Molecular Pathways Underlying Cholesterol Homeostasis

{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
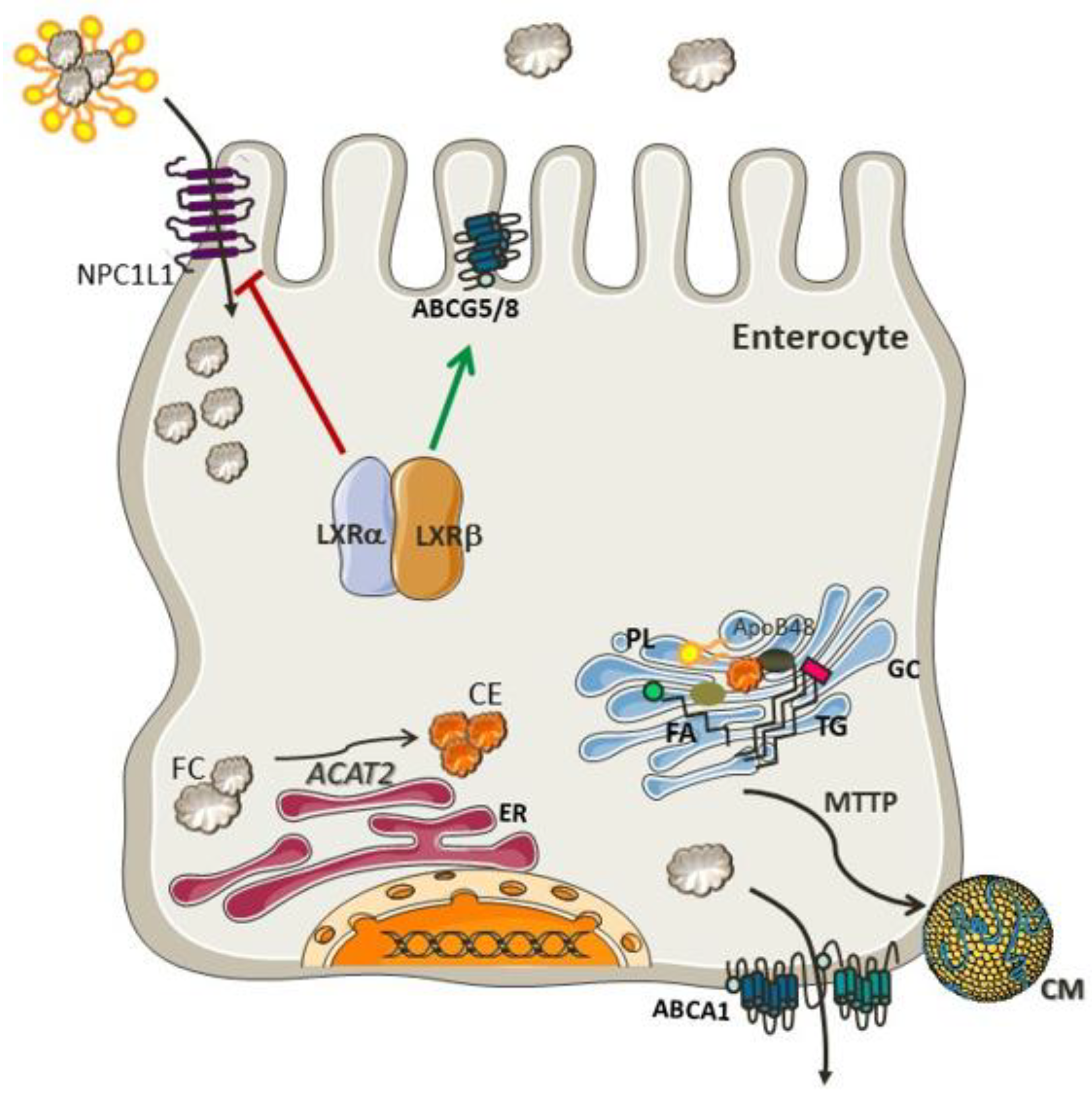
Cholesterol Absorption
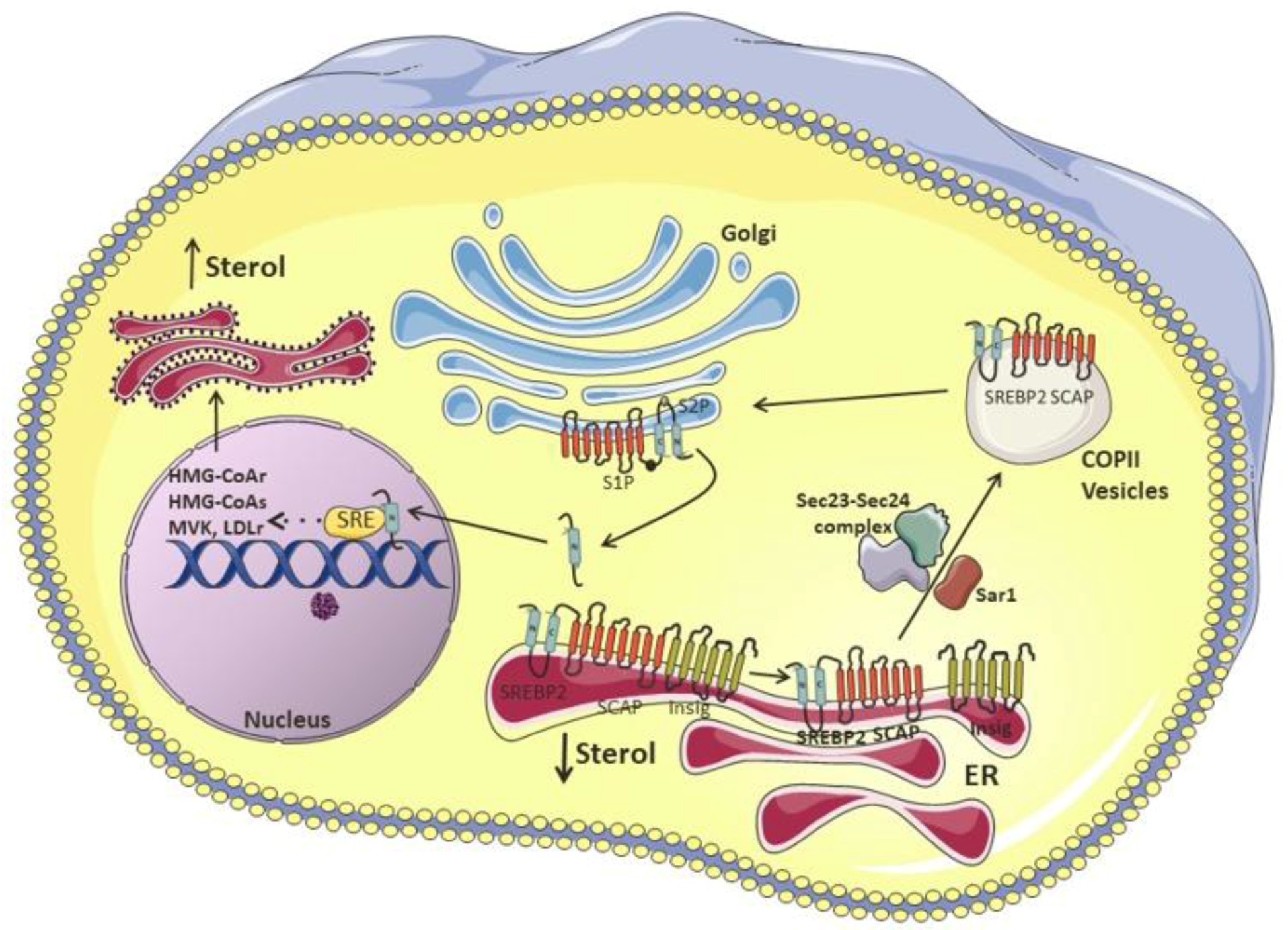
2. Cholesterol Biosynthesis and Uptake
3. Cholesterol Synthesis and Enzymatic Control
4. Cholesterol Balance
5. The LDLR and Cholesterol Uptake
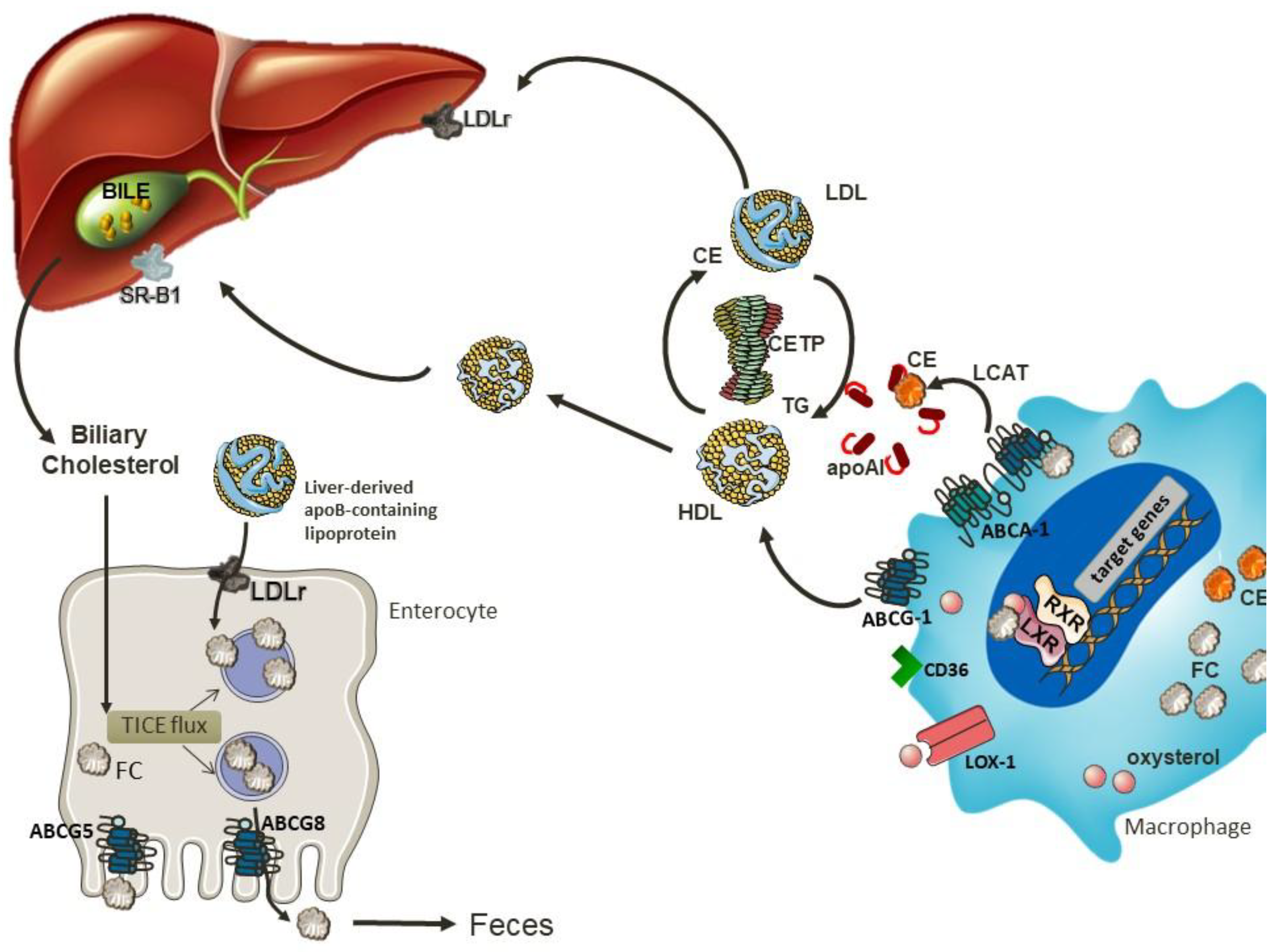
6. Reverse Cholesterol Transport
7. Conditions that Impair the Efficiency of the RCT
8. New Recommendations on Dietary Cholesterol
Author Contributions
Funding
Conflicts of Interest
References
- Ikonen, E. Cellular cholesterol trafficking and compartmentalization. Nat. Rev. Mol. Cell Biol. 2008, 9, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Lingwood, D.; Simons, K. Lipid rafts as a membrane-organizing principle. Science 2010, 327, 46–50. [Google Scholar] [CrossRef] [PubMed]
- Maxfield, F.R.; van Meer, G. Cholesterol, the central lipid of mammalian cells. Curr. Opin. Cell Biol. 2010, 22, 422–429. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, D.; Ashish, K.; Hajra, A.; Qureshi, A.; Ghosh, R.K. Cardiovascular Outcomes of PCSK9 Inhibitors: With Special Emphasis on Its Effect beyond LDL-Cholesterol Lowering. J. Lipids 2018, 2018, 3179201. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, E.J.; Virani, S.S.; Callaway, C.W.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Chiuve, S.E.; Cushman, M.; Delling, F.N.; Deo, R.; et al. Heart Disease and Stroke Statistics-2018 Update: A Report from the American Heart Association. Circulation 2018, 137, e67–e492. [Google Scholar] [CrossRef] [PubMed]
- Mok, H.Y.; von Bergmann, K.; Grundy, S.M. Effects of continuous and intermittent feeding on biliary lipid outputs in man: Application for measurements of intestinal absorption of cholesterol and bile acids. J. Lipid Res. 1979, 20, 389–398. [Google Scholar] [PubMed]
- Ikeda, I. Factors affecting intestinal absorption of cholesterol and plant sterols and stanols. J. Oleo Sci. 2015, 64, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Altmann, S.W.; Davis, H.R., Jr.; Zhu, L.J.; Yao, X.; Hoos, L.M.; Tetzloff, G.; Iyer, S.P.; Maguire, M.; Golovko, A.; Zeng, M.; et al. Niemann-Pick C1 Like 1 protein is critical for intestinal cholesterol absorption. Science 2004, 303, 1201–1204. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.M.; Sawyer, J.K.; Kelley, K.L.; Davis, M.A.; Rudel, L.L. Cholesterol esterification by ACAT2 is essential for efficient intestinal cholesterol absorption: Evidence from thoracic lymph duct cannulation. J. Lipid Res. 2012, 53, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, J.; Hussain, M.M. Intestinal lipid absorption. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E1183–E1194. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Hammer, R.E.; Li-Hawkins, J.; Von Bergmann, K.; Lutjohann, D.; Cohen, J.C.; Hobbs, H.H. Disruption of Abcg5 and Abcg8 in mice reveals their crucial role in biliary cholesterol secretion. Proc. Natl. Acad. Sci. USA 2002, 99, 16237–16242. [Google Scholar] [CrossRef] [PubMed]
- Sugizaki, T.; Watanabe, M.; Horai, Y.; Kaneko-Iwasaki, N.; Arita, E.; Miyazaki, T.; Morimoto, K.; Honda, A.; Irie, J.; Itoh, H. The Niemann-Pick C1 like 1 (NPC1L1) inhibitor ezetimibe improves metabolic disease via decreased liver X receptor (LXR) activity in liver of obese male mice. Endocrinology 2014, 155, 2810–2819. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; York, J.; von Bergmann, K.; Lutjohann, D.; Cohen, J.C.; Hobbs, H.H. Stimulation of cholesterol excretion by the liver X receptor agonist requires ATP-binding cassette transporters G5 and G8. J. Biol. Chem. 2003, 278, 15565–15570. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Steffensen, K.R.; Jiang, Z.Y.; Parini, P.; Gustafsson, J.Å.; Gåfvels, M.; Eggertsen, G. LXRβ activation increases intestinal cholesterol absorption, leading to an atherogenic lipoprotein profile. J. Intern. Med. 2012, 272, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Hui, D.Y. The good side of cholesterol: A requirement for maintenance of intestinal integrity. J. Lipid Res. 2017, 58, 1935–1936. [Google Scholar] [CrossRef] [PubMed]
