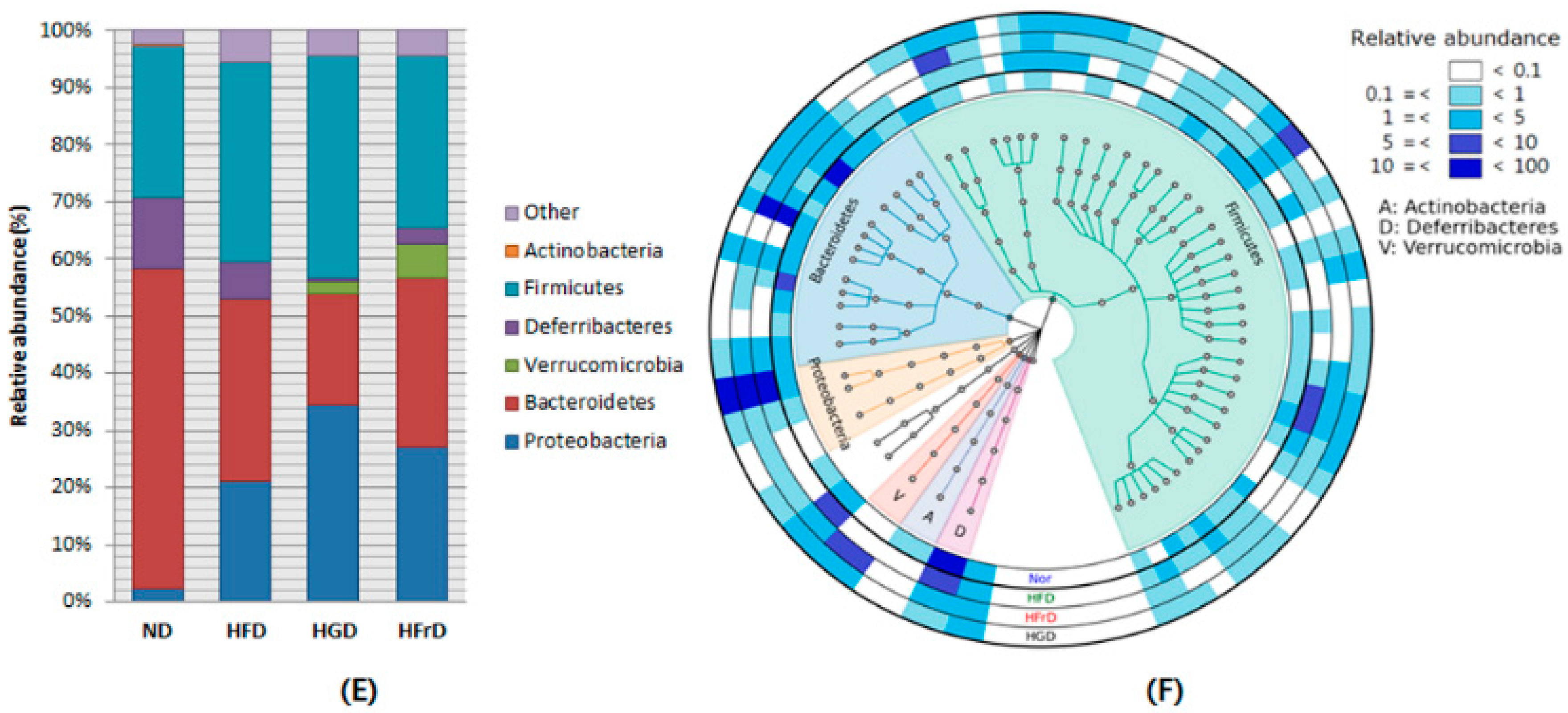
3.2. Effects of Diet on Gut Microbial Diversity and Composition
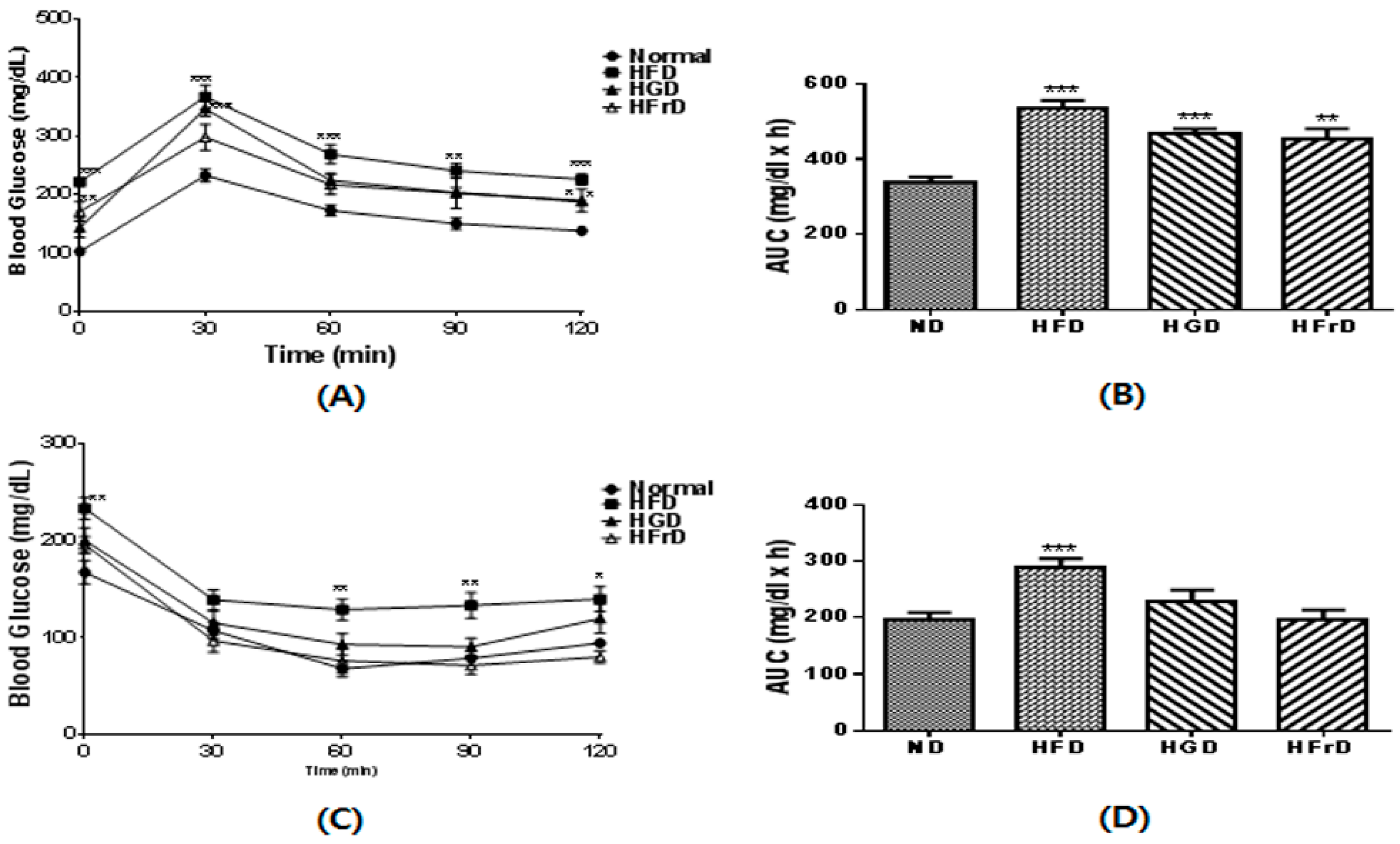
3.3. Effects of Diet on Glucose Intolerance and Insulin Resistance
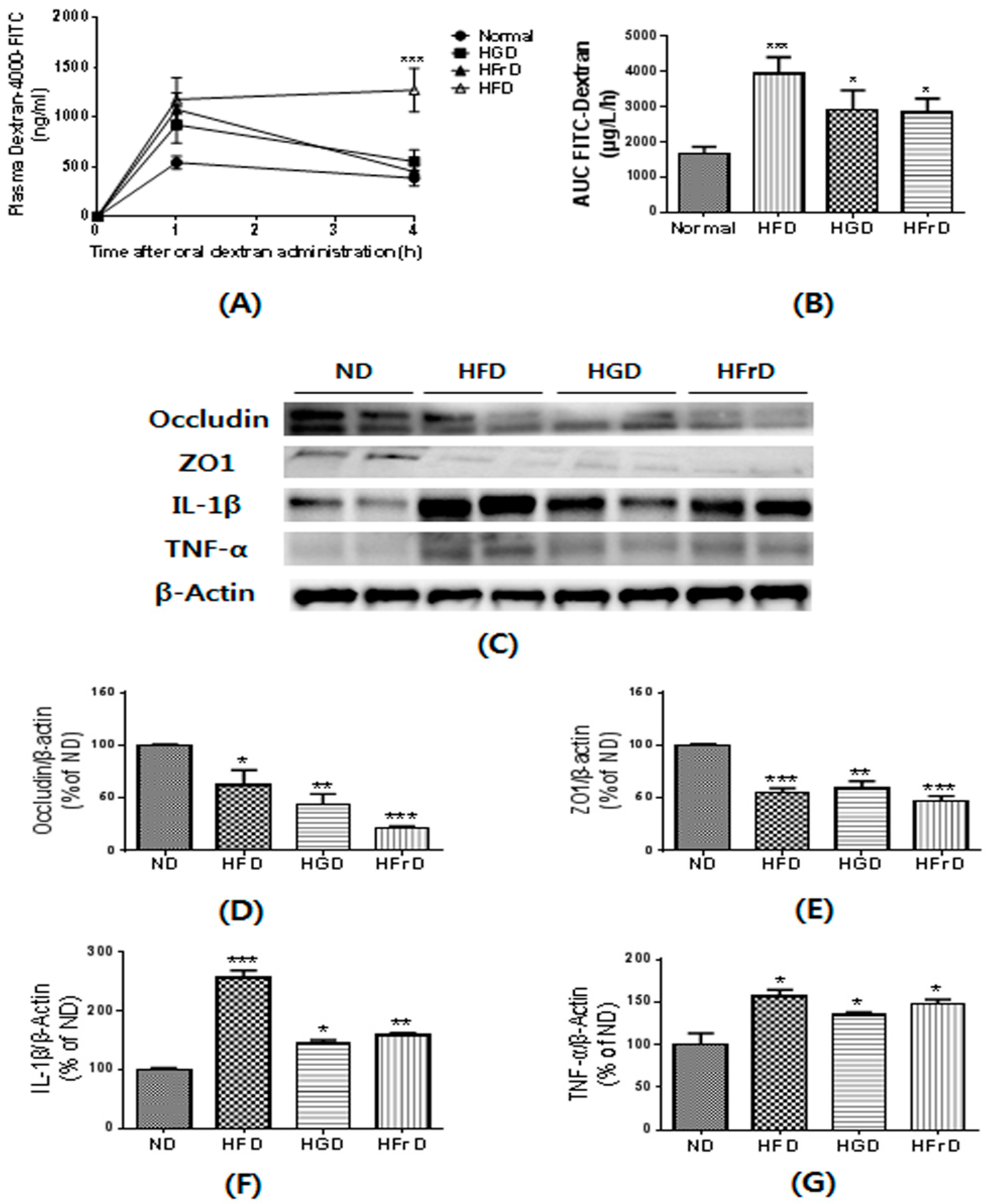
3.4. Effects of Diet on Gut Permeability and Inflammation
3.5. Effects of Diet on Liver Inflammation and Lipid Metabolism
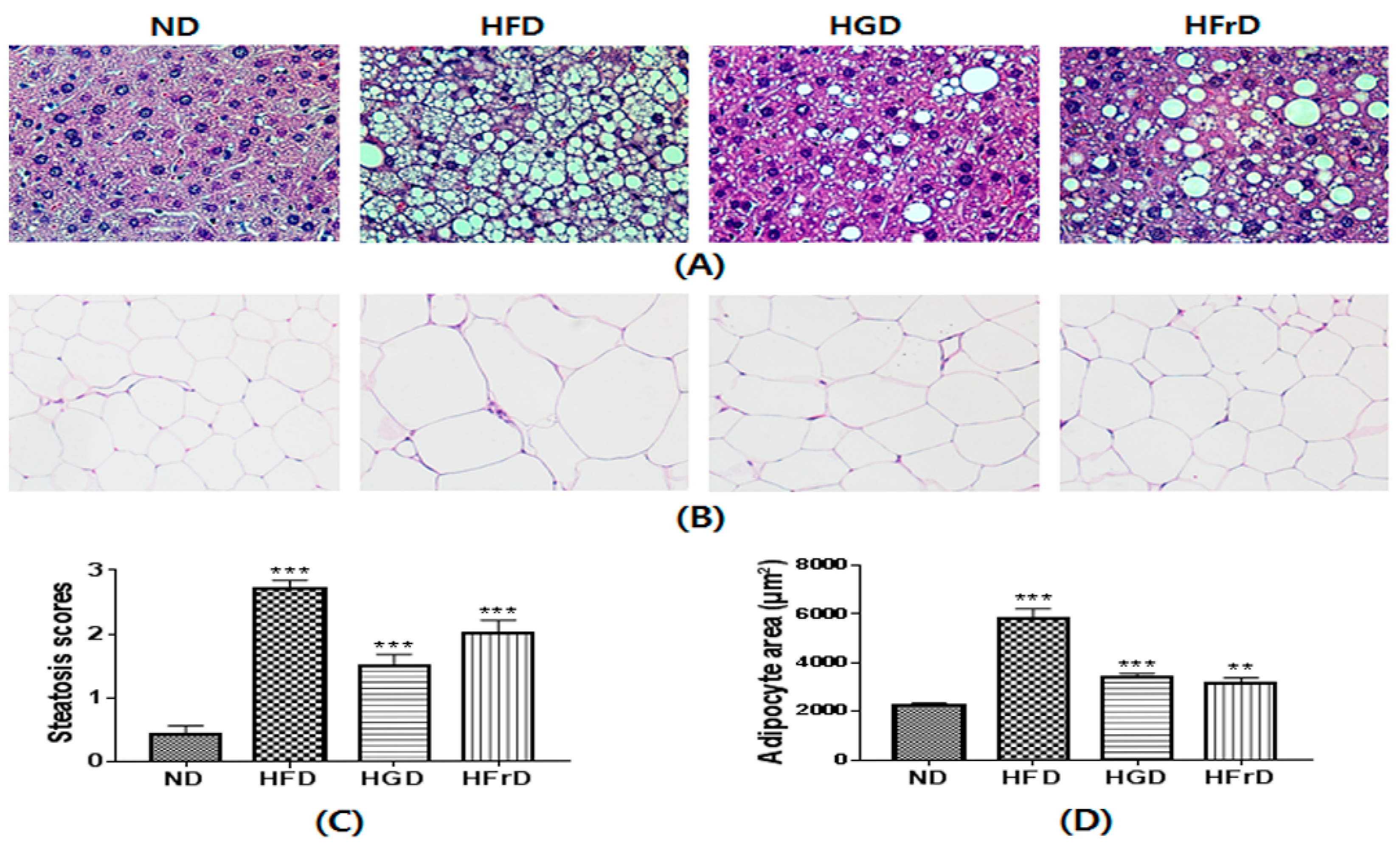
3.6. Histological Changes
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Williams, E.P.; Mesidor, M.; Winters, K.; Dubbert, P.M.; Wyatt, S.B. Overweight and obesity: Prevalence, consequences, and causes of a growing public health problem. Curr. Obes. Rep. 2015, 4, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Parsons, M.J.; Moffitt, T.E.; Gregory, A.; Goldman-Mellor, S.; Nolan, P.; Poulton, R.; Caspi, A. Social jetlag, obesity and metabolic disorder: Investigation in a cohort study. Int. J. Obes. 2015, 39, 842–848. [Google Scholar] [CrossRef] [PubMed]
- Quines, C.B.; Rosa, S.G.; Chagas, P.M.; Velasquez, D.; Prado, V.C.; Nogueira, C.W. (p-clphse) 2 stimulates carbohydrate metabolism and reverses the metabolic alterations induced by high fructose load in rats. Food Chem. Toxicol. 2017, 107, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Dorton, H.M.; Luo, S.; Monterosso, J.R.; Page, K.A. Influences of dietary added sugar consumption on striatal food cue reactivity and postprandial glp-1 response. Front. Psychiatry 2017, 8, 297. [Google Scholar] [CrossRef] [PubMed]
- Stanhope, K.L. Sugar consumption, metabolic disease and obesity: The state of the controversy. Crit. Rev. Clin. Lab. Sci. 2016, 53, 52–67. [Google Scholar] [CrossRef] [PubMed]
- Boulangé, C.L.; Neves, A.L.; Chilloux, J.; Nicholson, J.K.; Dumas, M.-E. Impact of the gut microbiota on inflammation, obesity, and metabolic disease. Genome Med. 2016, 8, 42. [Google Scholar] [CrossRef] [PubMed]