- Adams, C.M.; Reitz, J.; De Brabander, J.K.; Feramisco, J.D.; Li, L.; Brown, M.S.; Goldstein, J.L. Cholesterol and 25-hydroxycholesterol inhibit activation of SREBPs by different mechanisms, both involving SCAP and Insigs. J. Biol. Chem. 2004, 279, 52772–52780. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.S.; Radhakrishnan, A.; Goldstein, J.L. Retrospective on Cholesterol Homeostasis: The Central Role of Scap. Annu. Rev. Biochem. 2018, 87, 1.1–1.25. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Lee, J.N.; Lee, P.C.W.; Goldstein, J.L.; Brown, M.S.; Ye, J. Sterol-regulated ubiquitination and degradation of Insig-1 creates a convergent mechanism for feedback control of cholesterol synthesis and uptake. Cell Metab. 2006, 3, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.-P.; Li, L.; Goldstein, J.L.; Brown, M.S. Insig required for sterol-mediated inhibition of Scap/SREBP binding to COPII proteins in vitro. J. Biol. Chem. 2005, 280, 26483–26490. [Google Scholar] [CrossRef] [PubMed]
- Sato, K. COPII coat assembly and selective export from the endoplasmic reticulum. J. Biochem. 2004, 136, 755–760. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.-P.; Seemann, J.; Brown, M.S.; Goldstein, J.L. Sterol-regulated transport of SREBPs from endoplasmic reticulum to Golgi: Insig renders sorting signal in Scap inaccessible to COPII proteins. Proc. Natl. Acad. Sci. USA 2007, 104, 6519–6526. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.J.; Sun, L.; Feramisco, J.D.; Brown, M.S.; Goldstein, J.L. Cholesterol addition to ER membranes alters conformation of SCAP, the SREBP escort protein that regulates cholesterol metabolism. Mol. Cell 2002, 10, 237–245. [Google Scholar] [CrossRef]
- Goldstein, J.L.; DeBose-Boyd, R.A.; Brown, M.S. Protein sensors for membrane sterols. Cell 2006, 124, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Liao, H.; Liu, Y.; Lee, T.S.; Zhu, M.; Wang, X.; Stemerman, M.B.; Zhu, Y.; Shyy, J.Y. Sterol-responsive element-binding protein (SREBP) 2 down-regulates ATP-binding cassette transporter A1 in vascular endothelial cells: A novel role of SREBP in regulating cholesterol metabolism. J. Biol Chem. 2004, 279, 48801–48807. [Google Scholar] [CrossRef] [PubMed]
- Rayner, K.J.; Sheedy, F.J.; Esau, C.C.; Hussain, F.N.; Temel, R.E.; Parathath, S.; van Gils, J.M.; Rayner, A.J.; Chang, A.N.; Suarez, Y.; et al. Antagonism of miR-33 in mice promotes reverse cholesterol transport and regression of atherosclerosis. J. Clin. Investig. 2011, 121, 2921–2931. [Google Scholar] [CrossRef] [PubMed]
- Rayner, K.J.; Suárez, Y.; Dávalos, A.; Parathath, S.; Fitzgerald, M.L.; Tamehiro, N.; Fisher, E.A.; Moore, K.J.; Fernández-Hernando, C. MiR-33 contributes to the regulation of cholesterol homeostasis. Science 2010, 328, 1570–1573. [Google Scholar] [CrossRef] [PubMed]
- Feramisco, J.D.; Radhakrishnan, A.; Reitz, J.; Brown, M.S.; Goldstein, J.L. Intramembrane aspartic acid in SCAP protein governs cholesterol-induced conformational change. Proc. Natl. Acad. Sci. USA 2005, 102, 3242–3247. [Google Scholar] [CrossRef] [PubMed]
- Cook, E.C.; Nelson, J.K.; Sorrentino, V.; Koenis, D.; Moeton, M.; Scheij, S.; Ottenhoff, R.; Bleijlevens, B.; Loregger, A.; Zelcer, N. Identification of the ER-resident E3 ubiquitin ligase RNF145 as a novel LXR-regulated gene. PLoS ONE 2017, 12, e0172721. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Rajbhandari, P.; Priest, C.; Sandhu, J.; Wu, X.; Temel, R.; Castrillo, A.; de Aguiar Vallim, T.Q.; Sallam, T.; Tontonoz, P. Inhibition of cholesterol biosynthesis through RNF145-dependent ubiquitination of SCAP. eLife 2017, 6, e28766. [Google Scholar] [CrossRef] [PubMed]
- Alphonse, P.A.; Jones, P.J. Revisiting Human Cholesterol Synthesis and Absorption: The Reciprocity Paradigm and its Key Regulators. Lipids 2016, 51, 519–536. [Google Scholar] [CrossRef] [PubMed]
- Vaklavas, C.; Chatzizisis, Y.S.; Ziakas, A.; Zamboulis, C.; Giannoglou, G.D. Molecular basis of statin-associated myopathy. Atherosclerosis 2009, 202, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Berger, S.; Raman, G.; Vishwanathan, R.; Jacques, P.F.; Johnson, E.J. Dietary cholesterol and cardiovascular disease: A systematic review and meta-analysis. Am. J. Clin. Nutr. 2015, 102, 276–294. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.S.; Goldstein, J.L. Cholesterol feedback: From Schoenheimer’s bottle to Scap’s MELADL. J. Lipid Res. 2009, 50, S15–S27. [Google Scholar] [CrossRef] [PubMed]
- Sever, N.; Song, B.L.; Yabe, D.; Goldstein, J.L.; Brown, M.S.; DeBose-Boyd, R.A. Insig-dependent ubiquitination and degradation of mammalian 3-hydroxy-3-methylglutaryl-CoA reductase stimulated by sterols andgeranylgeraniol. J. Biol. Chem. 2003, 278, 52479–52490. [Google Scholar] [CrossRef] [PubMed]
- Jo, Y.; Lee, P.C.; Sguigna, P.V.; DeBose-Boyd, R.A. Sterol-induced degradation of HMG-CoA reductase depends on interplay of two Insigs and two ubiquitin ligases, gp78 and Trc8. Proc. Natl. Acad. Sci. USA 2011, 108, 20503–20508. [Google Scholar] [CrossRef] [PubMed]
- Song, B.L.; Sever, N.; DeBose-Boyd, R.A. Gp78, a membrane-anchored ubiquitin ligase, associates with Insig-1 and couples sterol-regulated ubiquitination to degradation of HMG CoA reductase. Mol. Cell 2005, 19, 829–840. [Google Scholar] [CrossRef] [PubMed]
- Hartman, I.Z.; Liu, P.; Zehmer, J.K.; Luby-Phelps, K.; Jo, Y.; Anderson, R.G.; DeBose-Boyd, R.A. Sterol-induced dislocation of 3-hydroxy-3-methylglutaryl coenzyme A reductase from endoplasmic reticulum membranes into the cytosol through a subcellular compartment resembling lipid droplets. J. Biol. Chem. 2010, 285, 19288–19298. [Google Scholar] [CrossRef] [PubMed]
- Elsabrouty, R.; Jo, Y.; Dinh, T.T.; DeBose-Boyd, R.A. Sterol-induced dislocation of 3-hydroxy-3-methylglutaryl coenzyme A reductase from membranes of permeabilized cells. Mol. Biol. Cell 2013, 24, 3300–3308. [Google Scholar] [CrossRef] [PubMed]
- DeBose-Boyd, R.A. Feedback regulation of cholesterol synthesis: Sterol-accelerated ubiquitination and degradation of HMG CoA reductase. Cell Res. 2008, 18, 609–621. [Google Scholar] [CrossRef] [PubMed]
- Radhakrishnan, A.; Ikeda, Y.; Kwon, H.J.; Brown, M.S.; Goldstein, J.L. Sterol-regulated transport of SREBPs from endoplasmic reticulum to Golgi: Oxysterols block transport by binding to Insig. Proc. Natl. Acad. Sci. USA 2007, 104, 6511–6518. [Google Scholar] [CrossRef] [PubMed]
- Song, B.L.; Javitt, N.B.; DeBose-Boyd, R.A. Insig-mediated degradation of HMG CoA reductase stimulated by lanosterol, an intermediate in the synthesis of cholesterol. Cell Metab. 2005, 1, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Lange, Y.; Ory, D.S.; Ye, J.; Lanier, M.H.; Hsu, F.F.; Steck, T.L. Effectors of rapid homeostatic responses of endoplasmic reticulum cholesterol and 3-hydroxy-3-methylglutaryl-CoA reductase. J. Biol. Chem. 2008, 283, 1445–1455. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, L.J.; Brown, A.J. Controlling cholesterol synthesis beyond 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR). J. Biol. Chem. 2013, 288, 18707–18715. [Google Scholar] [CrossRef] [PubMed]
- Sirtori, C.R. The pharmacology of statins. Pharmacol. Res. 2014, 88, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Lamon-Fava, S. Statins and lipid metabolism: An update. Curr. Opin. Lipidol. 2013, 24, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Scandinavian Simvastatin Study Group. Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: The Scandinavian Simvastatin Survival Study (4S). Lancet 1994, 344, 1383–1389. [Google Scholar] [CrossRef]
- Taylor, F.; Huffman, M.D.; Macedo, A.F.; Moore, T.H.; Burke, M.; Davey Smith, G.; Ward, K.; Ebrahim, S. Statins for the primary prevention of cardiovascular disease. Cochrane Database Syst. Rev. 2013, 1, CD004816. [Google Scholar] [CrossRef] [PubMed]
- Howe, V.; Chua, N.K.; Stevenson, J.; Brown, A.J. The Regulatory Domain of Squalene Monooxygenase Contains a Re-entrant Loop and Senses Cholesterol via a Conformational Change. J. Biol. Chem. 2015, 290, 27533–27544. [Google Scholar] [CrossRef] [PubMed]
- Gill, S.; Stevenson, J.; Kristiana, I.; Brown, A.J. Cholesterol-dependent degradation of squalene monooxygenase, a control point in cholesterol synthesis beyond HMG-CoA reductase. Cell Metab. 2011, 13, 260–273. [Google Scholar] [CrossRef] [PubMed]
- Zelcer, N.; Sharpe, L.J.; Loregger, A.; Kristiana, I.; Cook, E.C.; Phan, L.; Stevenson, J.; Brown, A.J. The E3 ubiquitin ligase MARCH6 degrades squalene monooxygenase and affects 3-hydroxy-3-methyl-glutaryl coenzyme A reductase and the cholesterol synthesis pathway. Mol. Cell. Biol. 2014, 34, 1262–1270. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, A.J.; Lamarche, B.; Lemelin, V.; Hoos, L.; Benjannet, S.; Seidah, N.G.; Davis, H.R., Jr.; Couture, P. Atorvastatin increases intestinal expression of NPC1L1 in hyperlipidemic men. J. Lipid Res. 2011, 52, 558–565. [Google Scholar] [CrossRef] [PubMed]
- Quintão, E.; Grundy, S.M.; Ahrens, E.H., Jr. Effects of dietary cholesterol on the regulation of total body cholesterol in man. J. Lipid Res. 1971, 12, 233–247. [Google Scholar] [PubMed]