- Boursier, J.; Mueller, O.; Barret, M.; Machado, M.; Fizanne, L.; Araujo-Perez, F.; Guy, C.D.; Seed, P.C.; Rawls, J.F.; David, L.A. The severity of nonalcoholic fatty liver disease is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology 2016, 63, 764–775. [Google Scholar] [CrossRef] [PubMed]
- Cardinelli, C.S.; Sala, P.C.; Alves, C.C.; Torrinhas, R.S.; Waitzberg, D.L. Influence of intestinal microbiota on body weight gain: A narrative review of the literature. Obes. Surg. 2015, 25, 346–353. [Google Scholar] [CrossRef] [PubMed]
- Ussar, S.; Griffin, N.W.; Bezy, O.; Fujisaka, S.; Vienberg, S.; Softic, S.; Deng, L.; Bry, L.; Gordon, J.I.; Kahn, C.R. Interactions between gut microbiota, host genetics and diet modulate the predisposition to obesity and metabolic syndrome. Cell Metab. 2015, 22, 516–530. [Google Scholar] [CrossRef] [PubMed]
- Murphy, E.; Cotter, P.; Healy, S.; Marques, T.; O’sullivan, O.; Fouhy, F.; Clarke, S.; O’toole, P.; Quigley, E.M.; Stanton, C. Composition and energy harvesting capacity of the gut microbiota: Relationship to diet, obesity and time in mouse models. Gut 2010. [Google Scholar] [CrossRef] [PubMed]
- Rabot, S.; Membrez, M.; Bruneau, A.; Gérard, P.; Harach, T.; Moser, M.; Raymond, F.; Mansourian, R.; Chou, C.J. Germ-free c57bl/6j mice are resistant to high-fat-diet-induced insulin resistance and have altered cholesterol metabolism. FASEB J. 2010, 24, 4948–4959. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1131. [Google Scholar] [CrossRef] [PubMed]
- Maslowski, K.M.; Mackay, C.R. Diet, gut microbiota and immune responses. Nat. Immunol. 2011, 12, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, Y.; Sekita, A.; Chiji, H.; Kato, N. Consumption of lily bulb modulates fecal ratios of firmicutes and bacteroidetes phyla in rats fed a high-fat diet. Food Sci. Biotechnol. 2016, 25, 153–156. [Google Scholar] [CrossRef]
- Williams, L.M.; Campbell, F.M.; Drew, J.E.; Koch, C.; Hoggard, N.; Rees, W.D.; Kamolrat, T.; Ngo, H.T.; Steffensen, I.-L.; Gray, S.R. The development of diet-induced obesity and glucose intolerance in c57bl/6 mice on a high-fat diet consists of distinct phases. PLoS ONE 2014, 9, e106159. [Google Scholar] [CrossRef] [PubMed]