- Maranhão, R.C.; Quintão, E.C. Long term steroid metabolism balance studies in subjects on cholesterol-free and cholesterol-rich diets: Comparison between normal and hypercholesterolemic individuals. J. Lipid Res. 1983, 24, 167–173. [Google Scholar] [PubMed]
- Duane, W.C. Effects of lovastatin and dietary cholesterol on sterol homeostasis in healthy human subjects. J. Clin. Investig. 1993, 92, 911–918. [Google Scholar] [CrossRef] [PubMed]
- Nestel, P.J.; Poyser, A. Changes in cholesterol synthesis and excretion when cholesterol intake is increased. Metabolism 1976, 25, 1591–1599. [Google Scholar] [CrossRef]
- McNamara, D.J.; Kolb, R.; Parker, T.S.; Batwin, H.; Samuel, P.; Brown, C.D.; Ahrens, E.H., Jr. Heterogeneity of cholesterol homeostasis in man. Response to changes in dietary fat quality and cholesterol quantity. J. Clin. Investig. 1987, 79, 1729–1739. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.Q. Regulation of intestinal cholesterol absorption. Annu. Rev. Physiol. 2007, 69, 221–248. [Google Scholar] [CrossRef] [PubMed]
- Chiang, J.Y. Bile acid metabolism and signaling. Compr. Physiol. 2013, 3, 1191–1212. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, J.L.; Brown, M.S. The LDL pathway in human fibroblasts: A Receptor-mediated mechanism for the regulation of cholesterol metabolism. Curr. Top. Cell. Regul. 1976, 11, 147–148. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, J.L.; Brown, M.S. The LDL receptor. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Poirier, S.; Mayer, G.; Poupon, V.; McPherson, P.S.; Desjardins, R.; Ly, K.; Asselin, M.C.; Day, R.; Duclos, F.J.; Witmer, M.; et al. Dissection of the endogenous cellular pathways of PCSK9-induced low density lipoprotein receptor degradation: Evidence for an intracellular route. J. Biol. Chem. 2009, 284, 28856–28864. [Google Scholar] [CrossRef] [PubMed]
- Abifadel, M.; Varret, M.; Rabès, J.P.; Allard, D.; Ouguerram, K.; Devillers, M.; Cruaud, C.; Benjannet, S.; Wickham, L.; Erlich, D.; et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat. Genet. 2003, 34, 154–156. [Google Scholar] [CrossRef] [PubMed]
- Zelcer, N.; Hong, C.; Boyadjian, R.; Tontonoz, P. LXR regulates cholesterol uptake through Idol-dependent ubiquitination of the LDL receptor. Science 2009, 325, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.; Duit, S.; Jalonen, P.; Out, R.; Scheer, L.; Sorrentino, V.; Boyadjian, R.; Rodenburg, K.W.; Foley, E.; Korhonen, L.; et al. The E3 ubiquitin ligase IDOL induces the degradation of the low density lipoprotein receptor family members VLDLR and ApoER2. J. Biol. Chem. 2010, 285, 19720–19726. [Google Scholar] [CrossRef] [PubMed]
- Bhakdi, S.; Dorweiler, B.; Kirchmann, R.; Torzewski, J.; Weise, E.; Tranum-Jensen, J.; Walev, I.; Wieland, E. On the pathogenesis of atherosclerosis: Enzymatic transformation of human low density lipoprotein to an atherogenic moiety. J. Exp. Med. 1995, 182, 1959–1971. [Google Scholar] [CrossRef] [PubMed]
- Parthasarathy, S.; Raghavamenon, A.; Garelnabi, M.O.; Santanam, N. Oxidized low-density lipoprotein. Methods Mol. Biol. 2010, 610, 403–417. [Google Scholar] [CrossRef] [PubMed]
- Arjuman, A.; Chandra, N.C. LOX-1: A potential target for therapy in atherosclerosis; an in vitro study. Int. J. Biochem. Cell Biol. 2017, 91, 65–80. [Google Scholar] [CrossRef] [PubMed]
- Sacks, F.M.; Jensen, M.K. From High-Density Lipoprotein Cholesterol to Measurements of Function: Prospects for the Development of Tests for High-Density Lipoprotein Functionality in Cardiovascular Disease. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 487–499. [Google Scholar] [CrossRef] [PubMed]
- Glomset, J.A. The plasma lecithins:cholesterol acyltransferase reaction. J. Lipid Res. 1968, 9, 155–167. [Google Scholar] [PubMed]
- Karathanasis, S.K.; Freeman, L.A.; Gordon, S.M.; Remaley, A.T. The Changing Face of HDL and the Best Way to Measure It. Clin. Chem. 2017, 63, 196–210. [Google Scholar] [CrossRef] [PubMed]
- Brufau, G.; Groen, A.K.; Kuipers, F. Reverse cholesterol transport revisited: Contribution of biliary versus intestinal cholesterol excretion. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1726–1733. [Google Scholar] [CrossRef] [PubMed]
- Olkkonen, V.M. Macrophage oxysterols and their binding proteins: Roles in atherosclerosis. Curr. Opin. Lipidol. 2012, 23, 462–470. [Google Scholar] [CrossRef] [PubMed]
- Huwait, E.A.; Singh, N.N.; Michael, D.R.; Davies, T.S.; Moss, J.W.; Ramji, D.P. Protein Kinase C is Involved in the Induction of ATP-Binding Cassette Transporter A1 Expression by Liver X Receptor/Retinoid X Receptor Agonist in Human Macrophages. J. Cell. Physiol. 2015, 116, 2032–2038. [Google Scholar] [CrossRef]
- Monzel, J.V.; Budde, T.; Meyer Zu Schwabedissen, H.E.; Schwebe, M.; Bien-Möller, S.; Lütjohann, D.; Kroemer, H.K.; Jedlitschky, G.; Grube, M. Doxorubicin enhances oxysterol levels resulting in a LXR-mediated upregulation of cardiac cholesterol transporters. Biochem. Pharmacol. 2017, 144, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Oram, J.F.; Vaughan, A.M. ABCA1-mediated transport of cellular cholesterol and phospholipids to HDL apolipoproteins. Curr. Opin. Lipidol. 2000, 11, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Costet, P.; Luo, Y.; Wang, N.; Tall, A.R. Sterol-dependent transactivation of the ABC1 promoter by the liver X receptor/retinoid X receptor. J. Biol. Chem. 2000, 275, 28240–28245. [Google Scholar] [CrossRef] [PubMed]
- Qian, H.; Zhao, X.; Cao, P.; Lei, J.; Yan, N.; Gong, X. Structure of the Human Lipid Exporter ABCA1. Cell 2017, 169, 1228–1239.e10. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, M.L.; Morris, A.L.; Chroni, A.; Mendez, A.J.; Zannis, V.I.; Freeman, M.W. ABCA1 and amphipathic apolipoproteins form high-affinity molecular complexes required for cholesterol efflux. J. Lipid Res. 2004, 45, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, Y.; Hayashi, M.; Abe-Dohmae, S.; Yokoyama, S. Apolipoprotein A-I activates protein kinase C alpha signaling to phosphorylate and stabilize ATP binding cassette transporter A1 for the high density lipoprotein assembly. J. Biol. Chem. 2003, 278, 47890–47897. [Google Scholar] [CrossRef] [PubMed]
- Casteleijn, M.G.; Parkkila, P.; Viitala, T.; Koivuniemi, A. Interaction of lecithin:cholesterol acyltransferase with lipid surfaces and apolipoprotein A-I-derived peptides. J. Lipid Res. 2018, 59, 670–683. [Google Scholar] [CrossRef] [PubMed]
- Jonas, A. Lecithin cholesterol acyltransferase. Biochim. Biophys. Acta 2000, 1529, 245–256. [Google Scholar] [CrossRef]
- Gunawardane, R.N.; Fordstrom, P.; Piper, D.E.; Masterman, S.; Siu, S.; Liu, D.; Brown, M.; Lu, M.; Tang, J.; Zhang, R.; et al. Agonistic Human Antibodies Binding to Lecithin-Cholesterol Acyltransferase Modulate High Density Lipoprotein Metabolism. J. Biol. Chem. 2016, 291, 2799–2811. [Google Scholar] [CrossRef] [PubMed]
- Peelman, F.; Verschelde, J.L.; Vanloo, B.; Ampe, C.; Labeur, C.; Tavernier, J.; Vandekerckhove, J.; Rosseneu, M. Effects of natural mutations in lecithin:cholesterol acyltransferase on the enzyme structure and activity. J. Lipid Res. 1999, 40, 59–69. [Google Scholar] [PubMed]
- Piper, D.E.; Romanow, W.G.; Gunawardane, R.N.; Fordstrom, P.; Masterman, S.; Pan, O.; Thibault, S.T.; Zhang, R.; Meininger, D.; Schwarz, M.; et al. The high-resolution crystal structure of human LCAT. J. Lipid Res. 2015, 56, 1711–1719. [Google Scholar] [CrossRef] [PubMed]
- Terasaka, N.; Wang, N.; Yvan-Charvet, L.; Tall, A.R. High-density lipoprotein protects macrophages from oxidized low-density lipoprotein-induced apoptosis by promoting efflux of 7-ketocholesterol via ABCG1. Proc. Natl. Acad. Sci. USA 2007, 104, 15093–15098. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Zhang, Y.; Ma, J.; Ling, W.; Xia, M. Adenosine monophosphate activated protein kinase regulates ABCG1-mediated oxysterol efflux from endothelial cells and protects against hypercholesterolemia-induced endothelial dysfunction. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1354–1362. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Xiu, Z.; Wang, R.; Yu, C.; Chi, Y.; Qin, J.; Fu, C.; Matsuura, E.; Liu, Q. The lipid moiety 7-ketocholesteryl-9-carboxynonanoate mediates binding interaction of oxLDL to LOX-1 and upregulates ABCA1 expression through PPARγ. Life Sci. 2017, 177, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yu, C.; Wang, R.; Xu, J.; Chi, Y.; Qin, J.; Liu, Q. The ω-carboxyl group of 7-ketocholesteryl-9-carboxynonanoate mediates the binding of oxLDL to CD36 receptor and enhances caveolin-1 expression in macrophages. Int. J. Biochem. Cell Biol. 2017, 90, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.L.; Windmueller, H.G. Relative contributions by liver and intestine to individual plasma apolipoproteins in the rat. J. Biol. Chem. 1979, 254, 7316–7322. [Google Scholar] [PubMed]
- Gerbod-Giannone, M.C.; Li, Y.; Holleboom, A.; Han, S.; Hsu, L.C.; Tabas, I.; Tall, A.R. TNF-alpha induces ABCA1 through NF-kappaB in macrophages and in phagocytes ingesting apoptotic cells. Proc. Natl. Acad. Sci. USA 2006, 103, 3112–3117. [Google Scholar] [CrossRef] [PubMed]