- Xue, L.; He, J.; Gao, N.; Lu, X.; Li, M.; Wu, X.; Liu, Z.; Jin, Y.; Liu, J.; Xu, J. Probiotics may delay the progression of nonalcoholic fatty liver disease by restoring the gut microbiota structure and improving intestinal endotoxemia. Sci. Rep. 2017, 7, 45176. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.-M.; Jeong, J.-J.; Woo, K.H.; Han, M.J.; Kim, D.-H. Lactobacillus sakei ok67 ameliorates high-fat diet–induced blood glucose intolerance and obesity in mice by inhibiting gut microbiota lipopolysaccharide production and inducing colon tight junction protein expression. Nutr. Res. 2016, 36, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Roquetto, A.R.; Monteiro, N.E.S.; Moura, C.S.; Toreti, V.C.; de Pace, F.; dos Santos, A.; Park, Y.K.; Amaya-Farfan, J. Green propolis modulates gut microbiota, reduces endotoxemia and expression of tlr4 pathway in mice fed a high-fat diet. Food Res. Int. 2015, 76, 796–803. [Google Scholar] [CrossRef] [PubMed]
- Leal-Díaz, A.M.; Noriega, L.G.; Torre-Villalvazo, I.; Torres, N.; Alemán-Escondrillas, G.; López-Romero, P.; Sánchez-Tapia, M.; Aguilar-López, M.; Furuzawa-Carballeda, J.; Velázquez-Villegas, L.A. Aguamiel concentrate from agave salmiana and its extracted saponins attenuated obesity and hepatic steatosis and increased akkermansia muciniphila in c57bl6 mice. Sci. Rep. 2016, 6, 34242. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Jiao, J.; Zhang, W.; Xu, J.; Wan, Z.; Zhang, W.; Gao, X.; Qin, L. Dietary fiber prevents obesity-related liver lipotoxicity by modulating sterol-regulatory element binding protein pathway in c57bl/6j mice fed a high-fat/cholesterol diet. Sci. Rep. 2015, 5, 15256. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Sugimoto, K.; Soejima, Y.; Kumagai, A.; Koeda, T.; Shojo, A.; Nakagawa, K.; Harada, N.; Yamaji, R.; Inui, H. Inhibitory effects of eucalyptus and banaba leaf extracts on nonalcoholic steatohepatitis induced by a high-fructose/high-glucose diet in rats. Biomed. Res. Int. 