- Shavva, V.S.; Mogilenko, D.A.; Nekrasova, E.V.; Trulioff, A.S.; Kudriavtsev, I.V.; Larionova, E.E.; Babina, A.V.; Dizhe, E.B.; Missyul, B.V.; Orlov, S.V.; et al. Tumor necrosis factor α stimulates endogenous apolipoprotein A-I expression and secretion by human monocytes and macrophages: Role of MAP-kinases, NF-κB, and nuclear receptors PPARα and LXRs. Mol. Cell. Biochem. 2018. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Kawakami, Y.; Yamauchi, K. Impact of TLR 2, TLR 4-activation on the Expression of ABCA1 and ABCG1 in Raw Cells. Ann. Clin. Lab. Sci. 2017, 47, 436–446. [Google Scholar] [PubMed]
- Takiguchi, S.; Ayaori, M.; Yakushiji, E.; Nishida, T.; Nakaya, K.; Sasaki, M.; Iizuka, M.; Uto-Kondo, H.; Terao, Y.; Yogo, M.; et al. Hepatic Overexpression of Endothelial Lipase Lowers HDL (High-Density Lipoprotein) but Maintains Reverse Cholesterol Transport in Mice: Role of SR-BI (Scavenger Receptor Class B Type I)/ABCA1 (ATP-Binding Cassette Transporter A1)-Dependent Pathways. Arterioscler. Thromb. Vasc. Biol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, S.; Wu, B.J.; Guiney, L.; Barter, P.J.; Rye, K.A. Cholesteryl ester transfer protein and its inhibitors. J. Lipid Res. 2018, 59, 772–783. [Google Scholar] [CrossRef] [PubMed]
- Masson, D.; Jiang, X.C.; Lagrost, L.; Tall, A.R. The role of plasma lipid transfer proteins in lipoprotein metabolism and atherogenesis. J. Lipid Res. 2009, 50, S201–S206. [Google Scholar] [CrossRef] [PubMed]
- Beamer, L.J.; Carroll, S.F.; Eisenberg, D. Crystal structure of human BPI and two bound phospholipids at 2.4 angstrom resolution. Science 1997, 276, 1861–1864. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.C.; Agellon, L.B.; Walsh, A.; Breslow, J.L.; Tall, A. Dietary cholesterol increases transcription of the human cholesteryl ester transfer protein gene in transgenic mice. Dependence on natural flanking sequences. J. Clin. Investig. 1992, 90, 1290–1295. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J.; Connelly, P.W.; Nancoo, D.; Wood, N.; Zhang, Z.J.; Maguire, G.; Quinet, E.; Tall, A.R.; Marcel, Y.L.; McPherson, R. Cholesteryl ester transfer protein and high density lipoprotein responses to cholesterol feeding in men: Relationship to apolipoprotein E genotype. J. Lipid Res. 1993, 34, 437–446. [Google Scholar] [PubMed]
- Qiu, X.; Mistry, A.; Ammirati, M.J.; Chrunyk, B.A.; Clark, R.W.; Cong, Y.; Culp, J.S.; Danley, D.E.; Freeman, T.B.; Geoghegan, K.F.; et al. Crystal structure of cholesteryl ester transfer protein reveals a long tunnel and four bound lipid molecules. Nat. Struct. Mol. Biol. 2007, 14, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Chiang, J.Y. Bile acids as metabolic regulators. Curr. Opin. Gastroenterol. 2015, 31, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Russell, D.W. The enzymes, regulation, and genetics of bile acid synthesis. Annu. Rev. Biochem. 2003, 72, 137–174. [Google Scholar] [CrossRef] [PubMed]
- Li-Hawkins, J.; Gåfvels, M.; Olin, M.; Lund, E.G.; Andersson, U.; Schuster, G.; Björkhem, I.; Russell, D.W.; Eggertsen, G. Cholic acid mediates negative feedback regulation of bile acid synthesis in mice. J. Clin. Investig. 2002, 110, 1191–1200. [Google Scholar] [CrossRef] [PubMed]
- Chiang, J.Y. Bile acids: Regulation of synthesis. J. Lipid Res. 2009, 50, 1955–1966. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Pandak, W.M.; Hylemon, P.B. LXR alpha is the dominant regulator of CYP7A1 transcription. Biochem. Biophys. Res. Commun. 2002, 293, 338–343. [Google Scholar] [CrossRef]
- Davis, R.A.; Miyake, J.H.; Hui, T.Y.; Spann, N.J. Regulation of cholesterol-7alpha-hydroxylase: BAREly missing a SHP. J. Lipid Res. 2002, 43, 533–543. [Google Scholar] [PubMed]
- Tarling, E.J.; Clifford, B.L.; Cheng, J.; Morand, P.; Cheng, A.; Lester, E.; Sallam, T.; Turner, M.; de Aguiar Vallim, T.Q. RNA-binding protein ZFP36L1 maintains posttranscriptional regulation of bile acid metabolism. J. Clin. Investig. 2017, 127, 3741–3754. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, B.; Jones, S.A.; Price, R.R.; Watson, M.A.; McKee, D.D.; Moore, L.B.; Galardi, C.; Wilson, J.G.; Lewis, M.C.; Roth, M.E.; et al. A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol. Cell 2000, 6, 517–526. [Google Scholar] [CrossRef]