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- Crescenzo, R.; Mazzoli, A.; Di Luccia, B.; Bianco, F.; Cancelliere, R.; Cigliano, L.; Liverini, G.; Baccigalupi, L.; Iossa, S. Dietary fructose causes defective insulin signalling and ceramide accumulation in the liver that can be reversed by gut microbiota modulation. Food Nutr. Res. 2017, 61, 1331657. [Google Scholar] [CrossRef] [PubMed]
- Jena, P.K.; Singh, S.; Prajapati, B.; Nareshkumar, G.; Mehta, T.; Seshadri, S. Impact of targeted specific antibiotic delivery for gut microbiota modulation on high-fructose-fed rats. Appl. Biochem. Biotechnol. 2014, 172, 3810–3826. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Possemiers, S.; Van de Wiele, T.; Guiot, Y.; Everard, A.; Rottier, O.; Geurts, L.; Naslain, D.; Neyrinck, A.; Lambert, D.M. Changes in gut microbiota control inflammation in obese mice through a mechanism involving glp-2-driven improvement of gut permeability. Gut 2009, 58, 1091–1103. [Google Scholar] [CrossRef] [PubMed]
- Magoč, T.; Salzberg, S.L. Flash: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [PubMed]
- Toma, I.; Siegel, M.O.; Keiser, J.; Yakovleva, A.; Kim, A.; Davenport, L.; Devaney, J.; Hoffman, E.P.; Alsubail, R.; Crandall, K.A. Single-molecule long-read 16s sequencing to characterize the lung microbiome from mechanically ventilated patients with suspected pneumonia. J. Clin. Microbiol. 2014, 52, 3913–3921. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Search and clustering orders of magnitude faster than blast. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I. Qiime allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed]
- Asnicar, F.; Weingart, G.; Tickle, T.L.; Huttenhower, C.; Segata, N. Compact graphical representation of phylogenetic data and metadata with Graphlan. PeerJ 2015, 3, e1029. [Google Scholar] [CrossRef] [PubMed]