- Reeskamp, L.F.; Meessen, E.C.E.; Groen, A.K. Transintestinal cholesterol excretion in humans. Curr. Opin. Lipidol. 2018, 29, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Le May, C.; Berger, J.M.; Lespine, A.L.; Pillot, B.; Prieur, X.; Letessier, E.; Hussain, M.M.; Collet, X.; Cariou, B.; Costet, P. Transintestinal cholesterol excretion is an active metabolic process modulated by PCSK9 and statin involving ABCB1. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1484–1493. [Google Scholar] [CrossRef] [PubMed]
- Vrins, C.L.; Ottenhoff, R.; van den Oever, K.; de Waart, D.R.; Kruyt, J.K.; Zhao, Y.; van Berkel, T.J.; Havekes, L.M.; Aerts, J.M.; van Eck, M.; et al. Trans-intestinal cholesterol efflux is not mediated through high density lipoprotein. J. Lipid Res. 2012, 53, 2017–2023. [Google Scholar] [CrossRef] [PubMed]
- Van der Velde, A.E.; Vrins, C.L.; van den Oever, K.; Seemann, I.; Oude Elferink, R.P.; van Eck, M.; Kuipers, F.; Groen, A.K. Regulation of direct transintestinal cholesterol excretion in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 295, G203–G208. [Google Scholar] [CrossRef] [PubMed]
- Talbot, C.P.J.; Plat, J.; Ritsch, A.; Mensink, R.P. Determinants of cholesterol efflux capacity in humans. Prog. Lipid Res. 2018, 69, 21–32. [Google Scholar] [CrossRef] [PubMed]
- McGillicuddy, F.C.; de la Llera Moya, M.; Hinkle, C.C.; Joshi, M.R.; Chiquoine, E.H.; Billheimer, J.T.; Rothblat, G.H.; Reilly, M.P. Inflammation impairs reverse cholesterol transport in vivo. Circulation 2009, 119, 1135–1145. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, M.; Dillon, E.; Guo, W.; Finucane, O.; McMorrow, A.; Murphy, A.; Lyons, C.; Jones, D.; Ryan, M.; Gibney, M.; et al. High-Density Lipoprotein Proteomic Composition, and not Efflux Capacity, Reflects Differential Modulation of Reverse Cholesterol Transport by Saturated and Monounsaturated Fat Diets. Circulation 2016, 133, 1838–1850. [Google Scholar] [CrossRef] [PubMed]
- Castilho, G.; Okuda, L.S.; Pinto, R.S.; Iborra, R.T.; Nakandakare, E.R.; Santos, C.X.; Laurindo, F.R.; Passarelli, M. ER stress is associated with reduced ABCA-1 protein levels in macrophages treated with advanced glycated albumin—Reversal by a chemical chaperone. Int. J. Biochem. Cell Biol. 2012, 44, 1078–1086. [Google Scholar] [CrossRef] [PubMed]
- De Souza Pinto, R.; Castilho, G.; Paim, B.A.; Machado-Lima, A.; Inada, N.M.; Nakandakare, E.R.; Vercesi, A.E.; Passarelli, M. Inhibition of macrophage oxidative stress prevents the reduction of ABCA-1 transporter induced by advanced glycated albumin. Lipids 2012, 47, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Tang, Y.; Jin, X.; Chen, C.; Lu, Y.; Liu, L.; Shen, C. Metformin Inhibits Advanced Glycation End Products-Induced Inflammatory Response in Murine Macrophages Partly through AMPK Activation and RAGE/NFκB Pathway Suppression. J. Diabetes Res. 2016, 2016, 4847812. [Google Scholar] [CrossRef] [PubMed]
- Iborra, R.T.; Machado-Lima, A.; Okuda, L.S.; Pinto, P.R.; Nakandakare, E.R.; Machado, U.F.; Correa-Giannella, M.L.; Pickford, R.; Woods, T.; Brimble, M.A.; et al. AGE-albumin enhances ABCA1 degradation by ubiquitin-proteasome and lysosomal pathways in macrophages. J. Diabetes Complicat. 2018, 32, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Daffu, G.; Shen, X.; Senatus, L.; Thiagarajan, D.; Abedini, A.; Hurtado Del Pozo, C.; Rosario, R.; Song, F.; Friedman, R.A.; Ramasamy, R.; et al. RAGE Suppresses ABCG1-Mediated Macrophage Cholesterol Efflux in Diabetes. Diabetes 2015, 64, 4046–4060. [Google Scholar] [CrossRef] [PubMed]
- Aleidi, S.M.; Howe, V.; Sharpe, L.J.; Yang, A.; Rao, G.; Brown, A.J.; Gelissen, I.C. The E3 ubiquitin ligases, HUWE1 and NEDD4-1, are involved in the post-translational regulation of the ABCG1 and ABCG4 lipid transporters. J. Biol. Chem. 2015, 290, 24604–24613. [Google Scholar] [CrossRef] [PubMed]
- Aleidi, S.M.; Yang, A.; Sharpe, L.J.; Rao, G.; Cochran, B.J.; Rye, K.A.; Kockx, M.; Brown, A.J.; Gelissen, I.C. The E3 ubiquitin ligase, HECTD1, is involved in ABCA1-mediated cholesterol export from macrophages. Biochim. Biophys. Acta 2018, 1863, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Clayton, Z.S.; Fusco, E.; Kern, M. Egg consumption and heart health: A review. Nutrition 2017, 37, 79–85. [Google Scholar] [CrossRef] [PubMed]
- US Department of Health and Human Services; US Department of Agriculture. 2015–2020 Dietary Guidelines for Americans, 8th ed.; US Department of Health and Human Services: Washington, DC, USA, 2015. Available online: http://www.health.gov/DietaryGuidelines (accessed on 20 May 2018).