- Kato, K.I.; Takeshita, Y.; Misu, H.; Zen, Y.; Kaneko, S.; Takamura, T. Liver steatosis is associated with insulin resistance in skeletal muscle rather than in the liver in japanese patients with non-alcoholic fatty liver disease. J. Diabetes Investig. 2015, 6, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Han, D.; Xu, R.; Li, S.; Wu, H.; Qu, C.; Wang, F.; Wang, X.; Zhao, Y. A model of metabolic syndrome and related diseases with intestinal endotoxemia in rats fed a high fat and high sucrose diet. PLoS ONE 2014, 9, e115148. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Bibiloni, R.; Knauf, C.; Waget, A.; Neyrinck, A.M.; Delzenne, N.M.; Burcelin, R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet–induced obesity and diabetes in mice. Diabetes 2008, 57, 1470–1481. [Google Scholar] [CrossRef] [PubMed]
- Anhê, F.F.; Roy, D.; Pilon, G.; Dudonné, S.; Matamoros, S.; Varin, T.V.; Garofalo, C.; Moine, Q.; Desjardins, Y.; Levy, E. A polyphenol-rich cranberry extract protects from diet-induced obesity, insulin resistance and intestinal inflammation in association with increased akkermansia spp. Population in the gut microbiota of mice. Gut 2015, 64, 872–883. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.W.; Lee, E.K.; Kim, D.H.; An, H.J.; Kim, N.D.; Im, D.S.; Lee, J.; Yu, B.P.; Chung, H.Y. Age-related sensitivity to endotoxin-induced liver inflammation: Implication of inflammasome/il-1β for steatohepatitis. Aging Cell 2015, 14, 524–533. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Tang, H.; Zhang, C.; Zhao, Y.; Derrien, M.; Rocher, E.; Vlieg, J.E.v.-H.; Strissel, K.; Zhao, L.; Obin, M. Modulation of gut microbiota during probiotic-mediated attenuation of metabolic syndrome in high fat diet-fed mice. ISME J. 2015, 9, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Daniel, H.; Gholami, A.M.; Berry, D.; Desmarchelier, C.; Hahne, H.; Loh, G.; Mondot, S.; Lepage, P.; Rothballer, M.; Walker, A. High-fat diet alters gut microbiota physiology in mice. ISME J. 2014, 8, 295. [Google Scholar] [CrossRef] [PubMed]
- Jung, U.J.; Cho, Y.-Y.; Choi, M.-S. Apigenin ameliorates dyslipidemia, hepatic steatosis and insulin resistance by modulating metabolic and transcriptional profiles in the liver of high-fat diet-induced obese mice. Nutrients 2016, 8, 305. [Google Scholar] [CrossRef] [PubMed]
- Cremonini, E.; Bettaieb, A.; Haj, F.G.; Fraga, C.G.; Oteiza, P.I. (−)-epicatechin improves insulin sensitivity in high fat diet-fed mice. Arch. Biochem. Biophys. 2016, 599, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Lecomte, V.; Kaakoush, N.O.; Maloney, C.A.; Raipuria, M.; Huinao, K.D.; Mitchell, H.M.; Morris, M.J. Changes in gut microbiota in rats fed a high fat diet correlate with obesity-associated metabolic parameters. PLoS ONE 2015, 10, e0126931. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Shen, D.; Fang, Z.; Jie, Z.; Qiu, X.; Zhang, C.; Chen, Y.; Ji, L. Human gut microbiota changes reveal the progression of glucose intolerance. PLoS ONE 2013, 8, e71108. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Guo, J.; Ji, K.; Zhang, P. Bamboo shoot fiber prevents obesity in mice by modulating the gut microbiota. Sci. Rep. 2016, 6, 32953. [Google Scholar] [CrossRef] [PubMed]
- Miura, K.; Ishioka, M.; Iijima, K. The roles of the gut microbiota and toll-like receptors in obesity and nonalcoholic fatty liver disease. J. Obes. Metab. Syndr. 2017, 26, 86–96. [Google Scholar] [CrossRef]
- Jang, C.; Hui, S.; Lu, W.; Cowan, A.J.; Morscher, R.J.; Lee, G.; Liu, W.; Tesz, G.J.; Birnbaum, M.J.; Rabinowitz, J.D. The small intestine converts dietary fructose into glucose and organic acids. Cell Metab. 2018, 27, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Szabo, G. Gut–liver axis in alcoholic liver disease. Gastroenterology 2015, 148, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Rainone, V.; Schneider, L.; Saulle, I.; Ricci, C.; Biasin, M.; Al-Daghri, N.; Giani, E.; Zuccotti, G.; Clerici, M.; Trabattoni, D. Upregulation of inflammasome activity and increased gut permeability are associated with obesity in children and adolescents. Int. J. Obes. 2016, 40, 1026–1033. [Google Scholar] [CrossRef] [PubMed]