- Virtanen, J.K.; Mursu, J.; Virtanen, H.E.; Fogelholm, M.; Salonen, J.T.; Koskinen, T.T.; Voutilainen, S.; Tuomainen, T.P. Associations of egg and cholesterol intakes with carotid intima-media thickness and risk of incident coronary artery disease according to apolipoprotein E phenotype in men: The Kuopio Ischaemic Heart Disease Risk Factor Study. Am. J. Clin. Nutr. 2016, 103, 895–901. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, S.; Gardener, H.; Tiozzo, E.; Ying Kuen, C.; Elkind, M.S.; Sacco, R.L.; Rundek, T. Egg consumption and carotid atherosclerosis in the Northern Manhattan study. Atherosclerosis 2014, 235, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Rhee, E.J.; Ryu, S.; Lee, J.Y.; Lee, S.H.; Cheong, E.; Park, S.E.; Park, C.Y.; Won, Y.S.; Kim, J.M.; Cho, D.S.; et al. The association between dietary cholesterol intake and subclinical atherosclerosis in Korean adults: The Kangbuk Samsung Health Study. J. Clin. Lipidol. 2017, 11, 432–441.e3. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Chang, Y.; Lee, J.E.; Chun, S.; Cho, J.; Sung, E.; Suh, B.S.; Rampal, S.; Zhao, D.; Zhang, Y.; et al. Egg consumption and coronary artery calcification in asymptomatic men and women. Atherosclerosis 2015, 241, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Blesso, C.N.; Fernandez, M.L. Dietary Cholesterol, Serum Lipids, and Heart Disease: Are Eggs Working for or Against You? Nutrients 2018, 10, 426. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.Y.; Xun, P.; Nakamura, Y.; He, K. Egg consumption in relation to risk of cardiovascular disease and diabetes: A systematic review and meta-analysis. Am. J. Clin. Nutr. 2013, 98, 146–159. [Google Scholar] [CrossRef] [PubMed]
- Barraj, L.; Tran, N.; Mink, P. A comparison of egg consumption with other modifiable coronary heart disease lifestyle risk factors: A relative risk apportionment study. Risk Anal. 2009, 29, 401–415. [Google Scholar] [CrossRef] [PubMed]
- Herron, K.L.; Vega-Lopez, S.; Conde, K.; Ramjiganesh, T.; Shachter, N.S.; Fernandez, M.L. Men classified as hypo- or hyperresponders to dietary cholesterol feeding exhibit differences in lipoprotein metabolism. J. Nutr. 2003, 133, 1036–1042. [Google Scholar] [CrossRef] [PubMed]
- García-Otín, A.L.; Cofán, M.; Junyent, M.; Recalde, D.; Cenarro, A.; Pocoví, M.; Ros, E.; Civeira, F. Increased intestinal cholesterol absorption in autosomal dominant hypercholesterolemia and no mutations in the low-density lipoprotein receptor or apolipoprotein B genes. J. Clin. Endocrinol. Metab. 2007, 92, 3667–3673. [Google Scholar] [CrossRef] [PubMed]
- Grundy, S.M. Does Dietary Cholesterol Matter? Curr. Atheroscler. Rep. 2016, 18, 68. [Google Scholar] [CrossRef] [PubMed]
- Ravera, A.; Carubelli, V.; Sciatti, E.; Bonadei, I.; Gorga, E.; Cani, D.; Vizzardi, E.; Metra, M.; Lombardi, C. Nutrition and Cardiovascular Disease: Finding the Perfect Recipe for Cardiovascular Health. Nutrients 2016, 8, 363. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Afonso, M.S.; Machado, R.M.; Lavrador, M.S.; Quintao, E.C.R.; Moore, K.J.; Lottenberg, A.M. Molecular Pathways Underlying Cholesterol Homeostasis. Nutrients 2018, 10, 760. https://doi.org/10.3390/nu10060760
Afonso MS, Machado RM, Lavrador MS, Quintao ECR, Moore KJ, Lottenberg AM. Molecular Pathways Underlying Cholesterol Homeostasis. Nutrients. 2018; 10(6):760. https://doi.org/10.3390/nu10060760
Chicago/Turabian StyleAfonso, Milessa Silva, Roberta Marcondes Machado, Maria Silvia Lavrador, Eder Carlos Rocha Quintao, Kathryn J. Moore, and Ana Maria Lottenberg. 2018. "Molecular Pathways Underlying Cholesterol Homeostasis" Nutrients 10, no. 6: 760. https://doi.org/10.3390/nu10060760
APA StyleAfonso, M. S., Machado, R. M., Lavrador, M. S., Quintao, E. C. R., Moore, K. J., & Lottenberg, A. M. (2018). Molecular Pathways Underlying Cholesterol Homeostasis. Nutrients, 10(6), 760. https://doi.org/10.3390/nu10060760