- Kirpich, I.A.; Marsano, L.S.; McClain, C.J. Gut–liver axis, nutrition, and non-alcoholic fatty liver disease. Clin. Biochem. 2015, 48, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Mazzon, E.; Cuzzocrea, S. Role of tnf-α in ileum tight junction alteration in mouse model of restraint stress. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G1268–G1280. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.-H.; Liu, X.-Q.; Zhang, C.; He, W.; Wang, H.; Chen, Y.-H.; Liu, X.-J.; Chen, X.; Xu, D.-X. Tlr4-mutant mice are resistant to acute alcohol-induced sterol-regulatory element binding protein activation and hepatic lipid accumulation. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Sheedfar, F.; Sung, M.M.; Aparicio-Vergara, M.; Kloosterhuis, N.J.; Miquilena-Colina, M.E.; Vargas-Castrillón, J.; Febbraio, M.; Jacobs, R.L.; de Bruin, A.; Vinciguerra, M. Increased hepatic cd36 expression with age is associated with enhanced susceptibility to nonalcoholic fatty liver disease. Aging 2014, 6, 281. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.-S.; Lee, S.; Park, M.-A.; Siempos, I.I.; Haslip, M.; Lee, P.J.; Yun, M.; Kim, C.K.; Howrylak, J.; Ryter, S.W. Ucp2-induced fatty acid synthase promotes nlrp3 inflammasome activation during sepsis. J. Clin. Investig. 2015, 125, 665. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Zheng, X.; Qiu, C.; Dongol, S.; Lv, Q.; Jiang, J.; Kong, B.; Wang, C. Sirt1 promotes endometrial tumor growth by targeting srebp1 and lipogenesis. Oncol. Rep. 2014, 32, 2831–2835. [Google Scholar] [CrossRef] [PubMed]
- Hersoug, L.G.; Møller, P.; Loft, S. Gut microbiota-derived lipopolysaccharide uptake and trafficking to adipose tissue: Implications for inflammation and obesity. Obes. Rev. 2016, 17, 297–312. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Ridaura, V.K.; Faith, J.J.; Rey, F.E.; Knight, R.; Gordon, J.I. The effect of diet on the human gut microbiome: A metagenomic analysis in humanized gnotobiotic mice. Sci. Transl. Med. 2009, 1, 6ra14–16ra14. [Google Scholar] [CrossRef] [PubMed]
- Everard, A.; Belzer, C.; Geurts, L.; Ouwerkerk, J.P.; Druart, C.; Bindels, L.B.; Guiot, Y.; Derrien, M.; Muccioli, G.G.; Delzenne, N.M. Cross-talk between akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc. Natl. Acad. Sci. USA 2013, 110, 9066–9071. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.K.; Han, K.; Kwon, H.S.; Song, K.H.; Yim, H.W.; Lee, W.C.; Park, Y.M. Normal weight obesity in korean adults. Clin. Endocrinol. 2014, 80, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Moy, F.M.; Loh, D.A. Cardiometabolic risks profile of normal weight obese and multi-ethnic women in a developing country. Maturitas 2015, 81, 389–393. [Google Scholar] [CrossRef] [PubMed]







© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Do, M.H.; Lee, E.; Oh, M.-J.; Kim, Y.; Park, H.-Y. High-Glucose or -Fructose Diet Cause Changes of the Gut Microbiota and Metabolic Disorders in Mice without Body Weight Change. Nutrients 2018, 10, 761. https://doi.org/10.3390/nu10060761
Do MH, Lee E, Oh M-J, Kim Y, Park H-Y. High-Glucose or -Fructose Diet Cause Changes of the Gut Microbiota and Metabolic Disorders in Mice without Body Weight Change. Nutrients. 2018; 10(6):761. https://doi.org/10.3390/nu10060761
Chicago/Turabian StyleDo, Moon Ho, Eunjung Lee, Mi-Jin Oh, Yoonsook Kim, and Ho-Young Park. 2018. "High-Glucose or -Fructose Diet Cause Changes of the Gut Microbiota and Metabolic Disorders in Mice without Body Weight Change" Nutrients 10, no. 6: 761. https://doi.org/10.3390/nu10060761
APA StyleDo, M. H., Lee, E., Oh, M.-J., Kim, Y., & Park, H.-Y. (2018). High-Glucose or -Fructose Diet Cause Changes of the Gut Microbiota and Metabolic Disorders in Mice without Body Weight Change. Nutrients, 10(6), 761. https://doi.org/10.3390/nu10060761